Emerging Role and Therapeutic Potential of lncRNAs in Colorectal Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
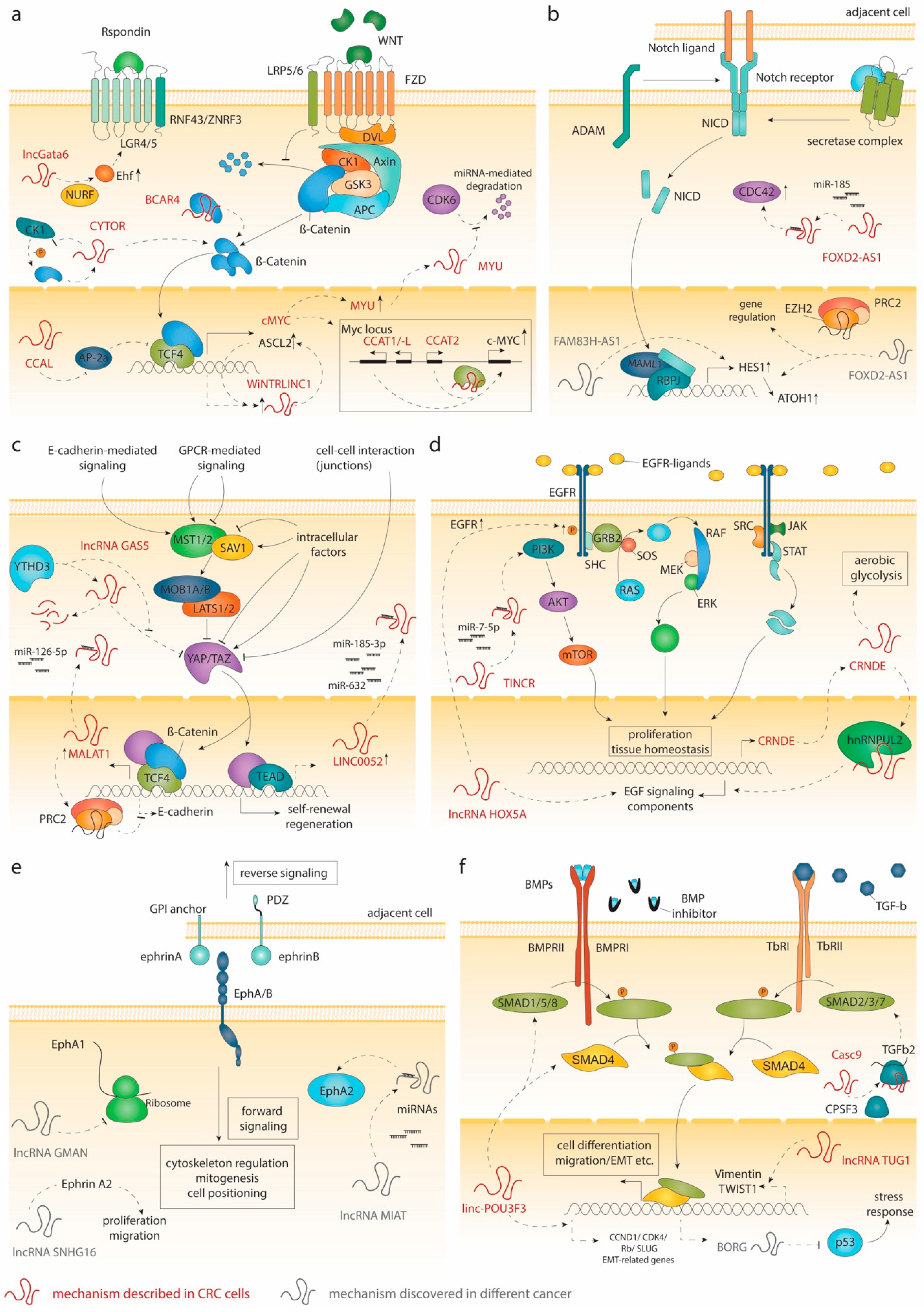
2. Signaling in the Intestinal Crypt and lncRNAs
2.1. Wnt Signaling
2.2. Notch Signaling
2.3. The Hippo Pathway
2.4. EGF Signaling
2.5. EphB Signaling
2.6. BMP and TGF-β Signaling
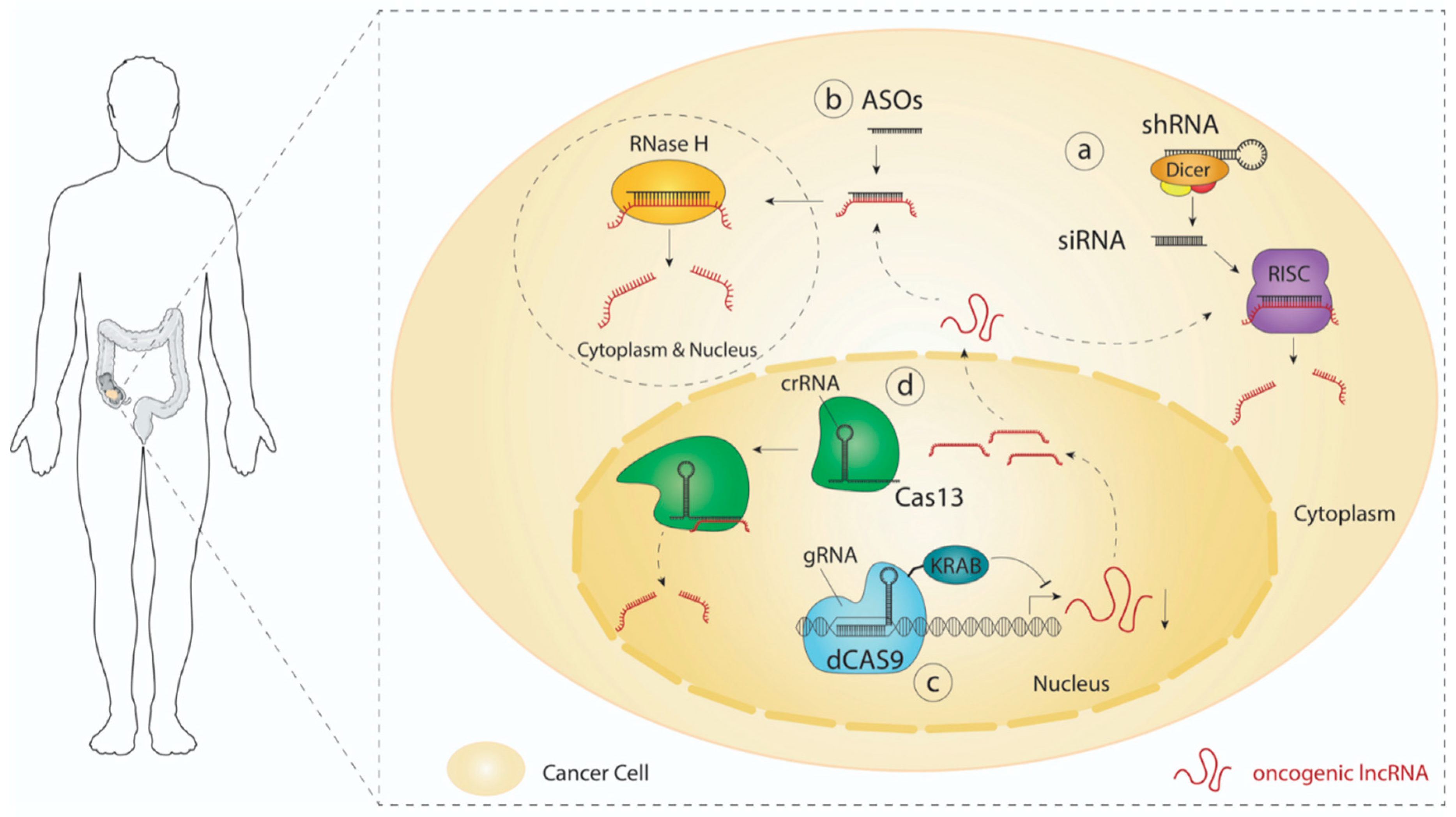
3. LncRNA-Directed Therapeutic Approaches
3.1. RNAi
3.2. Antisense Oligonucleotides (ASOs)
3.3. CRISPR Technology
4. Conclusions
Funding
Conflicts of Interest
References
- Lieberkühn, J.N. Dissertationes quatuor: Nimirum: De Valvula Coli & Usu Processus Vermicularis. De Fabrica & Actione Villorum INTESTINORUM tenuium Hominis. Sur les MOYENS propres à Decouvrir la Construction des Visceres. Description d’un Microscope Anatomique; Thomas Cadell: London, UK, 1745. [Google Scholar]
- Darwich, A.S.; Aslam, U.; Ashcroft, D.M.; Rostami-Hodjegan, A. Meta-analysis of the turnover of intestinal epithelia in pre-clinical animal species and human. Drug Metabol. Disposit. 2014, 18. [Google Scholar] [CrossRef] [Green Version]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R. A Genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Fumagalli, A.; Drost, J.; Sujekerbujik, S.J.E.; van Boxtel, R.; De Ligt, J.; Offerhaus, G.J.; Beghtel, H.; Beerling, E.; Hong Tang, E.; Sansom, O.J. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Nat. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [Green Version]
- Reed, K.R.; Owen, J.S.; Hayes, A.J.; Gescher, A.J.; Winton, D.J.; Peters, J.M.; Clarske, A.R. PPARδ status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene 2004, 23, 8992–8996. [Google Scholar] [CrossRef] [Green Version]
- Miyaki, M. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 1999, 18, 3098–3103. [Google Scholar] [CrossRef] [Green Version]
- Koyama, M.; Ito, M.; Nagai, H.; Emi, M.; Moriyama, Y. Inactivation of both alleles of the DPC4/SMAD4 gene in advanced colorectal cancers: Identification of seven novel somatic mutations in tumors from Japanese patients. Mutat. Res. Mutat. Res. Genom. 1999, 406, 71–77. [Google Scholar] [CrossRef]
- Markowitz, S. Inactivation of the Type 11 TGF-f receptor in colon cancer cells with microsatellite instability. Sci. 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, A.C.; Gingeras, T.R. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guerncec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Gen. Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Cabili, M.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Develop. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Brunner, A.D.; Cogan, J.Z.; Nunez, J.K.; Fields, A.P. Pervasive functional translation of non-canonical human open reading frames. HHS Public Access Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef]
- Kay, G.F. Expression of Xist during mouse development suggests a role in the initiation of X chromosome inactivation. Cell 1993, 72, 171–182. [Google Scholar] [CrossRef]
- Léveillé, N.; Melo, C.A.; Rooijers, K.; Diaz-Lagares, A.; Melo, S.A. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 2015, 6, 6520. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Abudayyeh, O.O. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Chen, J. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar]
- Engreitz, J.M.; Pandya Jones, A.; McDonel, P.; Shiskin, A.; Sirokman, K.; Surka, C. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341. [Google Scholar] [CrossRef] [Green Version]
- Kretz, M.; Siprashvili, S.; Chu, C.; Webster, D.E.; Zehnder, A. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Vrielink, J.A.O.; Elkon, R.; Melo, S.A.; Léveillé, N.; Kalluri, R.; et al. ERNAs Are Required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.P.; Eko, N.G.; Jin, H. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010, 31, 350–358. [Google Scholar] [CrossRef]
