TMEFF2: A Transmembrane Proteoglycan with Multifaceted Actions in Cancer and Disease


Abstract
:Simple Summary
Abstract

1. Introduction
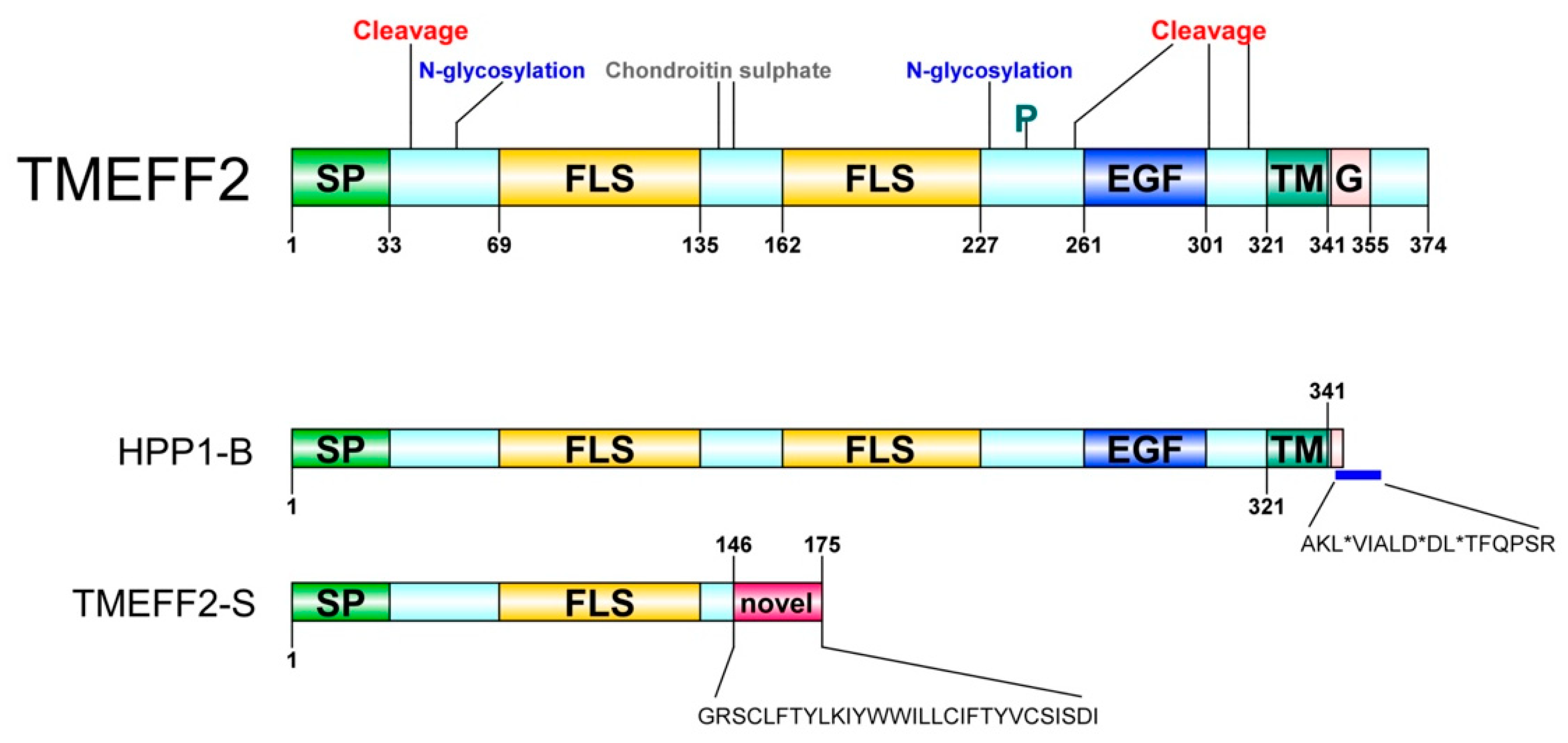
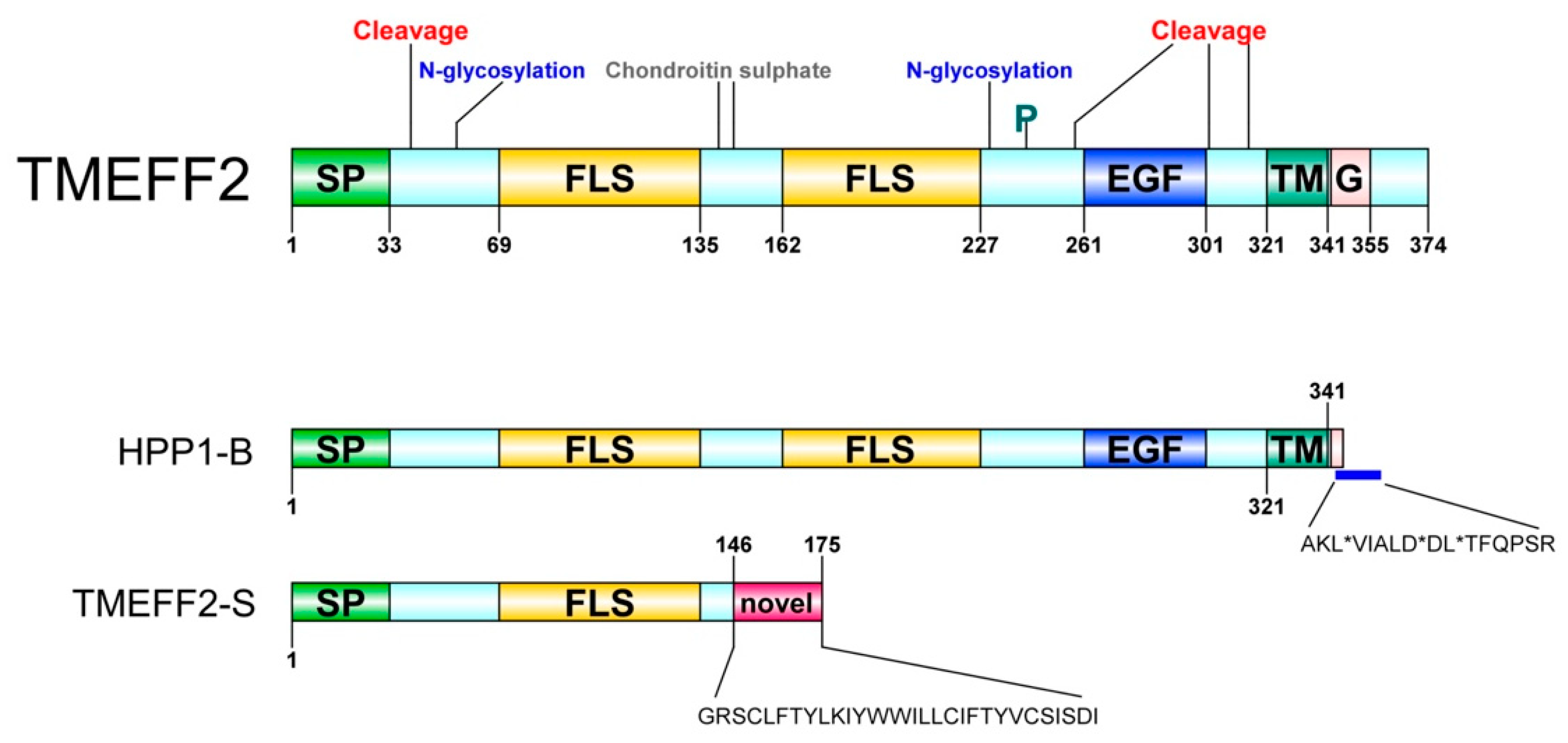
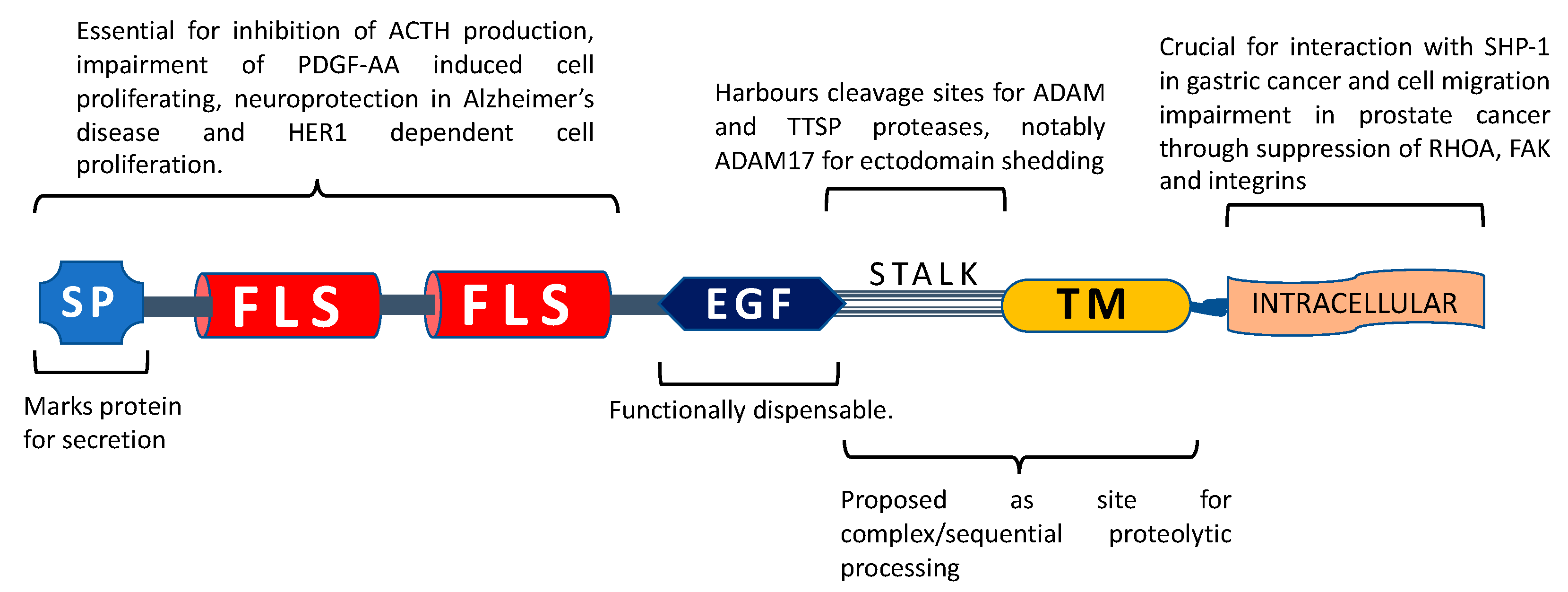
