Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia

 , , , and
, , , and 
Abstract
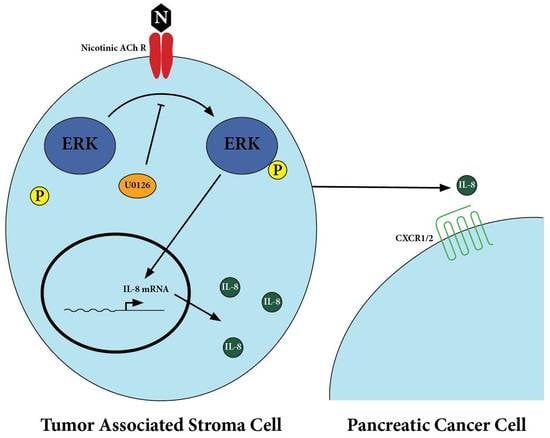
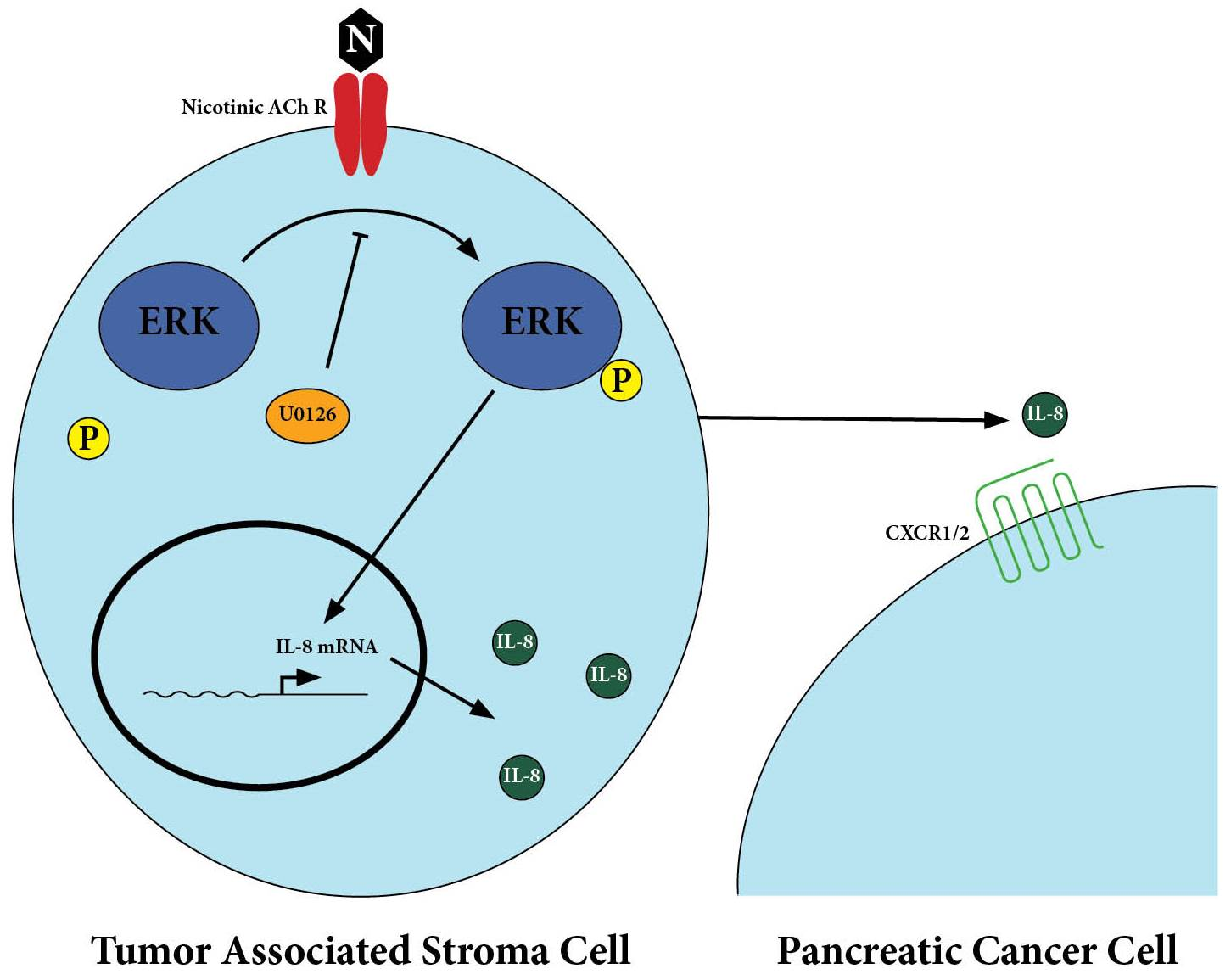
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
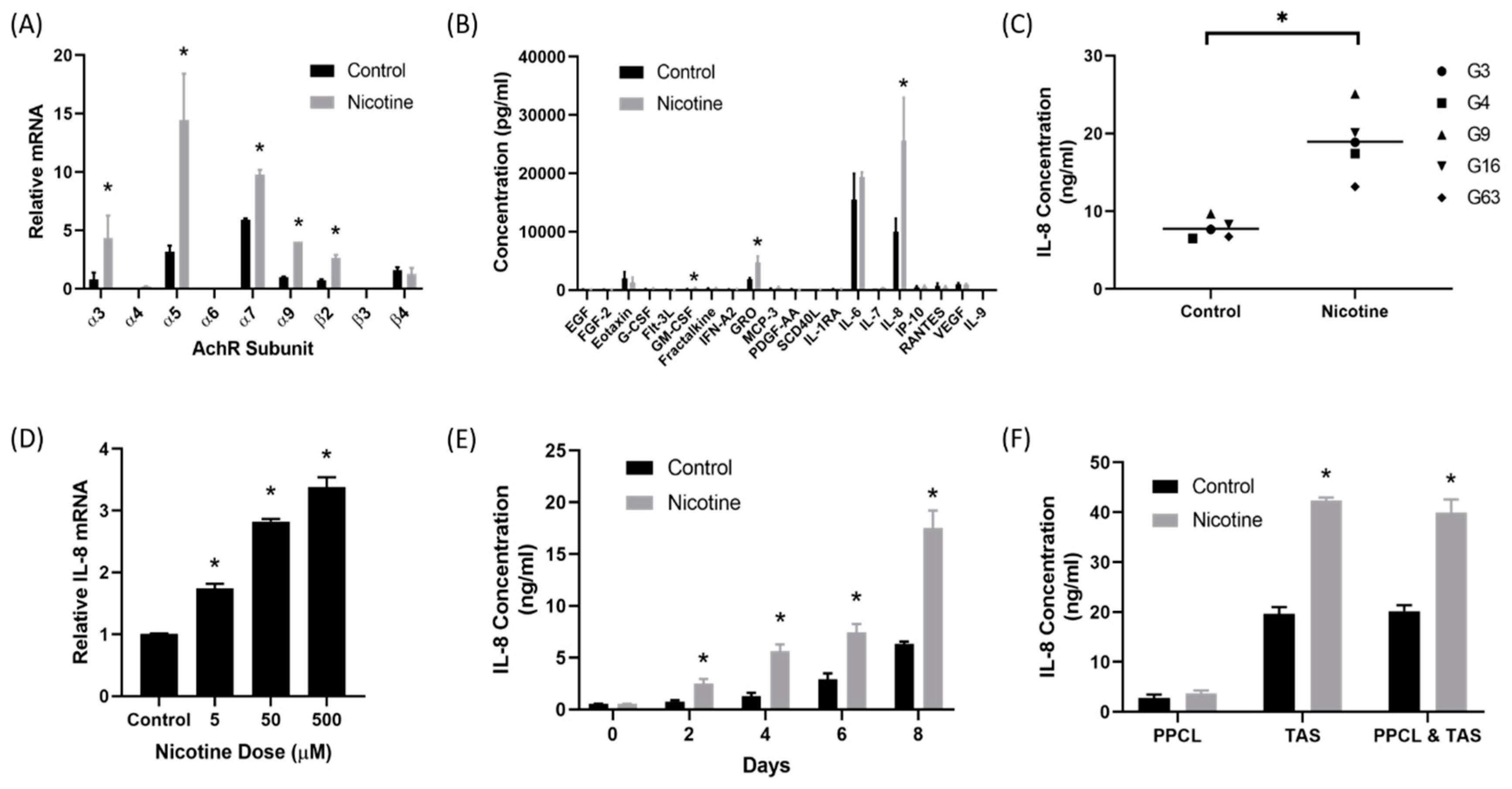
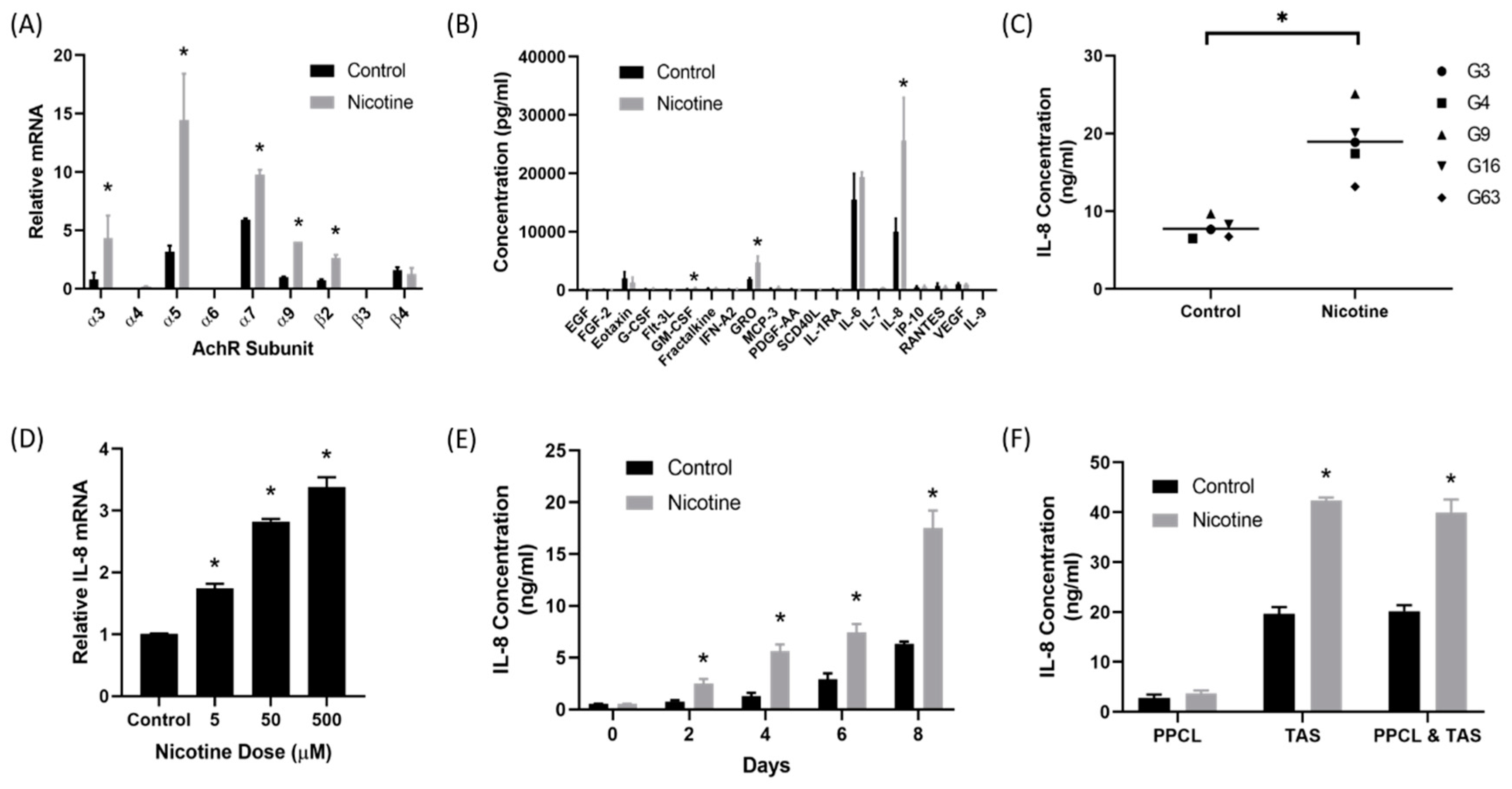
2.1. Nicotine Induced Secretion of IL-8 by Tumor-Associated Stromal Cells
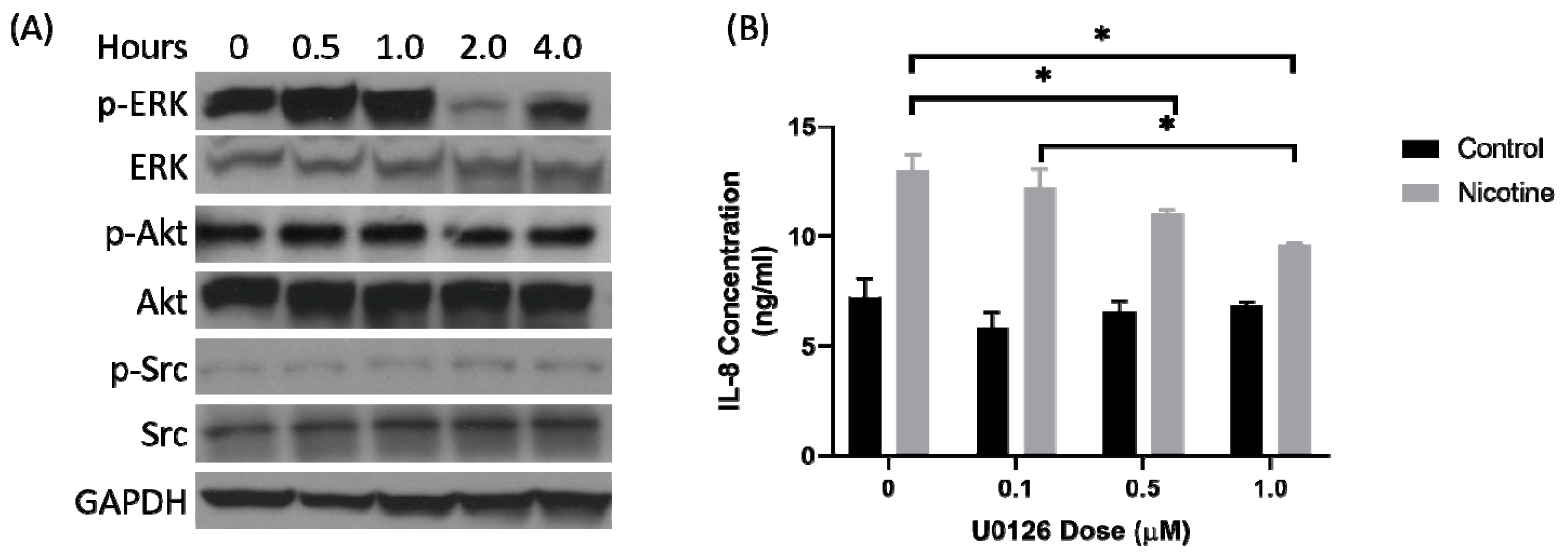
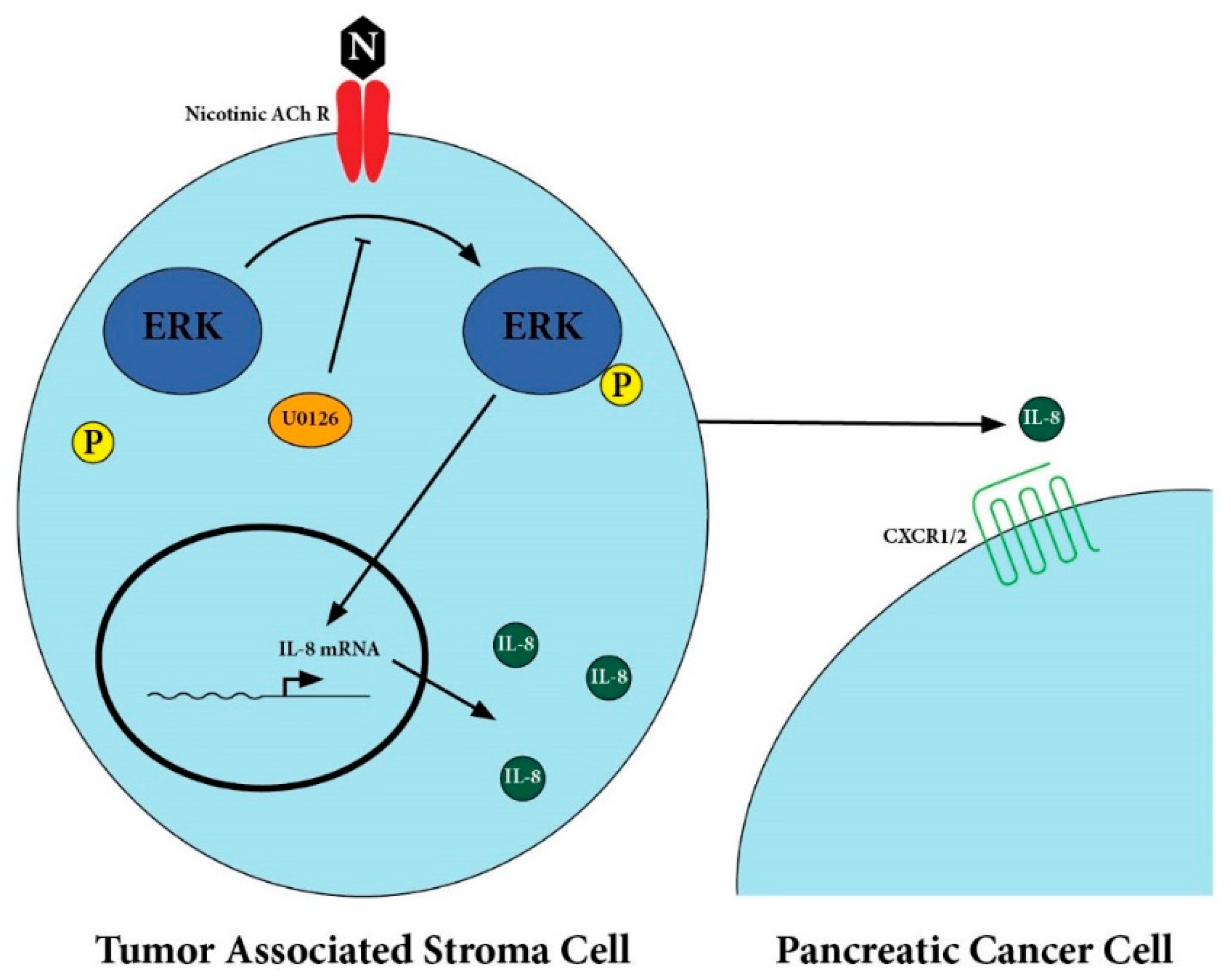
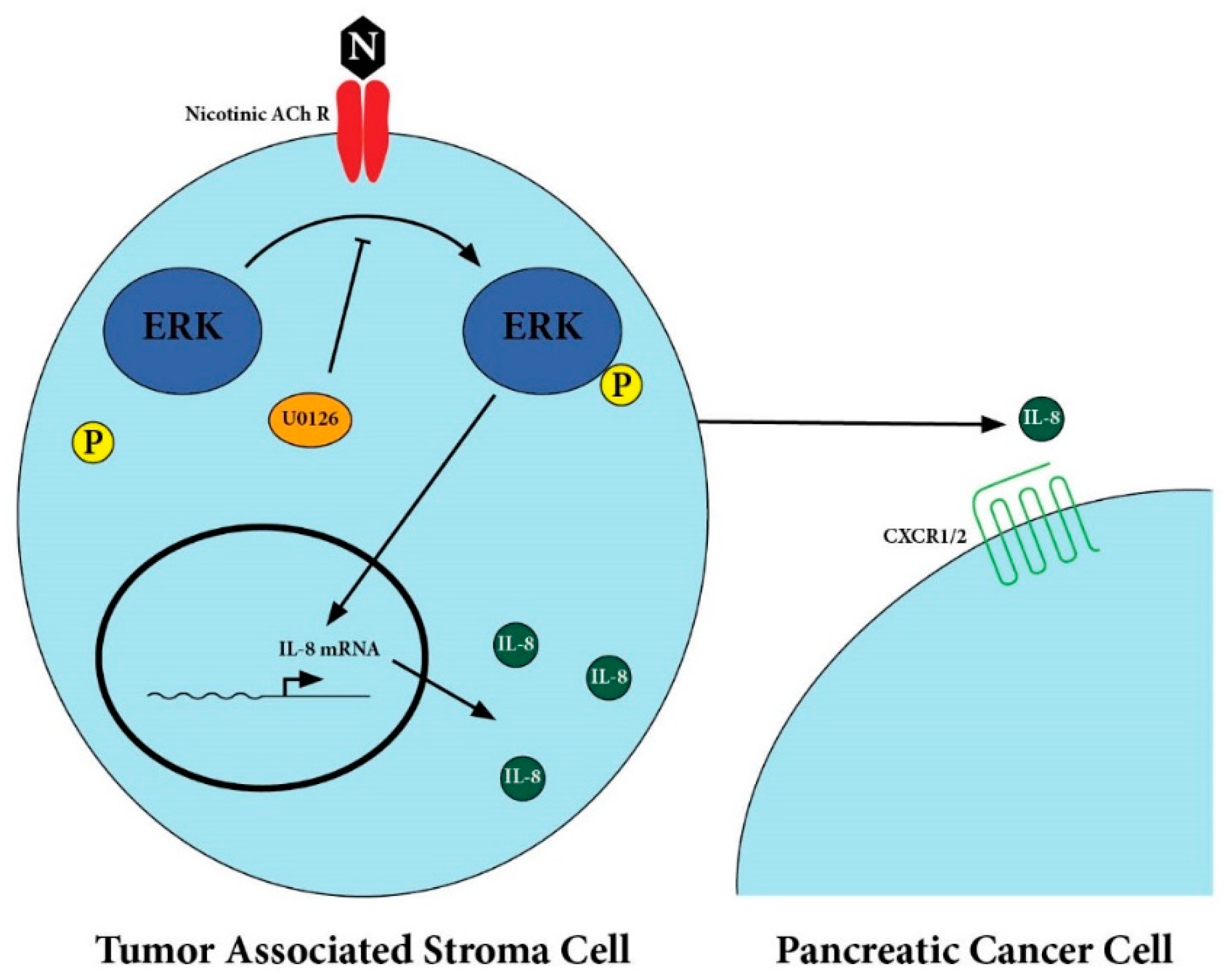
2.2. Secretion of IL-8 in Nicotine-Treated Tumor-Associated Stroma Cells Was Dependent on the ERK Pathway
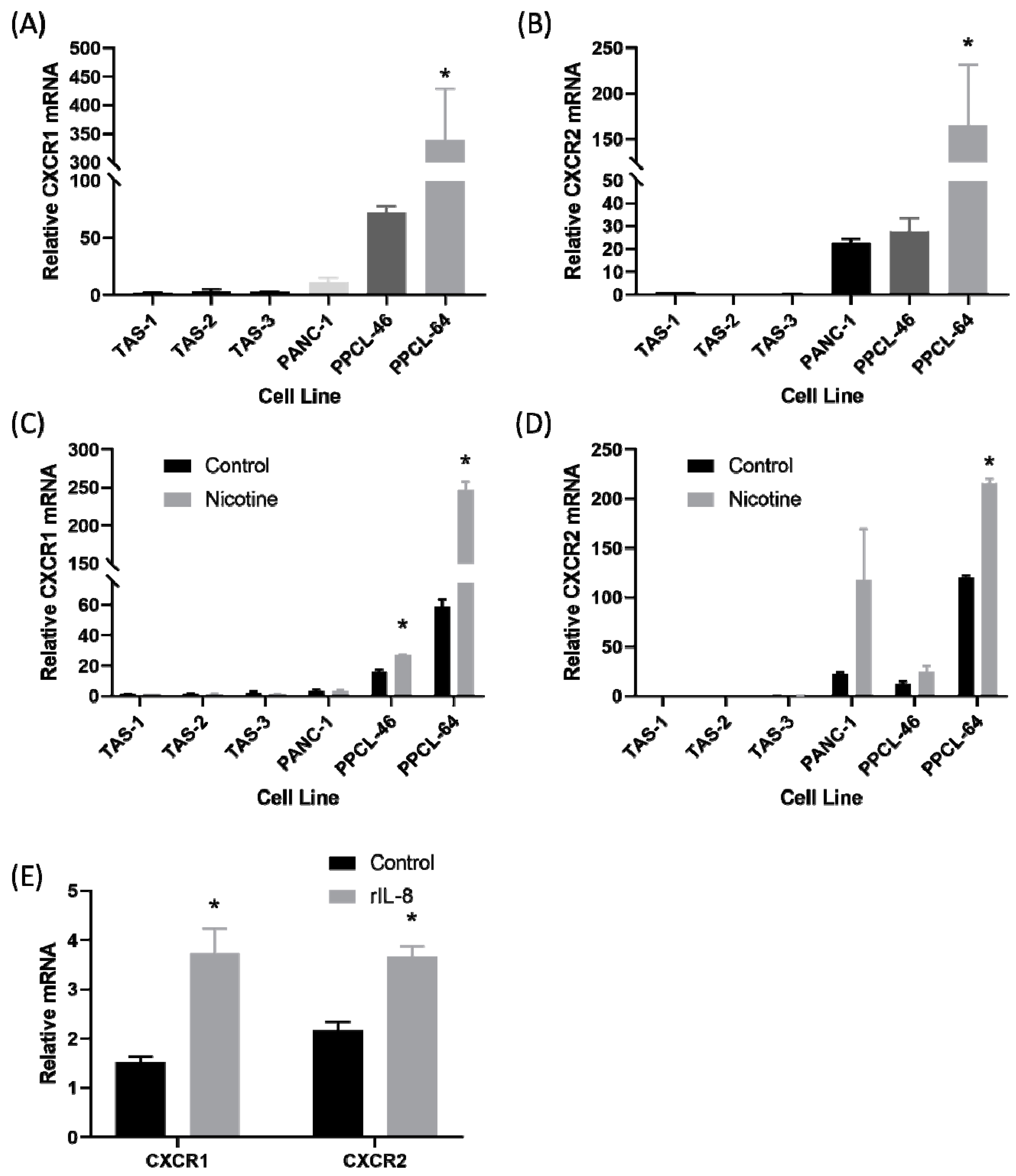
2.3. There Was Upregulation of IL-8 Receptors on Pancreatic Cancer Cells After Treatment with Nicotine
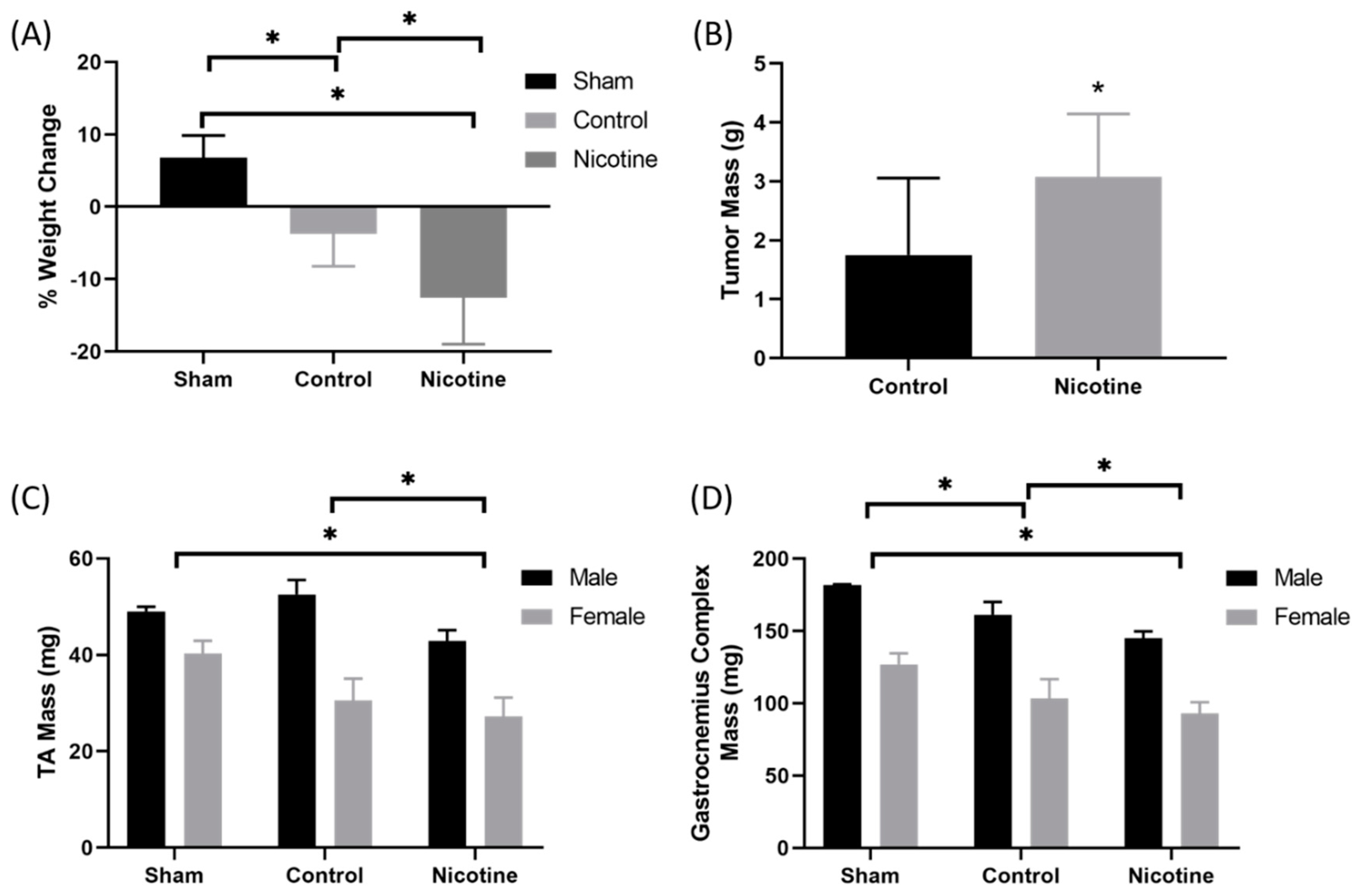
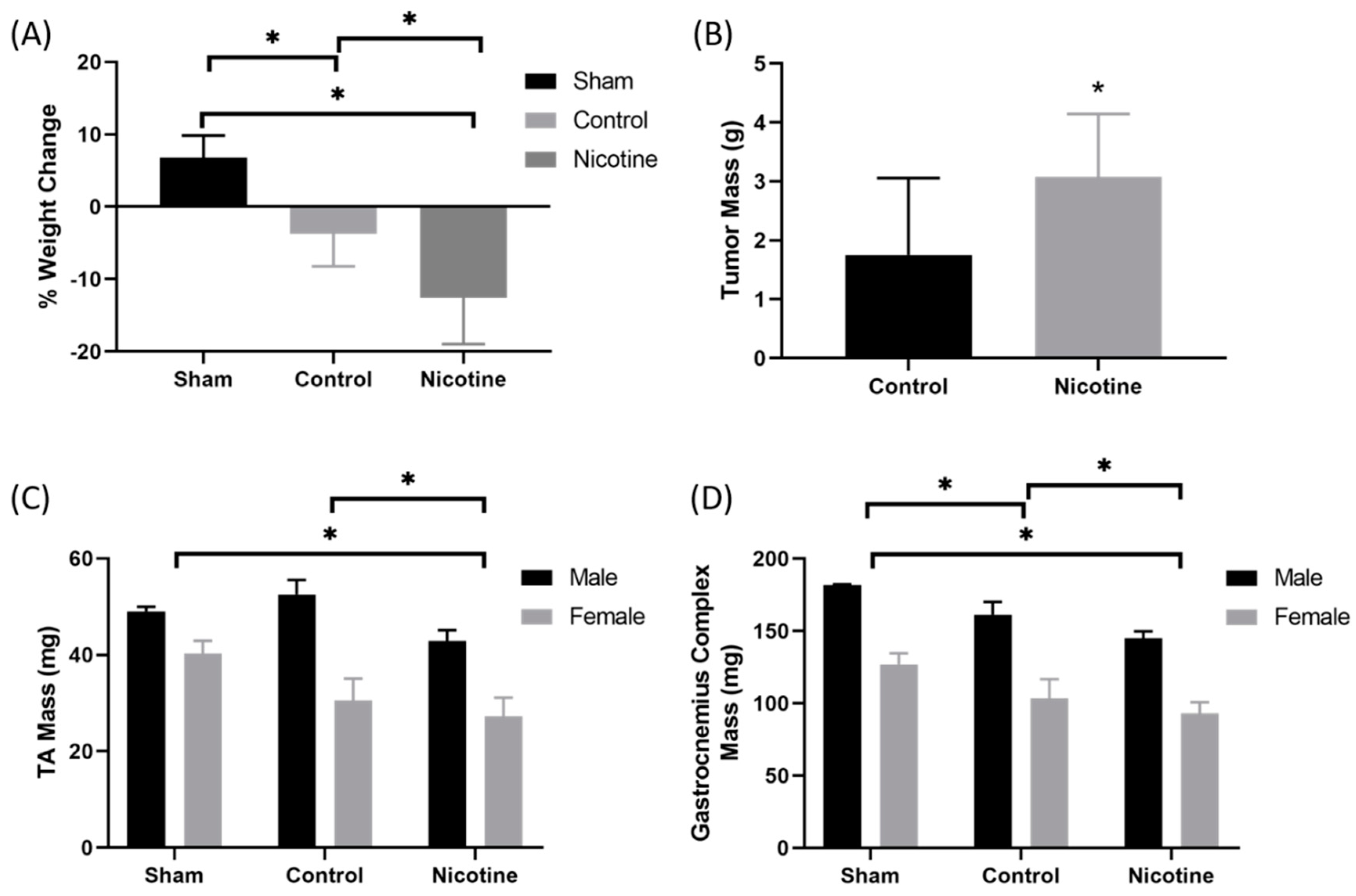
2.4. PDAC-Bearing Mice Treated with Nicotine Had Worsened Cancer-Induced Cachexia
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Quantitative RT-PCR
4.3. ELISA and 41-Plex Protein Assay
4.4. Western Blot Analysis
4.5. Patient-Derived Xenografts