- Bian, Z.; Jin, L.; Zhang, J.; Yin, Y.; Quan, C.; Hu, Y.; Feng, Y. LncRNA—UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting MIR-204-5p. Sci. Rep. 2016, 6, 23892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Sun, L.; Liu, D.; Liu, C.; Sun, L. Long non-coding RNA lincRNA-ROR promotes the progression of colon cancer and holds prognostic value by associating with miR-145. Pathol. Oncol. Res. 2016, 22, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Ni, Y.; He, X.; Wu, D.; Li, M.; Wu, S.; Zhang, R.; Guo, M. Decreasing lncRNA HOTAIR expression inhibits human colorectal cancer stem cells. Am. J. Transl. Res. 2016, 8, 98–108. [Google Scholar] [PubMed]
- Lu, M.; Liu, Z.; Li, B.; Zhu, Y. The high expression of long non-coding RNA PANDAR indicates a poor prognosis for colorectal cancer and promotes metastasis by EMT pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 71–81. [Google Scholar] [PubMed]
- He, F.; Song, Z.; Chen, H.; Yang, P.; Li, W.; Yang, Z. Long noncoding RNA PVT1-214 promotes proliferation and invasion of colorectal cancer by stabilizing Lin28 and interacting with miR-128. Oncogene 2019, 38, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Shen, X. Long noncoding RNAs: Functions and mechanisms in colon cancer. Mol. Cancer 2020, 19, 167. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D. Functional interaction of β-catenin with the transcription factor LEF- 1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M. XTcf-3 transcription factor mediates β-catenin-induced axis formation in xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Schuijers, J.; Mokry, M.; Hatzis, P.; Cuppen, E.; Clevers, H. Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J. 2014, 33, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Lustig, B.; Jerchow, B.; Sachs, M.; Weiler, S. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Langenbeck’s Arch. Surg. 2001, 386, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nature 1998, 19, 379–383. [Google Scholar] [CrossRef] [PubMed]
- van Es, J.H.; Haegebarth, A.; Kujala, P. A critical role for the wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 2012, 32, 1918–1927. [Google Scholar] [CrossRef] [Green Version]
- Ireland, H.; Kemp, R.; Houghton, C.; Howard, L.; Clarke, A.R. Inducible cre-mediated control of gene expression in the murine gastrointestinal tract: Effect of loss of-catenin. Gastroenterolgy 2004, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sachs, N.; Wiebrands, K. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc. Natl. Acad. Sci. USA 2016, 113, E5399–E5407. [Google Scholar]
- Sato, T.; Van Es, J.H.; Snippert, H.J.; Stange, D.E. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Stzepourginski, I.; Nigro, G.; Jacob, J.M. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc. Natl. Acad. Sci. USA 2016, 114, E506–E513. [Google Scholar] [CrossRef] [Green Version]
- Valenta, T.; Degrimenci, B.; Moor, A.E.; Herr, P.; Zimmerli, D.; Moor, M.B.; Hausmann, G. Wnt ligands secreted by subepithelial mesenchymal cells are essential for the survival of intestinal stem cells and gut homeostasis. Cell Rep. 2016, 15, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar]
- de Lau, W.; Barcker, N.; Low, T.J.; Koo, B.K.; Li, V.S.W.; Teunissen, H. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [PubMed] [Green Version]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Avello, M. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Develop. 2014, 28, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Gregorieff, A.; Begthel, H.; Clevers, H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Develop. 2003, 17, 1709–1713. [Google Scholar] [CrossRef] [Green Version]