2. Identification and Characterization
3. TMEFF2 Promoter Methylation in Cancer
4. Tissue Distribution
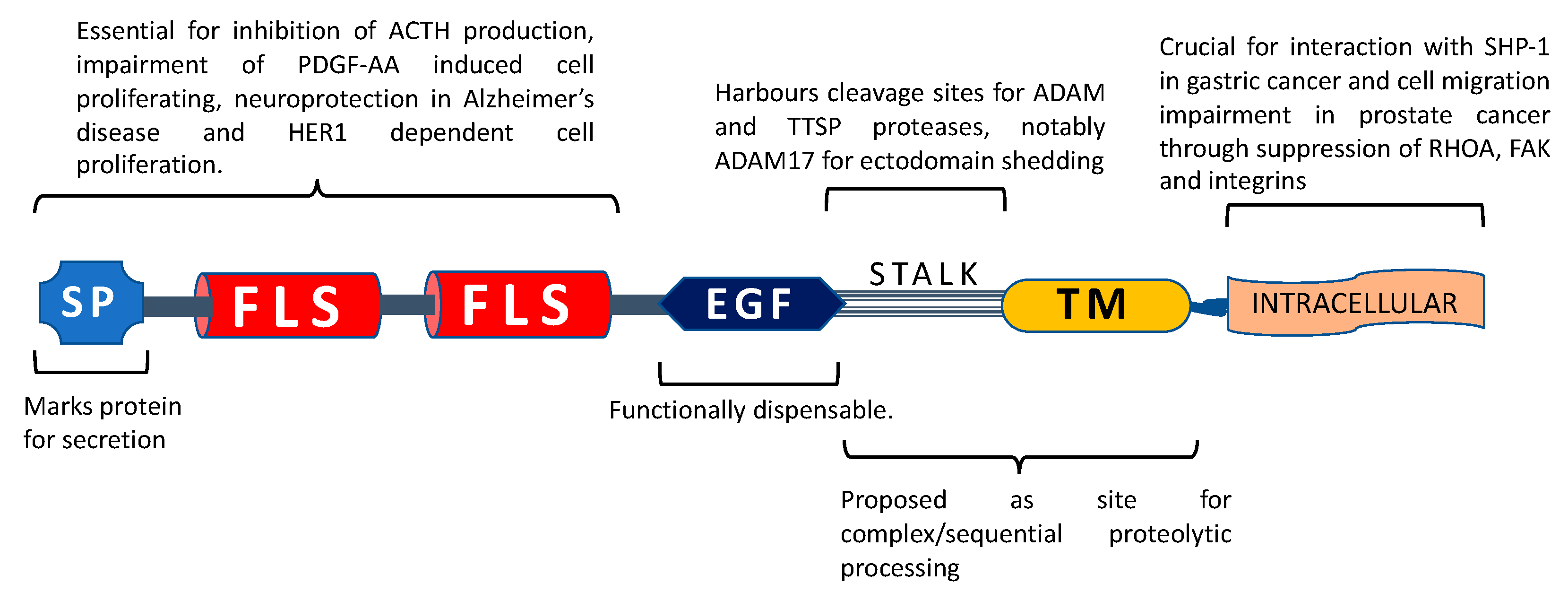
5. Proposed Functions
6. TMEFF2 in Prostate Cancer
7. TMEFF2 in Gastric and Colorectal Cancers
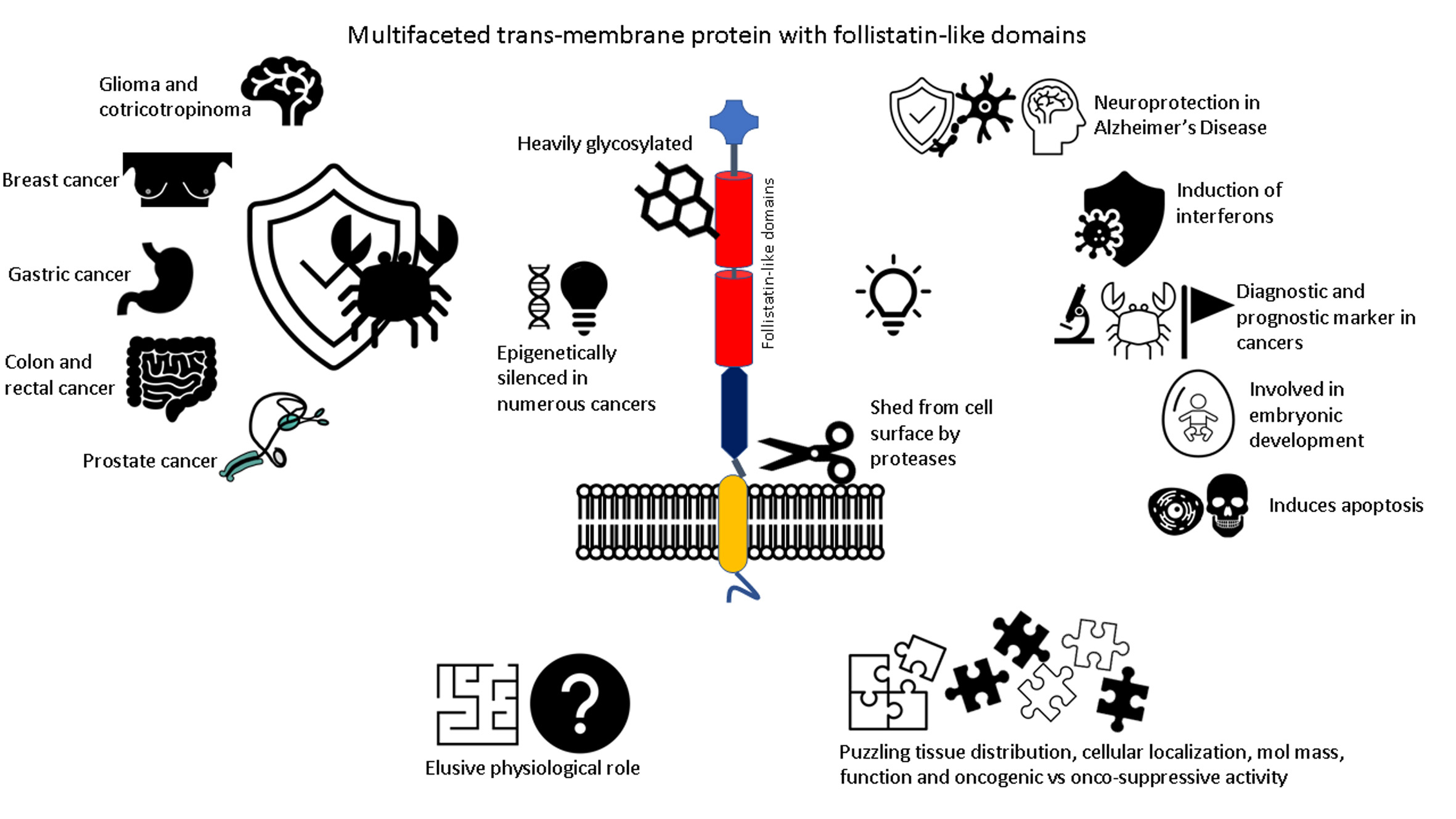
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res 2020. [Google Scholar] [CrossRef] [PubMed]
- HGNC Database, HUGO Gene Nomenclature Committee (HGNC), European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus. Available online: https://www.genenames.org/ (accessed on 16 December 2020).