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Howlader, N.N.A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; Chen, H.S.; et al. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Lucenteforte, E.; Silverman, D.T.; Petersen, G.; Bracci, P.M.; Ji, B.T.; Negri, E.; Li, D.; Risch, H.A.; Olson, S.H.; et al. Cigarette smoking and pancreatic cancer: An analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann. Oncol. 2012, 23, 1880–1888. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, H.; Yang, X.; Guo, J. Cigarette smoking and the risk of pancreatic cancer: A case-control study. Med. Oncol. 2014, 31, 184. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Delitto, D.; Zhang, D.; Han, S.; Black, B.S.; Knowlton, A.E.; Vlada, A.C.; Sarosi, G.A.; Behrns, K.E.; Thomas, R.M.; Lu, X.; et al. Nicotine Reduces Survival via Augmentation of Paracrine HGF-MET Signaling in the Pancreatic Cancer Microenvironment. Clin. Cancer Res. 2016, 22, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Sancho, P.; Canamero, M.; Martinelli, P.; Madriles, F.; Michl, P.; Gress, T.; de Pascual, R.; Gandia, L.; Guerra, C.; et al. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 2014, 147, 1119–1133.e1114. [Google Scholar] [CrossRef]
- Bersch, V.P.; Osvaldt, A.B.; Edelweiss, M.I.; Schumacher Rde, C.; Wendt, L.R.; Abreu, L.P.; Blom, C.B.; Abreu, G.P.; Costa, L.; Piccinini, P.; et al. Effect of nicotine and cigarette smoke on an experimental model of intraepithelial lesions and pancreatic adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice. Pancreas 2009, 38, 65–70. [Google Scholar] [CrossRef]
- Hanaki, T.; Horikoshi, Y.; Nakaso, K.; Nakasone, M.; Kitagawa, Y.; Amisaki, M.; Arai, Y.; Tokuyasu, N.; Sakamoto, T.; Honjo, S.; et al. Nicotine enhances the malignant potential of human pancreatic cancer cells via activation of atypical protein kinase C. Biochim. Biophys. Acta 2016, 1860, 2404–2415. [Google Scholar] [CrossRef] [Green Version]
- Momi, N.; Ponnusamy, M.P.; Kaur, S.; Rachagani, S.; Kunigal, S.S.; Chellappan, S.; Ouellette, M.M.; Batra, S.K. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through alpha7nAChR-mediated MUC4 upregulation. Oncogene 2013, 32, 1384–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevino, J.G.; Pillai, S.; Kunigal, S.; Singh, S.; Fulp, W.J.; Centeno, B.A.; Chellappan, S.P. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia 2012, 14, 1102–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, J.; Al-Wadei, H.A.; Schuller, H.M. Chronic nicotine inhibits the therapeutic effects of gemcitabine