- Kuhnert, F.; Davis, C.R.; Wang, H.T.; Chu, P. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc. Nat. Acad. Sci. USA 2004, 101, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.M.; Zitz, N.; Barclay-Beazer, Y.; Bryan, T.M. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Nishisho, I.; Nakumara, Y.; Miyoshi, M.; Miki, Y. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC(-/-) colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Wetering, M.; Sancho, E.; Verweij, C. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wu, J.; Wang, Y.; Zhu, X.; Lu, T.; Liu, B.; He, L. LncGata6 maintains stemness of intestinal stem cells and promotes intestinal tumorigenesis. Nat. Cell Biol. 2018, 20, 1134–1144. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, Y.; Wang, F.; Moyer, M.P.; Wei, Q.; Zhang, P. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/beta-catenin signalling pathway via suppression of activator protein 2alpha. Gut 2016, 65, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Zheng, X.; Zhou, X.; Chen, Z.; Yang, X.; Xie, M. LncRNA BCAR4 promotes colon cancer progression via activating Wnt/beta-catenin signaling. Oncotarget 2017, 8, 92815–92826. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Liu, C.; Sun, H.; Liu, M.; Song, C.; Cui, R.; Qiu, S. A positive feed-forward loop between LncRNA-CYTOR and Wnt/beta-Catenin signaling promotes metastasis of colon cancer. Mol. Ther. 2018, 26, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- van der Flier, L.G.; van Gijin, M.E.; Hatzis, P.; Kujala, P. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell 2009, 136, 903–912. [Google Scholar]
- Giakountis, A.; Moulos, P.; Zarkou, V.; Oikonomou, C.; Harokopos, V.; Hatzigeorgiou, A.G.; Reczko1, M.; Hatzis, P. A positive regulatory loop between a Wnt-regulated non-CODING RNA and ASCL2 controls intestinal stem cell fate. Cell Rep. 2016, 15, 2588–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; Da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiyeh, N.; Pomerantz, M.I.M. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Nat. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuupanen, S.; Turuner, M.; Lehtonen, R.; Halikas, O. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Gen. 2009, 41, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Sur, I.K.; Halikas, O.; Vaharautio, A.; Yan, J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 2012, 338, 1360–1363. [Google Scholar] [CrossRef]
- Alaiyan, B.; Ilyayev, N.; Stojadinovic, A.; Izadjoo, M. Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer 2013, 13, 196. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Gen. Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef] [Green Version]
- Nissan, A.; Stojadinovic, A. Colon cancer associated transcript-1: A novel RNA expressed in malignant and pre-malignant human tissues. Int. J. Cancer 2012, 130, 1598–1606. [Google Scholar] [CrossRef]
- Ozawa, T.; Matsuyama, T.; Toiyama, Y.; Takahashi, N.; Ishikawa, T.; Uetake, H.; Yamada, Y.; Kusunoki, M.; Calin, G.; Goel, A. CCAT1 and CCAT2 long noncoding RNAs, located within the 8q.24.21 “gene desert”, serve as important prognostic biomarkers in colorectal cancer. Ann. Oncol. 2017, 28, 1882–1888. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yan, T.; Bao, Y.; Shen, C.; Yu, C.; Zhu, X.; Tian, X.; Guo, F. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Komiya, M.; Matsumura, K.; Negishi, L.; Suda, S.; Okuno, M.; Yokota, N. MYU, a Target lncRNA for Wnt/c-Myc signaling, mediates induction of CDK6 to promote cell cycle progression. Cell Rep. 2016, 16, 2554–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Bone 2008, 23, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinson, K.E.; George, D.C.; Fender, A.W.; Bertrand, F.E.; Sigounas, G. The Notch pathway in colorectal cancer. Int. J. Cancer 2016, 138, 1835–1842. [Google Scholar] [CrossRef]
- Ghaleb, A.M.; Aggarwal, G.; Bialkowska, A.B.; Nandan, M.O.; Yang, V.W. Notch inhibits expression of the Krüppel-like factor 4 tumor suppressor in the intestinal epithelium. Mol. Cancer Res. 2008, 6, 1920–1927. [Google Scholar] [CrossRef] [Green Version]