- Liang, G.; Robertson, K.D.; Talmadge, C.; Sumegi, J.; Jones, P.A. The Gene for a Novel Transmembrane Protein Containing Epidermal Growth Factor and Follistatin Domains Is Frequently Hypermethylated in Human Tumor Cells. Cancer Res. 2000, 60, 4907–4912. [Google Scholar] [PubMed]
- Horie, M.; Mitsumoto, Y.; Kyushiki, H.; Kanemoto, N.; Watanabe, A.; Taniguchi, Y.; Nishino, N.; Okamoto, T.; Kondo, M.; Mori, T.; et al. Identification and Characterization of TMEFF2, a Novel Survival Factor for Hippocampal and Mesencephalic Neurons. Genomics 2000, 67, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Knaüper, V. Phorbol Ester-induced Shedding of the Prostate Cancer Marker Transmembrane Protein with Epidermal Growth Factor and Two Follistatin Motifs 2 Is Mediated by the Disintegrin and Metalloproteinase-17. J. Biol. Chem. 2007, 282, 37378–37388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labeur, M.; Wolfel, B.; Stalla, J.; Stalla, G.K. TMEFF2 is an endogenous inhibitor of the CRH signal transduction pathway. J. Mol. Endocrinol. 2015, 54, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.S.; Maezawa, I.; Petrlova, J.; Zhao, X.Y.; John, C.V.; Jin, L.W. Tomoregulin (TMEFF2) Binds Alzheimer’s Disease Amyloid-beta (Abeta) Oligomer and AbetaPP and Protects Neurons from Abeta-Induced Toxicity. J. Alzheimers Dis. 2015, 48, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Taylor, J.R., Jr.; Wu, T.D.; Gutierrez, J.; Elliott, J.M.; Vernes, J.-M.; Koeppen, H.; Phillips, H.S.; de Sauvage, F.J.; Meng, Y.G. TMEFF2 is a PDGF-AA binding protein with methylation-associated gene silencing in multiple cancer types including glioma. PLoS ONE 2011, 6, e18608. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. DOG 1.0: Illustrator of protein domain structures. Cell Res. 2009, 19, 271–273. [Google Scholar] [CrossRef]
- Young, J.; Biden, K.G.; Simms, L.A.; Huggard, P.; Karamatic, R.; Eyre, H.J.; Sutherland, G.R.; Herath, N.; Barker, M.; Anderson, G.J.; et al. HPP1: A transmembrane protein-encoding gene commonly methylated in colorectal polyps and cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 265–270. [Google Scholar] [CrossRef]
- Yonezawa, M.; Uchida, T.; Wada, K.; Akamatsu, T.; Mizoguchi, A.; Sakamoto, C.; Tsukui, T.; Sinoki, K.; Satou, J. A novel epidermal growth factor-like molecule containing two follistatin modules stimulates tyrosine phosphorylation of ERBB-4 in MKN28 gastric cancer cells. Gastroenterology 2000, 118, A557. [Google Scholar] [CrossRef]
- Glynne-Jones, E.; Harper, M.E.; Seery, L.T.; James, R.; Anglin, I.; Morgan, H.E.; Taylor, K.M.; Gee, J.M.; Nicholson, R.I. TENB2, a proteoglycan identified in prostate cancer that is associated with disease progression and androgen independence. Int. J. Cancer 2001, 94, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Quayle, S.N.; Sadar, M.D. A truncated isoform of TMEFF2 encodes a secreted protein in prostate cancer cells. Genomics 2006, 87, 633–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, T.; Wada, K.; Akamatsu, T.; Yonezawa, M.; Noguchi, H.; Mizoguchi, A.; Kasuga, M.; Sakamoto, C. A Novel Epidermal Growth Factor-like Molecule Containing Two Follistatin Modules Stimulates Tyrosine Phosphorylation of erbB-4 in MKN28 Gastric Cancer Cells. Biochem. Biophys. Res. Commun. 1999, 266, 593–602. [Google Scholar] [CrossRef]
- Zhao, X.-Y.; Schneider, D.; Biroc, S.L.; Parry, R.; Alicke, B.; Toy, P.; Xuan, J.-A.; Sakamoto, C.; Wada, K.; Schulze, M.; et al. Targeting Tomoregulin for Radioimmunotherapy of Prostate Cancer. Cancer Res. 2005, 65, 2846–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Salem, C.E.; Yu, M.C.; Nguyen, H.D.; Gonzales, F.A.; Nguyen, T.T.; Nichols, P.W.; Jones, P.A. DNA Methylation Differences Associated with Tumor Tissues Identified by Genome Scanning Analysis. Genomics 1998, 53, 260–268. [Google Scholar] [CrossRef]
- Gawel-Beben, K.; Ali, N.; Ellis, V.; Velasco, G.; Poghosyan, Z.; Ager, A.; Knauper, V. TMEFF2 shedding is regulated by oxidative stress and mediated by ADAMs and transmembrane serine proteases implicated in prostate cancer. Cell Biol. Int. 2018, 42, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, J.L.; Morris, T.L.; Ford, O.H., 3rd; Alvey, R.F.; Sakamoto, C.; Gregory, C.W. Identification of differentially expressed genes associated with androgen-independent growth of prostate cancer. Prostate 2002, 51, 247–255. [Google Scholar] [CrossRef]
- Chen, Q.; Watson, J.T.; Marengo, S.R.; Decker, K.S.; Coleman, I.; Nelson, P.S.; Sikes, R.A. Gene expression in the LNCaP human prostate cancer progression model: Progression associated expression in vitro corresponds to expression changes associated with prostate cancer progression in vivo. Cancer Lett. 2006, 244, 274–288. [Google Scholar] [CrossRef]
- Green, T.; Chen, X.; Ryan, S.; Asch, A.S.; Ruiz-Echevarría, M.J. TMEFF2 and SARDH cooperate to modulate one-carbon metabolism and invasion of prostate cancer cells. Prostate 2013, 73, 1561–1575. [Google Scholar] [CrossRef] [Green Version]