on pancreatic cancer in vitro and in mouse xenografts. Eur. J. Cancer 2013, 49, 1152–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, K.L.; Delitto, D.; Judge, S.M.; Gerber, M.H.; George, T.J., Jr.; Behrns, K.E.; Hughes, S.J.; Judge, A.R.; Trevino, J.G. Orthotopic Patient-Derived Pancreatic Cancer Xenografts Engraft Into the Pancreatic Parenchyma, Metastasize, and Induce Muscle Wasting to Recapitulate the Human Disease. Pancreas 2017, 46, 813–819. [Google Scholar] [CrossRef] [PubMed]
- St Helen, G.; Havel, C.; Dempsey, D.A.; Jacob, P., 3rd; Benowitz, N.L. Nicotine delivery, retention and pharmacokinetics from various electronic cigarettes. Addiction 2016, 111, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Wang, C.J.; Chao, Y.J.; Chen, H.Y.; Wang, H.C.; Tung, H.L.; Lin, J.T.; Shan, Y.S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Zhang, D.; Wang, S.; Zheng, H.; Wang, X. Association of interleukin-8 with cachexia from patients with low-third gastric cancer. Comp. Funct. Genom. 2009, 212345. [Google Scholar] [CrossRef] [Green Version]
- Krzystek-Korpacka, M.; Matusiewicz, M.; Diakowska, D.; Grabowski, K.; Blachut, K.; Kustrzeba-Wojcicka, I.; Banas, T. Impact of weight loss on circulating IL-1, IL-6, IL-8, TNF-alpha, VEGF-A, VEGF-C and midkine in gastroesophageal cancer patients. Clin. Biochem. 2007, 40, 1353–1360. [Google Scholar] [CrossRef]
- Pfitzenmaier, J.; Vessella, R.; Higano, C.S.; Noteboom, J.L.; Wallace, D., Jr.; Corey, E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer 2003, 97, 1211–1216. [Google Scholar] [CrossRef]
- Cheng, Y.A.; Shiue, L.F.; Yu, H.S.; Hsieh, T.Y.; Tsai, C.C. Interleukin-8 secretion by cultured oral epidermoid carcinoma cells induced with nicotine and/or arecoline treatments. Kaohsiung J. Med. Sci. 2000, 16, 126–133. [Google Scholar]
- Tsunoda, K.; Tsujino, I.; Koshi, R.; Sugano, N.; Sato, S.; Asano, M. Nicotine-Mediated Ca(2+)-Influx Induces IL-8 Secretion in Oral Squamous Cell Carcinoma Cell. J. Cell. Biochem. 2016, 117, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, Y.; Zhou, Z.; Liu, Y.; Bai, Y.; Xing, X.; Wang, X. Nicotine induces the production of IL-1beta and IL-8 via the alpha7 nAChR/NF-kappaB pathway in human periodontal ligament cells: An in vitro study. Cell. Physiol. Biochem. 2014, 34, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.K.; Lee, H.F.; Lin, A.H.; Liu, M.H.; Liu, C.I.; Lee, T.S.; Kou, Y.R. Regulation of Cigarette Smoke Induction of IL-8 in Macrophages by AMP-activated Protein Kinase Signaling. J. Cell. Physiol. 