- Riccio, O.; van Gijn, M.E.; Bezdek, A.C.; Pellegrinet, R.; van Es, J.H. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008, 9, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Fre, S.; Huyghe, M.; Mouriks, P. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Datar, R.; Murtaugh, L.C.; Melton, D.A. Direct regulation of intestinal fate by Notch. Proc. Nat. Acad. Sci. USA 2005, 102, 12443–12448. [Google Scholar]
- Chu, D.; Zhang, Z.; Zhou, Y.; Wang, W.; Li, Y.; Zhang, H.; Dong, G.; Zhao, Q.; Ji, G. Notch1 and notch2 have opposite prognostic effects on patients with colorectal cancer. Ann. Oncol. 2011, 22, 2440–2447. [Google Scholar] [CrossRef]
- Katsushima, K.; Natsume, A.; Ohka, F.; Shunjo, K.; Hatanaka, A.; Ichimura, N.; Sato, S. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nature Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Wei, R.; Chen, Y.; Zhao, Z.; Gu, Q.; Wu, J. LncRNA FAM83H-AS1 induces nucleus pulposus cell growth via targeting the Notch signaling pathway. J. Cell. Physiol. 2019, 234, 22163–22171. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P. Genome-wide mapping and characterization of notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Duan, B.; Zhou, X. Long non-coding RNA FOXD2-AS1 functions as a tumor promoter in colorectal cancer by regulating EMT and Notch signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3586–3591. [Google Scholar] [PubMed]
- Ren, Z.; Hu, Y.; Li, G.; Kang, Y.; Liu, Y.; Zhao, H. HIF-1α induced long noncoding RNA FOXD2-AS1 promotes the osteosarcoma through repressing p21. Biomed. Pharmacother. 2019, 117, 109104. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, W.; Ma, P.; Shuai, Y.; Zhao, K.; Wang, Y.; Li, W. Upregulation of the long noncoding RNA FOXD2-AS1 promotes carcinogenesis by epigenetically silencing EphB3 through EZH2 and LSD1, and predicts poor prognosis in gastric cancer. Oncogene 2018, 37, 5020–5036. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qiao, L.; Zhou, Y.; Wang, C.; Zhou, J. Long non-coding RNA FOXD2-AS1 contributes to colorectal cancer proliferation through its interaction with microRNA-185-5p. Cancer Sci. 2018, 109, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Dong, W.; Zhao, P.; Liu, Z. LncRNA FAM83H AS1 is associated with the prognosis of colorectal carcinoma and promotes cell proliferation by targeting the notch signaling pathway. Oncol. Lett. 2018, 15, 1861–1868. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, L.; Lao, X.; Qian, Z. Hes1 is associated with long non-coding RNAs in colorectal cancer. Ann. Transl. Med. 2019, 7, 459. [Google Scholar] [CrossRef]
- Hong, A.W.; Meng, Z.; Guan, K.L. The Hippo pathway in intestinal regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 324–337. [Google Scholar]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W. YAP1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Current Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhang, N.; Zheng, Y.; De Wilde, R.F. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Develop. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Ou, C.; Liu, J.; Chen, C.; Zhou, Q.; Yang, S.; Li, G. YAP1-induced MALAT1 promotes epithelial–mesenchymal transition and angiogenesis by sponging miR-126-5p in colorectal cancer. Oncogene 2019, 38, 2627–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoda, M.; Moroishi, T. The emerging link between the Hippo pathway and non-coding RNA. Biol. Pharma. Bull. 2020, 43, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.; Xie, Z.; Chen, J.; Ni, W.; Ma, Y.; Huang, K. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m6A reader YTHDF3. Mol. Cancer 2019, 18, 1–20. [Google Scholar]