- Overcash, R.F.; Chappell, V.A.; Green, T.; Geyer, C.B.; Asch, A.S.; Ruiz-Echevarria, M.J. Androgen signaling promotes translation of TMEFF2 in prostate cancer cells via phosphorylation of the alpha subunit of the translation initiation factor 2. PLoS ONE 2013, 8, e55257. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Corbin, J.M.; Tipton, G.J.; Yang, L.V.; Asch, A.S.; Ruiz-Echevarría, M.J. The TMEFF2 tumor suppressor modulates integrin expression, RhoA activation and migration of prostate cancer cells. Biochim. Biophys. Acta 2014, 1843, 1216–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrecilla, D.; Lozano, M.V.; Lallana, E.; Neissa, J.I.; Novoa-Carballal, R.; Vidal, A.; Fernandez-Megia, E.; Torres, D.; Riguera, R.; Alonso, M.J.; et al. Anti-tumor efficacy of chitosan-g-poly(ethylene glycol) nanocapsules containing docetaxel: Anti-TMEFF-2 functionalized nanocapsules vs. non-functionalized nanocapsules. Eur. J. Pharm. Biopharm. 2013, 83, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Shibata, D.; Harpaz, N.; Xu, Y.; Yin, J.; Mori, Y.; Wang, S.; Olaru, A.; Deacu, E.; Selaru, F.M.; et al. Aberrant methylation of the HPP1 gene in ulcerative colitis-associated colorectal carcinoma. Cancer Res. 2002, 62, 6820–6822. [Google Scholar] [PubMed]
- Lee, S.M.; Park, J.Y.; Kim, D.S. Methylation of TMEFF2 gene in tissue and serum DNA from patients with non-small cell lung cancer. Mol. Cells 2012, 34, 171–176. [Google Scholar] [CrossRef]
- Young, J.; Barker, M.; Fraser, L.; Walsh, M.D.; Spring, K.; Biden, K.G.; Hopper, J.L.; Leggett, B.A.; Jass, J.R. Mutation searching in colorectal cancer studies: Experience with a denaturing high-pressure liquid chromatography system for exon-by-exon scanning of tumour suppressor genes. Pathology 2002, 34, 529–533. [Google Scholar] [CrossRef]
- Fackler, M.J.; Umbricht, C.B.; Williams, D.; Argani, P.; Cruz, L.A.; Merino, V.F.; Teo, W.W.; Zhang, Z.; Huang, P.; Visvananthan, K.; et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011, 71, 6195–6207. [Google Scholar] [CrossRef] [Green Version]
- Fackler, M.J.; Lopez Bujanda, Z.; Umbricht, C.; Teo, W.W.; Cho, S.; Zhang, Z.; Visvanathan, K.; Jeter, S.; Argani, P.; Wang, C.; et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014, 74, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kwon, H.J.; Lee, H.E.; Ryu, H.S.; Kim, S.-W.; Kim, J.H.; Kim, I.A.; Jung, N.; Cho, N.-Y.; Kang, G.H. Promoter CpG island hypermethylation during breast cancer progression. Virchows Arch. 2011, 458, 73–84. [Google Scholar] [CrossRef]
- de Groot, J.S.; Pan, X.; Meeldijk, J.; van der Wall, E.; van Diest, P.J.; Moelans, C.B. Validation of DNA promoter hypermethylation biomarkers in breast cancer—A short report. Cell. Oncol. 2014, 37, 297–303. [Google Scholar] [CrossRef]
- Suzuki, M.; Shigematsu, H.; Shames, D.S.; Sunaga, N.; Takahashi, T.; Shivapurkar, N.; Iizasa, T.; Frenkel, E.P.; Minna, J.D.; Fujisawa, T.; et al. DNA methylation-associated inactivation of TGFβ-related genes DRM/Gremlin, RUNX3, and HPP1 in human cancers. Br. J. Cancer 2005, 93, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhao, H.; Li, J.; Liu, H.; Wang, F.; Wei, Y.; Su, J.; Zhang, D.; Liu, T.; Zhang, Y. The Identification of Specific Methylation Patterns across Different Cancers. PLoS ONE 2015, 10, e0120361. [Google Scholar] [CrossRef] [PubMed]
- Belshaw, N.J.; Elliott, G.O.; Williams, E.A.; Bradburn, D.M.; Mills, S.J.; Mathers, J.C.; Johnson, I.T. Use of DNA from Human Stools to Detect Aberrant CpG Island Methylation of Genes Implicated in Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1495–1501. [Google Scholar]
- Wynter, C.V.; Walsh, M.D.; Higuchi, T.; Leggett, B.A.; Young, J.; Jass, J.R. Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Gut 2004, 53, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Shivapurkar, N.; Riquelme, E.; Shigematsu, H.; Reddy, J.; Suzuki, M.; Miyajima, K.; Zhou, X.; Bekele, B.N.; Gazdar, A.F.; et al. Aberrant promoter hypermethylation of multiple genes in gallbladder carcinoma and chronic cholecystitis. Clin. Cancer Res. 2004, 10, 6126–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Zheng, F.; Yuan, X.; Ye, Y.; Li, X.; Dai, Y.; Chen, L. The effect of TMEFF2 methylation on the tumor stage and survival outcome of clear cell renal cell carcinoma. Cancer Biomark. 2017, 19, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Geddert, H.; Kiel, S.; Iskender, E.; Florl, A.R.; Krieg, T.; Vossen, S.; Gabbert, H.E.; Sarbia, M. Correlation of hMLH1 and HPP1 hypermethylation in gastric, but not in esophageal and cardiac adenocarcinoma. Int. J. Cancer 2004, 110, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Hadjinicolaou, A.V.; van Munster, S.N.; Achilleos, A.; Santiago Garcia, J.; Killcoyne, S.; Ragunath, K.; Bergman, J.J.G.H.M.; Fitzgerald, R.C.; di Pietro, M. Aneuploidy in targeted endoscopic biopsies outperforms other tissue biomarkers in the prediction of histologic progression of Barrett’s oesophagus: A multi-centre prospective cohort study. EBioMedicine 2020, 56, 102765. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.M.; Sato, F.; Mori, Y.; Perry, K.; Yin, J.; Wang, S.; Xu, Y.; Olaru, A.; Selaru, F.; Spring, K.; et al. Hypermethylation of HPP1 is associated with hMLH1 hypermethylation in gastric adenocarcinomas. Cancer Res. 2002, 62, 5637–5640. [Google Scholar]