2015, 230, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arcos, I.; Geraghty, P.; Baumlin, N.; Campos, M.; Dabo, A.J.; Jundi, B.; Cummins, N.; Eden, E.; Grosche, A.; Salathe, M.; et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 2016, 71, 1119–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iho, S.; Tanaka, Y.; Takauji, R.; Kobayashi, C.; Muramatsu, I.; Iwasaki, H.; Nakamura, K.; Sasaki, Y.; Nakao, K.; Takahashi, T. Nicotine induces human neutrophils to produce IL-8 through the generation of peroxynitrite and subsequent activation of NF-kappaB. J. Leukoc. Biol. 2003, 74, 942–951. [Google Scholar] [CrossRef]
- Kim, J.H.; Frantz, A.M.; Anderson, K.L.; Graef, A.J.; Scott, M.C.; Robinson, S.; Sharkey, L.C.; O’Brien, T.D.; Dickerson, E.B.; Modiano, J.F. Interleukin-8 promotes canine hemangiosarcoma growth by regulating the tumor microenvironment. Exp. Cell Res. 2014, 323, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Choi, I.; Ning, Y.; Kim, N.Y.; Khatchadourian, V.; Yang, D.; Chung, H.K.; Choi, D.; LaBonte, M.J.; Ladner, R.D.; et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br. J. Cancer 2012, 106, 1833–1841. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Bhardwaj, A.; Arora, S.; Tyagi, N.; Singh, A.P.; Carter, J.E.; Scammell, J.G.; Fodstad, O.; Singh, S. Interleukin-8 is a key mediator of FKBP51-induced melanoma growth, angiogenesis and metastasis. Br. J. Cancer 2015, 112, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Bo, S.; Dianliang, Z.; Hongmei, Z.; Xinxiang, W.; Yanbing, Z.; Xiaobo, L. Association of interleukin-8 gene polymorphism with cachexia from patients with gastric cancer. J. Interferon Cytokine Res. 2010, 30, 9–14. [Google Scholar] [CrossRef]
- Pham, K.; Delitto, D.; Knowlton, A.E.; Hartlage, E.R.; Madhavan, R.; Gonzalo, D.H.; Thomas, R.M.; Behrns, K.E.; George, T.J., Jr.; Hughes, S.J.; et al. Isolation of Pancreatic Cancer Cells from a Patient-Derived Xenograft Model Allows for Practical Expansion and Preserved Heterogeneity in Culture. Am. J. Pathol. 2016, 186, 1537–1546. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Delitto, D.; Zhang, D.; Sorenson, H.L.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Wallet, S.M.; Trevino, J.G.; Hughes, S.J. Primary outgrowth cultures are a reliable source of human pancreatic stellate cells. Lab. Investig. 