- Ou, C.; Sun, Z.; He, X.; Li, X.; Fan, S.; Zheng, X. Targeting YAP1/LINC00152/FSCN1 signaling axis prevents the progression of colorectal cancer. Adv. Sci. 2020, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, F.; Peng, L.; Li, Z.; Tan, G.; Chen, S. YAP triggers the Wnt/β-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after DSS-induced injury. Cell Death Dis. 2018, 9, 153. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Wang, H.; Liu, T.; Du, L. MALAT1 is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol. Cancer Ther. 2017, 16, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.; Thomas, P.J.; Ward, C.; Colin, W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003. [Google Scholar] [CrossRef]
- Yang, Y.P.; Ma, H.; Starchenko, A.; Huh, W.J.; Li, W.; Hickman, F.E.; Zhang, Q.; Franklin, J.L.; Mortlock, D.P.; Fuhrmann , S.; et al. A chimeric egfr protein reporter mouse reveals egfr localization and trafficking in vivo. Cell Rep. 2017, 19, 1257–1267. [Google Scholar]
- van Landeghem, L.; Chevalier, J.; Mahe, M.M.; Wedel, T. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am. J. Physiol. Gastro. Liver Physiol. 2011, 300, G976–G987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, O.; Beumer, J.; Wiebrands, K. Induced quiescence of Lgr5+ stem cells in intestinal organoids enables differentiation of hormone-producing enteroendocrine cells. Cell Stem Cell 2017, 20, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.Y.; Stange, D.E.; Page, M.E.; Buzacki, S. Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat. Cell Biol. 2012, 14, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reynies, A. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Schepers, A.G.; van Es, J.H.; Simons, B.D.; Clevers, H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014, 15, 62–69. [Google Scholar] [CrossRef]
- Vermeulen, L.; Morrissey, E.; van der Heijden, M.; Nicholson, A.M.; Sottoriva, A. Defining stem cell dynamics in models of intestinal tumor initiation. Science 2013, 342, 995–998. [Google Scholar] [CrossRef]
- de Bony, E.J.; Jonckheere, N.; Vincent, A.; Seuningen, I.V. Comprehensive identification of long noncoding RNAs in colorectal cancer. Oncotarget 2018, 9, 27605–27629. [Google Scholar]
- Dou, Y.; Cha, D.J.; Franklin, J.N.; Higginbotham, J.N. Circular RNAs are down-regulated in KRAS mutant colon cancer cells and can be transferred to exosomes. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Hinger, S.A.; Cha, D.J.; Franlin, J.L.; Higginbotham, J.N. Diverse long RNAs are differentially sorted into extracellular vesicles secreted by colorectal cancer cells. Cell Rep. 2018, 25, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Da Wang, Y.S.; Zhang, T.; Xie, H.; Jiang, X. SP1-induced lncRNA TINCR overexpression contributes to colorectal cancer progression by sponging miR-7-5p. Aging (Albany NY) 2019, 11, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.D.; Pedersen, S.K.; Brown, G.S. Colorectal neoplasia differentially expressed (CRNDE), a novel gene with elevated expression in colorectal adenomas and adenocarcinomas. Genes Cancer 2011, 2, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, B.C.; Graham, L.D.; Molloy, P.L. CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wang, Y.; Wang, H.; Duan, Z.; Wang, H. Long noncoding RNA CRNDE stabilized by hnRNPUL2 accelerates cell proliferation and migration in colorectal carcinoma via activating Ras/MAPK signaling pathways. Cell Death Dis. 2017, 8, e2862. [Google Scholar] [CrossRef] [PubMed]
- Saijo, S.; Kuwano, Y.; Tange, S.; Rokutan, K.; Nishida, K. A novel long non-coding RNA from the HOXA6-HOXA5 locus facilitates colon cancer cell growth. BMC Cancer 2019, 19, 532. [Google Scholar]
- Committee, E.N. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Eph nomenclature committee. Cell 1997, 90, 403–404. [Google Scholar] [CrossRef] [Green Version]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef]
- Batlle, E.; Henderson, J.T.; Beghtel, H.; van den Born, M.M.W. β-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/EphrinB. Cell 2002, 111, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, J.; Henderson, J.T.; Beghtel, H.; van den Born, M.M.W. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell 2006, 125, 1151–1163. [Google Scholar] [CrossRef] [Green Version]