- Suzuki, M.; Toyooka, S.; Shivapurkar, N.; Shigematsu, H.; Miyajima, K.; Takahashi, T.; Stastny, V.; Zern, A.L.; Fujisawa, T.; Pass, H.I.; et al. Aberrant methylation profile of human malignant mesotheliomas and its relationship to SV40 infection. Oncogene 2005, 24, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Du, W.; Xiong, H.; Yu, Y.; Weng, Y.; Ren, L.; Zhao, H.; Wang, Y.; Chen, Y.; Xu, J.; et al. TMEFF2 deregulation contributes to gastric carcinogenesis and indicates poor survival outcome. Clin. Cancer Res. 2014, 20, 4689–4704. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.-T.; Tang, J.-Y.; Du, W.; Zhao, H.-J.; Zhao, G.; Yang, S.-L.; Chen, H.-Y.; Hong, J.; Fang, J.-Y. Bidirectional regulation between TMEFF2 and STAT3 may contribute to Helicobacter pylori-associated gastric carcinogenesis. Int. J. Cancer 2015, 136, 1053–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbioni, S.; Miotto, E.; Veronese, A.; Sattin, E.; Gramantieri, L.; Bolondi, L.; Calin, G.A.; Gafa, R.; Lanza, G.; Carli, G.; et al. Multigene methylation analysis of gastrointestinal tumors: TPEF emerges as a frequent tumor-specific aberrantly methylated marker that can be detected in peripheral blood. Mol. Diagn. 2003, 7, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Vdovin, N.; Gacesa, S.; Philipp, A.; Nagel, D.; Holdt, L.M.; op den Winkel, M.; Heinemann, V.; Stieber, P.; Graeven, U.; et al. Methylated free-circulating HPP1 DNA is an early response marker in patients with metastatic colorectal cancer. Int. J. Cancer 2017, 140, 2134–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Elahi, A.; Ajidahun, A.; Clark, W.; Hernandez, J.; Achille, A.; Hao, J.H.; Seto, E.; Shibata, D. The interplay between histone deacetylases and c-Myc in the transcriptional suppression of HPP1 in colon cancer. Cancer Biol. Ther. 2014, 15, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gery, S.; Sawyers, C.L.; Agus, D.B.; Said, J.W.; Koeffler, H.P. TMEFF2 is an androgen-regulated gene exhibiting antiproliferative effects in prostate cancer cells. Oncogene 2002, 21, 4739–4746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afar, D.E.; Bhaskar, V.; Ibsen, E.; Breinberg, D.; Henshall, S.M.; Kench, J.G.; Drobnjak, M.; Powers, R.; Wong, M.; Evangelista, F.; et al. Preclinical validation of anti-TMEFF2-auristatin E-conjugated antibodies in the treatment of prostate cancer. Mol. Cancer Ther. 2004, 3, 921–932. [Google Scholar] [PubMed]
- Han, H.; Zhan, Z.; Xu, J.; Song, Z. TMEFF2 inhibits pancreatic cancer cells proliferation, migration, and invasion by suppressing phosphorylation of the MAPK signaling pathway. Onco Targets Ther. 2019, 12, 11371–11382. [Google Scholar] [CrossRef] [Green Version]
- Corbin, J.M.; Overcash, R.F.; Wren, J.D.; Coburn, A.; Tipton, G.J.; Ezzell, J.A.; McNaughton, K.K.; Fung, K.-M.; Kosanke, S.D.; Ruiz-Echevarria, M.J. Analysis of TMEFF2 allografts and transgenic mouse models reveals roles in prostate regeneration and cancer. Prostate 2016, 76, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Kanemoto, N.; Horie, M.; Omori, K.; Nishino, N.; Kondo, M.; Noguchi, K.; Tanigami, A. Expression of TMEFF1 mRNA in the mouse central nervous system: Precise examination and comparative studies of TMEFF1 and TMEFF2. Brain Res. Mol. Brain Res. 2001, 86, 48–55. [Google Scholar] [CrossRef]
- Chen, T.R.; Wang, P.; Carroll, L.K.; Zhang, Y.J.; Han, B.X.; Wang, F. Generation and characterization of TMEFF2 mutant mice. Biochem. Biophys. Res. Commun. 2012, 425, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Liu, H.L.; Liu, B.; Willuda, J.; Siemeister, G.; Mahmoudi, M.; Dinter, H. Tomoregulin internalization confers selective cytotoxicity of immunotoxins on prostate cancer cells. Transl. Oncol. 2008, 1, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Overcash, R.; Green, T.; Hoffman, D.; Asch, A.S.; Ruiz-Echevarria, M.J. The tumor suppressor activity of the transmembrane protein with epidermal growth factor and two follistatin motifs 2 (TMEFF2) correlates with its ability to modulate sarcosine levels. J. Biol. Chem. 2011, 286, 16091–16100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Teng, P.; Mei, R.; Yang, A.; Zhang, Z.; Zhao, X.; Qiu, M. TMEFF2 is expressed in differentiating oligodendrocytes but dispensable for their differentiation in vivo. Sci. Rep. 2017, 7, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elahi, A.; Zhang, L.; Yeatman, T.J.; Gery, S.; Sebti, S.; Shibata, D. HPP1-mediated tumor suppression requires activation of STAT1 pathways. Int. J. Cancer 2008, 122, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.M.; Elahi, A.; Clark, W.; Humphries, L.A.; Wang, J.; Achille, A.; Seto, E.; Shibata, D. The Tumor Suppressive Effects of HPP1 Are Mediated Through JAK-STAT-Interferon Signaling Pathways. DNA Cell Biol. 2015, 34, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K. Follistatin. Int. J. Biochem. Cell Biol. 1998, 30, 1087–1093. [Google Scholar] [CrossRef]
- Gao, L.; Nie, X.; Zheng, M.; Li, X.; Guo, Q.; Liu, J.; Liu, Q.; Hao, Y.; Lin, B. TMEFF2 is a novel prognosis signature and target for endometrial carcinoma. Life Sci. 2020, 243, 116910. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Y.; Cheng, H.; Tian, J.; Yang, S. Roles of a TMPO-AS1/microRNA-200c/TMEFF2 ceRNA network in the malignant behaviors and 5-FU resistance of ovarian cancer cells. Exp. Mol. Pathol. 2020, 115, 104481. [Google Scholar] [CrossRef]
- Li, K.; Han, H.; Gu, W.; Cao, C.; Zheng, P. Long non-coding RNA LINC01963 inhibits progression of pancreatic carcinoma by targeting miR-641/TMEFF2. Biomed. Pharmacother. 2020, 129, 110346. [Google Scholar] [CrossRef]