2015, 95, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Underwood, P.W.; Zhang, D.Y.; Cameron, M.E.; Gerber, M.H.; Delitto, D.; Maduka, M.U.; Cooper, K.J.; Han, S.; Hughes, S.J.; Judge, S.M.; et al. Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia. Cancers 2020, 12, 329. https://doi.org/10.3390/cancers12020329
Underwood PW, Zhang DY, Cameron ME, Gerber MH, Delitto D, Maduka MU, Cooper KJ, Han S, Hughes SJ, Judge SM, et al. Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia. Cancers. 2020; 12(2):329. https://doi.org/10.3390/cancers12020329
Chicago/Turabian StyleUnderwood, Patrick W., Dong Yu Zhang, Miles E. Cameron, Michael H. Gerber, Daniel Delitto, Michael U. Maduka, Kyle J. Cooper, Song Han, Steven J. Hughes, Sarah M. Judge, and et al. 2020. "Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia" Cancers 12, no. 2: 329. https://doi.org/10.3390/cancers12020329
APA StyleUnderwood, P. W., Zhang, D. Y., Cameron, M. E., Gerber, M. H., Delitto, D., Maduka, M. U., Cooper, K. J., Han, S., Hughes, S. J., Judge, S. M., Judge, A. R., & Trevino, J. G. (2020). Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia. Cancers, 12(2), 329. https://doi.org/10.3390/cancers12020329