- Genander, M.; Halford, M.M.; Xu, N.J.; Eriksson, M.; Yu, Z.; Qiu, Z. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell 2009, 139, 679–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Bacani, J.; Beghtel, H.; Jonkeer, S.; Greogorieff, A. EphB receptor activity suppresses colorectal cancer progression. Nature 2005, 435, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Zhong, F.; Bheddah, S.; Grabsch, H.I.; Frantz, G.D.; Mueller, W.; Kavi, V.; Quirke, P.; Polakis, P.; Koeppen, H. EphB2 is a prognostic factor in colorectal cancer. Clin. Cancer Res. 2005, 11, 5181–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugli, A.; Spichtin, H.; Maurer, R.; Mirlacher, M.; Kiefer, J. EphB2 expression across 138 human tumor types in a tissue microarray: High levels of expression in gastrointestinal cancers. Clin. Cancer Res. 2005, 11, 6450–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Batlle, E. EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer Res. 2006, 66, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortina, C.; Palomo Ponce, S.; Iglesias, M. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat. Gen. 2007, 39, 1376–1383. [Google Scholar]
- Zhuo, W.; Liu, Y.; Li, S.; Guo, D.; Sun, Q.; Jin, J.; Rao, X.; Li, M.; Sun, M.; Jiang, M.; et al. Long Noncoding RNA GMAN, Up-regulated in Gastric Cancer Tissues, Is Associated With Metastasis in Patients and Promotes Translation of Ephrin A1 by Competitively Binding GMAN-AS. Gastroenterology 2019, 156, 676–691. [Google Scholar]
- Xiang, Y.; Huang, Y.; Sun, H.; Pan, Y.; Wu, M.; Zhang, J. Deregulation of miR-520d-3p promotes hepatocellular carcinoma development via lncRNA MIAT regulation and EPHA2 signaling activation. Biomed. Pharmacother. 2019, 109, 1630–1639. [Google Scholar] [CrossRef]
- Yu, L.; Chen, D.; Song, J. LncRNA SNHG16 promotes non-small cell lung cancer development through regulating EphA2 expression by sponging miR-520a-3p. Thoracic Cancer 2020, 11, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Christensen, L.L. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol. Oncol. 2016, 10, 1266–1282. [Google Scholar] [CrossRef]
- Li, S.; Zhang, S.; Chen, J. c-Myc induced upregulation of long non-coding RNA SNHG16 enhances progression and carcinogenesis in oral squamous cell carcinoma. Cancer Gene Ther. 2019, 26, 400–410. [Google Scholar] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harbor Perspect. Biol. 2016, 8, 1–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, P.L.; Rosenberg, I.M.; Xavier, R.J.; Koh, T. Transforming growth factor-β mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am. J. Pathol. 2003, 162, 597–608. [Google Scholar] [CrossRef]
- Wang, X. LncRNA SNHG6 promotes proliferation, invasion and migration in colorectal cancer cells by activating TGF-β/smad signaling pathway via targeting upf1 and inducing EMT via regulation of ZEB1. Int. J. Med. Sci. 2019, 16, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zheng, L.; Yuan, Q.; Crane, J.L.; Zhou, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.; Hill, C.S. How the smads regulate transcription. Int. J. Biochem. Cell Biol. 2008, 40, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.M.; Calabrese, P.P.; Miller, A.J.; Muñoz, N.M.; Grady, W.M.; Shibata, D.; Liskay, R.M. Single cell lineage tracing reveals a role for TgfβR2 in intestinal stem cell dynamics and differentiation. Proc. Nat. Acad. Sci. USA 2016, 113, 12192–12197. [Google Scholar] [CrossRef] [Green Version]
- Múnera, J.O. Differentiation of human pluripotent stem cells into colonic organoids via transient activation of BMP signaling. Cell Stem Cell 2017, 21, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, H.; Ito, N.; Tamura, S.; Matsuda, Y.; Inada, M. Circulating transforming growth factor β1 as a predictor of liver metastasis after resection in colorectal cancer. Clin. Cancer Res. 2001, 7, 1258–1262. [Google Scholar]