- Fan, J.M.; Zheng, Z.R.; Zeng, Y.M.; Chen, X.Y. MiR-323-3p Targeting Transmembrane Protein with EGF-Like and 2 Follistatin Domain (TMEFF2) Inhibits Human Lung Cancer A549 Cell Apoptosis by Regulation of AKT and ERK Signaling Pathways. Med. Sci. Monit. 2020, 26, e919454. [Google Scholar] [CrossRef]
- Georgescu, C.; Corbin, J.M.; Thibivilliers, S.; Webb, Z.D.; Zhao, Y.D.; Koster, J.; Fung, K.-M.; Asch, A.S.; Wren, J.D.; Ruiz-Echevarría, M.J. A TMEFF2-regulated cell cycle derived gene signature is prognostic of recurrence risk in prostate cancer. BMC Cancer 2019, 19, 423. [Google Scholar] [CrossRef] [PubMed]
- Pitale, P.M.; Gorbatyuk, O.; Gorbatyuk, M. Neurodegeneration: Keeping ATF4 on a Tight Leash. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Lin, H.; Wada, K.; Yonezawa, M.; Shinoki, K.; Akamatsu, T.; Tsukui, T.; Sakamoto, C. Tomoregulin ectodomain shedding by proinflammatory cytokines. Life Sci. 2003, 73, 1617–1627. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Elahi, A.; Ajidahun, A.; Hendrick, L.; Getun, I.; Humphries, L.A.; Hernandez, J.; Shibata, D. HPP1 Ectodomain Shedding is Mediated by ADAM17 and is Necessary for Tumor Suppression in Colon Cancer. J. Surg. Res. 2020, 254, 183–190. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Cells | Reducing/Non-Reducing | Endogenous/Overexpressed | Antibody Used | Band Sizes kDa | Detail | Ref. |
|---|---|---|---|---|---|---|
| N2a | Not mentioned | Overexpressed | Anti-V5 | 17, 50, 56 | Duplet band between 50 and 56 kDa, plus a single band just below 17 kDa (C-terminal band) | [7] |
| CWR22 | Not mentioned | Endogenous | 2H-8 a | 51, 58 | Duplet band at 51–58 kDa | [18] |
| LNCap, C4-2 | Not mentioned | Endogenous | TMEFF-CP b | 44 | Single 44 kDa band in both LNCap and C4-2 cells | [19] |
| 22Rv1, LNCap | Not mentioned | Endogenous | Abcam | ~ 48 | Single band just under 50 kDa marker (size not mentioned in text) | [20] |
| 22Rv1, LNCap | Reducing | Endogenous | Abcam | 40 to 55 | Single smear ranging from 40 to 55 kDa in LNCap and doublet band between 40 and 55 kDa in 22Rv1 cells | [21] |
| RWPE1, RWPE2 | Not mentioned | Overexpressed | Abcam | 40 to 56 | A smear from 40–56 kDa in RWPE1 and RWPE2 cells, a heavily glycosylated band can be observed above 90 kDa in only the TMEFF2_ΔGA form | [22] |
| HEK293 | Reducing | Overexpressed | 11D1C1 c | 58 | Full length at 58 kDa, ECD only at 54 kDa (bands appear as duplets) | [23] |
| PC3, LNCap | Non-reducing | Both (see detail) | 2H-8 | 43, 48, 52, 97 | Endogenous TMEFF2 was detected at a single band of 43 kDa in LNCaP cells. V5/His tagged TMEFF2 overexpressing PC3 cells showed a major band at 48 kDa, a minor one at approximately 52kDa and another (smear) band at approximately 97 kDa. ECD-only overexpressing PC3 cells showed a band at 40 kDa. | [15] |
| CHO | Reducing | Overexpressed | Anti-V5 | 14, 71 | V5/His tagged overexpressing TMEFF2 in CHO cells showed two distinct bands: one at 71 kDa and another at 14 kDa. For ECD-only overexpressing CHO cells, a duplet band at 54–60 was detected in cell lysate while 4 bands were detected in the medium, at 99, 69, 63 and 52 kDa. | [12] |
| CHO, A172 | Both (see detail) | Both (see detail) | 2H-8, aTR-C d | 51 | In A172 cells, endogenous TRc was detected at 51 kDa under non-reducing conditions (2H8 antibody). For TRc-overexpressing CHO cells, the band was observed at 51 kDa under non-reducing conditions and at 58 kDa under reducing conditions in cell lysates (pAb-aTRc antibody). In the media of TRc-overexpressing CHO cells, under non-reducing conditions, a smear was observed at 39–45 kDa (2H8 antibody). | [11] |
| HEK293, LNCaP, CHO, PC3 | Not mentioned | Both (see detail) | Anti-V5, Anti-TMEFF2 (details not mentioned) | 60, 75, 10, 14, 22 | Endogenous TMEFF2 from LNCap cells, the shed form from media, showed a duplet band at 60–75 kDa. In TMEFF2/V5/His overexpressing HEK293, a duplet band at 60–75 kDa was detected in conditioned media (atni-TMEFF2 antibody) and 10, 14 and 22 kDa distinct bands were detected in the cell lysate (anti-V5 antibody). Similarly, in TMEFF2/V5/His overexpressing CHO and PC3 cells, 10, 14 and 22 kDa c-terminal bands were detected in the cell lysate using V5 antibody. | [5] |
| HEK293 | Not mentioned | Overexpressed | Anti-V5 | 15, 17, 20, 24, 28 | Only C terminal bands were mentioned. In HEK293 cells stably transfected with AP-TMEFF2-V5/His, bands changed upon overexpression of various proteinases: (Matriptase-1) 24 and 28 kDa, and (Hepsin) 15 and 20 kDa. A 17-kDa band was observed without overexpression of any protease and was proposed to be a result of native ADAM activity. | [17] |
| Cells | Antibody or Detection Method Used | Intracellular Location | Ref. |
|---|---|---|---|
| PC3 | Pr1#19 | Membranous and punctate-cytoplasmic | [47] |
| PC3 | 2H8 | Membranous and punctate-cytoplasmic upon internalization, co-localizes with acidic organelles | [52] |
| MC65 | 48G2 | Perinuclear and punctate-cytoplasmic, not membranous | [7] |
| 22Rv1 | Abcam (details not mentioned) | Punctate-cytoplasmic along the rim of the nucleus when tested though immunofluorescence; membranous, cytoplasmic and nuclear in cell fractionation | [21] |
| HEK293T | Not mentioned | Membranous and diffused-cytoplasmic upon immunofluorescence but exhibits mitochondrial appearance when fusion constructs were used | [53] |
| 22Rv1 | Abcam (details not mentioned) | Membranous, cytoplasmic, nuclear; colocalization with β-actin and α-tubulin | [20] |