- Oft, M.; Akhurst, R.J.; Balmain, A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat. Cell Biol. 2002, 4, 487–494. [Google Scholar]
- Grady, W.M.; Willis, J.E.; Trobridge, P. Proliferation and Cdk4 expression in microsatellite unstable colon cancers with TGFBR2 mutations. Int. J. Cancer 2006, 118, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Halder, S.K.; Kashikar, N.D.; Cho, Y.J.; Datta, A. Antimetastatic role of smad4 signaling in colorectal cancer. Gastroenterology 2010, 138, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Geng, J.; Zhang, Y. LncRNA CASC9 interacts with CPSF3 to regulate TGF-β signaling in colorectal cancer. J. Experiment. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, M.; Gro, M.; Goyal, A. The long noncoding RNA cancer susceptibility 9 and RNA binding protein heterogeneous nuclear ribonucleoprotein L form a complex and coregulate genes linked to AKT signaling. Hepatology 2018, 68, 1817–1832. [Google Scholar] [CrossRef]
- Liang, Y.; Chen, X.; Wu, Y.; Li, J.; Zhang, S.; Wang, K. LncRNA CASC9 promotes esophageal squamous cell carcinoma metastasis through upregulating LAMC2 expression by interacting with the CREB-binding protein. Cell Death Different. 2018, 25, 1980–1995. [Google Scholar] [CrossRef]
- Shang, C.; Sun, L.; Zhang, J.; Zhao, B.; Chen, X. Silence of cancer susceptibility candidate 9 inhibits gastric cancer and reverses chemoresistance. Oncotarget 2017, 8, 15393–15398. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Hu, X.; Mao, J.; Wu, Y.; Liu, H.; Shen, J.; Yu, J. The long noncoding RNA TUG1 is required for TGF-β/TWIST1/EMT-mediated metastasis in colorectal cancer cells. Cell Death Dis. 2020, 11, 1–10. [Google Scholar]
- Gooding, A.J.; Zhang, B.; Jahanbani, F.K.; Gilmore, H.L. The lncRNA BORG drives breast cancer metastasis and disease recurrence. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ma, J.; Zhang, X.; Tai, X.; Liu, L. Long non-coding RNA (lncRNA) BMP/OPResponsive Gene (BORG) promotes development of chemoresistance of colorectal cancer cells to carboplatin. Med. Sci. Monitor 2020, 26, 1–7. [Google Scholar]
- Shan, T.D.; Xu, J.H.; Yu, T.; Li, J.Y.; Zhao, L.N. Knockdown of linc-POU3F3 suppresses the proliferation, apoptosis, and migration resistance of colorectal cancer. Oncotarget 2016, 7, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhao, Y.; Zhang, W.; Bai, Y. Whole transcriptome sequencing identifies crucial genes associated with colon cancer and elucidation of their possible mechanisms of action. OncoTargets Ther. 2019, 12, 2737–2747. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [PubMed] [Green Version]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006, 124, 1283–1298. [Google Scholar]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Lee, S.-K.; Wang, J.; Ince, N.; Ouyang, N.; Min, J.; Chen, J.; Shankar, P.; Lieberman, J. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Med. 2003, 9, 347–351. [Google Scholar]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Mueller, A.C.; Cichewicz, M.A. MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol. Cell. Biol. 2015, 35, 498–513. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.T.Y.; Cho, H.; Lesch, H.P.; Gosselin, D.; Heinz, S. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013, 498, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current development of siRNA bioconjugates: From research to the clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Weng, J.; Peng, L. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. New England J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-targeted therapeutics. Cell Metabol. 2018, 27, 714–739. [Google Scholar] [CrossRef] [Green Version]
- Quemener, A.M.; Bachelot, L.; Forestier, A. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdis. Rev. 2020, 11, 1–22. [Google Scholar] [CrossRef]
- Crooke, S.T. Progress in antisense therapeutics. Med. Res. Rev. 1996, 16, 319–344. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Del. Rev. 2015, 87, 46–51. [Google Scholar]
- Gaus, H.J.; Gupta, R.; Chapell, A.E. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucl. Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef]
- Shen, W.; de Hoyos, C.L.; Migawa, M.T.; Vickers, T.A. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 2019, 37, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucl. Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Diermeir, S.; Akerman, M. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Develop. 2016, 30, 34–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschner, T.; Hammerle, M.; Eimann, M.; Hsu, J. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Dhuri, K. Antisense oligonucleotides: An emerging area in drug discovery and development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinksi, K.; Fonfara, I. A Programmable Dual-RNA—Guided. Res. Article 2012, 337, 816–822. [Google Scholar]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery approaches for therapeutic genome editing and challenges. Genes 2020, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering crispr: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nature Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| LncRNA | Phenotype | Signaling Pathway | Mechanism | References |
|---|---|---|---|---|
| lncGata6 | Stem cells, Tumorigenesis | Wnt | Enhances WNT activity | [57] |
| CCAL | Proliferation, Invasion, Migration | Wnt | Stabilizes β-Catenin/TCF-4 complex by suppression of AP-2alpha | [58] |
| BCAR4 | Proliferation, Migration | Wnt | Stabilization of β-Catenin | [59] |
| CYTOR | Proliferation, EMT | Wnt | Stabilization of β-Catenin | [60] |
| MYC locus (CCAT1, CCAT1-L, CCAT2) | Proliferation, Metastasis, Chromosomal instability | Wnt | Regulation of c-MYC expression | [64,65,66,67,68,69,70,71] |
| GLCC1 | Proliferation, Survival, Glycolysis | Wnt | Stabilization of c-Myc protein | [72] |
| MYU | Cell cycle, Proliferation | Wnt | Interacts with hnRNP-K and stabilizes CDK6 mRNA | [73] |
| FOXD2-AS1 | Proliferation, Invasion, Migration | Notch | Regulation of EMT and Notch signaling | [84] |
| FAM83H-AS1 | Proliferation, Migration | Notch | Modulates Notch signaling | [88,89] |
| LINC00152 | Cell motility, Migration | Hippo | miRNA sponging | [97] |
| MALAT1 | Survival, Migration, EMT | Hippo | miRNA sponging; Epigenetic silencing of targets | [94,99] |
| TINCR | Proliferation, Metastasis formation | EGF | miRNA sponging | [112] |
| CRNDE | Warburg effect, Proliferation | EGF | Activates RAS\MAPK pathway; Interacts with hnRNPUL2 | [115,116] |
| HOXA5 | Proliferation | EGF | Modulates EGF signaling | [117] |
| CASC9 | Anti-apoptotic, Proliferation | BMP/TGF-β | Interacts with endonuclease CPSF3 | [145] |
| TUG1 | EMT, Metastasis formation | BMP/TGF-β | Regulates expression of Vimentin and TWIST1 | [149] |
| linc-POU3F3 | Survival, Proliferation, Migration | BMP/TGF-β | Regulates EMT-associated genes, Inhibits BMP signaling | [152] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarzmueller, L.; Bril, O.; Vermeulen, L.; Léveillé, N. Emerging Role and Therapeutic Potential of lncRNAs in Colorectal Cancer. Cancers 2020, 12, 3843. https://doi.org/10.3390/cancers12123843
Schwarzmueller L, Bril O, Vermeulen L, Léveillé N. Emerging Role and Therapeutic Potential of lncRNAs in Colorectal Cancer. Cancers. 2020; 12(12):3843. https://doi.org/10.3390/cancers12123843
Chicago/Turabian StyleSchwarzmueller, Laura, Oscar Bril, Louis Vermeulen, and Nicolas Léveillé. 2020. "Emerging Role and Therapeutic Potential of lncRNAs in Colorectal Cancer" Cancers 12, no. 12: 3843. https://doi.org/10.3390/cancers12123843