| LnCaP | GFP-fusion construct | Membranous and diffused-cytoplasmic for full length and punctate-cytoplasmic for short secreted isoform | [13] |
| Function | Oncogenic vs. Onco-Suppressive | Disease or Process | Reference |
|---|---|---|---|
| TMEFF2 binds and inhibits PDGF-AA through its follistatin-like domains. | Onco-suppressive | Several cancers | [8] |
| TMEFF2 promotes survival of hippocampal and mesencephalic neurons in primary culture. | Neither but pro-growth | Neuronal development | [4] |
| Tomoregulin mildly activates erbB-4 receptors in MKN28 gastric cancer cells. | Oncogenic | Gastric cancer | [11] |
| TMEFF2 inhibits Akt phosphorylation and production of ACTH in response to CRH stimulation in AtT20 glioma cells. TMEFF2 decreases the proliferation of AtT20 cells. | Onco-suppressive | Glioma | [6] |
| (i) TMEFF2 is specifically upregulated in OPCs at onset of cell differentiation, but it is dispensable in the process. (ii) TMEFF2 weakly interacts with PDGFA but does not prevent the action of PDGFA on its receptor in HEK293T cells. (iii) TMEFF2 overexpression increases ERK1/2 phosphorylation in HEK293T cells. | Neither but pro-growth | Development | [54] |
| Tomoregulin mRNA is expressed in mouse embryo at E-11, increases gradually till E-15 and stabilizes through to E-17. | Neither | Development | [11] |
| TMEFF2 is regulated by androgens in prostate cancer and inhibits the proliferation of DU145 and PC3 prostate cancer cells. | Onco-suppressive | Prostate cancer | [46] |
| TMEFF2mAb conjugated cytotoxic drug Pr1-vcMMAE effectively reduced the tumour size in LNCap and CWR22 xenograft with minimal toxicity. | Potentially oncogenic | Prostate cancer | [47] |
| β-emitting isotope 90Y-labelled TMEFF2 mAB 2H8 effectively reduces LNCaP xenograft size and is well tolerated. | Potentially oncogenic | Prostate cancer | [15] |
| TMEFF2 is expressed on the cell surface, and it is internalized and recycled in lysosomes. TMEFF2 mAB is complexed with cytotoxin saporin, and it causes death of TMEFF2-Stable PC3 cells. | Potentially oncogenic | Prostate cancer | [52] |
| TMEFF2 binds amyloid-β protein, its precursor AβPP and the neurotoxic oligomeric forms of amyloid-β protein AβPOs in vitro and in vivo. TMEFF2 protects N2a cells from neurotoxicity induced by AβPO. | None | Alzheimer’s disease | [7] |
| TMEFF2 ectodomain shedding is triggered by TNFα, is brought about by ADAM10 and ADAM17, and increases proliferation of HEK293T cells. TMEFF2 causes phosphorylation of ERK1/2. | Neither but pro-growth | Prostate cancer | [5] |
| TMEFF2 is a substrate for membrane anchored serine proteases (TTSPs) matriptase-1 and hepsin. | Depends on proteolytic processing of TMEFF2 | Prostate cancer | [17] |
| Full-length TMEFF2 reduces cell proliferation and sensitises HEK293T cells to apoptosis, binds and augments sarcosine dehydrogenase (SRDH) activity and reduces sarcosine levels, leading to inhibition of cell migration. The TMEFF2 ectodomain does the opposite. | Ectodomain: oncogenic Full-length: onco-suppressive | Prostate cancer | [53] |
| Full-length TMEFF2 (i) reduces the expression of integrins αv, β1 and β3; (ii) inhibits RHOA activation; (iii) inhibits phosphorylation of the focal adhesion kinase (FAK); and (iv) reduces cell migration. Deletion of putative G-protein domain prevents all of the above. | Onco-suppressive | Prostate cancer | [22] |
| TMEFF2 reduces invasiveness of 22Rv1 prostate cancer cells through modulation of one-carbon metabolism. TMEFF2 interacts with the cytoskeleton. | Onco-suppressive | Prostate cancer | [20] |
| HPP1 induces apoptosis and causes changes in morphology and reduction in cell proliferation in HCT-116 colorectal cells through activation of the STAT1 pathway. | Onco-suppressive | Colorectal cancer | [55] |
| HPP1 activates JAK-STAT interferon pathways to induce apoptosis and to reduce proliferation of HCT-116 cells. TMEFF2 overexpression marginally sensitized HCT-116 cells to INF-α-induced cell death. | Onco-suppressive | Colorectal cancer | [56] |
| TMFF2 decreases STAT3 phosphorylation through its interaction with SHP1 in gastric cancer. STAT3 directly binds to the TMEFF2 promoter and represses its transcription. | Onco-suppressive | Gastric cancer | [42] |
| TMEF2 binds SHP-1 in gastric cancer, induces cell cycle arrest and apoptosis and prevents DNA damage. | Onco-suppressive | Gastric cancer | [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masood, M.; Grimm, S.; El-Bahrawy, M.; Yagüe, E. TMEFF2: A Transmembrane Proteoglycan with Multifaceted Actions in Cancer and Disease. Cancers 2020, 12, 3862. https://doi.org/10.3390/cancers12123862
Masood M, Grimm S, El-Bahrawy M, Yagüe E. TMEFF2: A Transmembrane Proteoglycan with Multifaceted Actions in Cancer and Disease. Cancers. 2020; 12(12):3862. https://doi.org/10.3390/cancers12123862
Chicago/Turabian StyleMasood, Motasim, Stefan Grimm, Mona El-Bahrawy, and Ernesto Yagüe. 2020. "TMEFF2: A Transmembrane Proteoglycan with Multifaceted Actions in Cancer and Disease" Cancers 12, no. 12: 3862. https://doi.org/10.3390/cancers12123862
APA StyleMasood, M., Grimm, S., El-Bahrawy, M., & Yagüe, E. (2020). TMEFF2: A Transmembrane Proteoglycan with Multifaceted Actions in Cancer and Disease. Cancers, 12(12), 3862. https://doi.org/10.3390/cancers12123862