Overcoming the Hurdles of Autologous T-Cell-Based Therapies in B-Cell Non-Hodgkin Lymphoma

 ,
,
Abstract
Simple Summary
Abstract

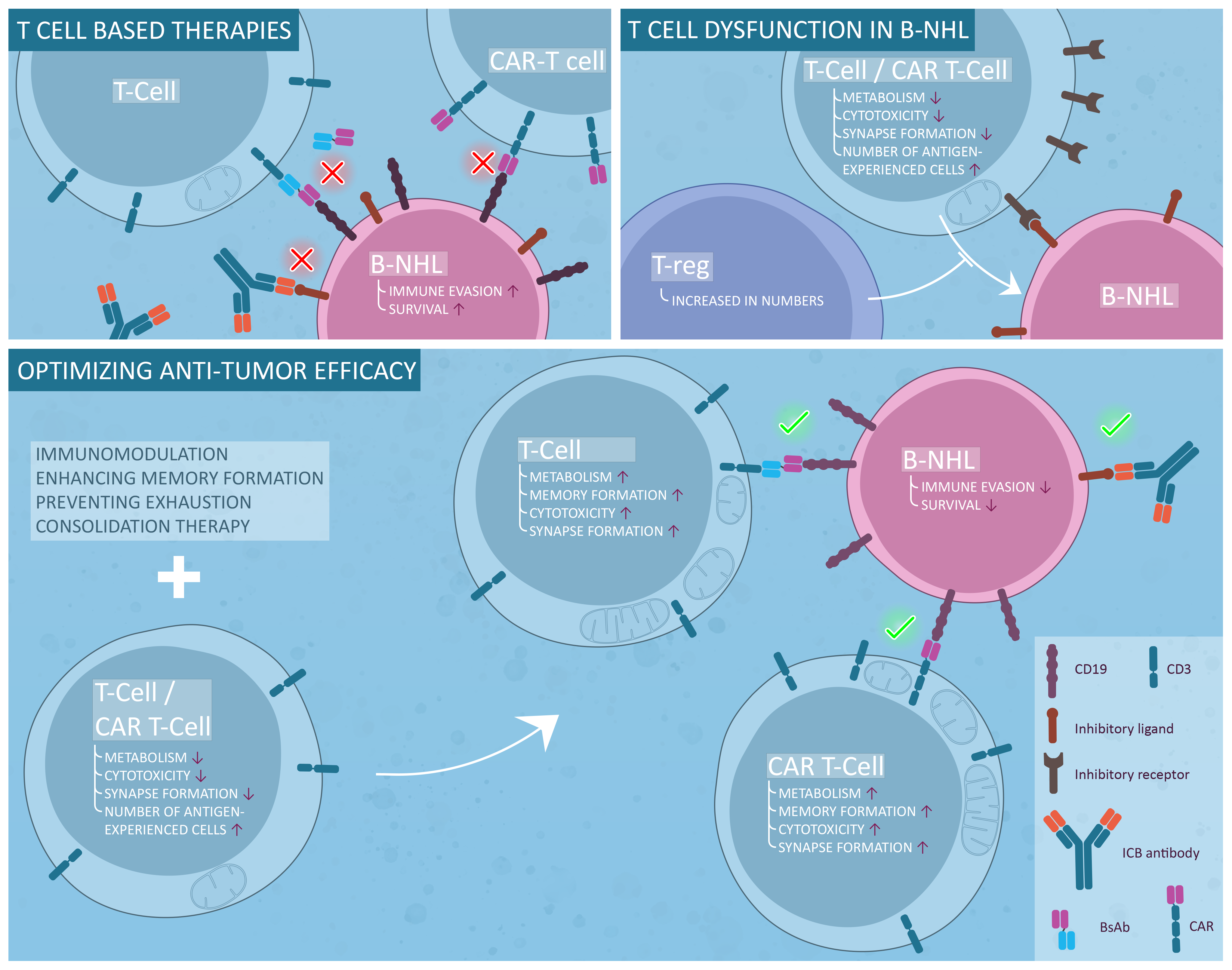
1. Introduction
2. Clinical Data
2.1. Immune Checkpoint Blockade
2.2. Bispecific Antibody Therapy
2.3. Chimeric Antigen Receptor Therapy
3. T-Cell Immune Surveillance in NHL
3.1. Mutational Load in B-NHL
3.2. T-Cell Skewing
3.3. Inhibitory Receptor Expression and Exhaustion in B-NHL
3.4. Functional Defects
3.5. T-Cell Metabolism in B-NHL
3.6. Regulatory T-Cells and T Follicular Helper Cells
4. Possible Solutions
4.1. Combination Therapy Improves ICB and BsAbs and Potentially CARs
4.2. Consolidation Therapy
4.3. Improving CAR T-Cell Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet 2017, 390, 298–310. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Flinn, I.W.; Advani, R.; Sehn, L.H.; Diefenbach, C.; Kolibaba, K.; Press, O.W.; Salles, G.; Tilly, H.; Chen, A.I.; et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: Final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019, 6, e254–e265. [Google Scholar] [CrossRef]
- Palanca-Wessels, M.C.A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L.H.; Flinn, I.; Patel, M.R.; Sangha, R.; Hagenbeek, A.; Advani, R.; et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: A phase 1 study. Lancet Oncol. 2015, 16, 704–715. [Google Scholar] [CrossRef]
- Jacobsen, E.D.; Sharman, J.P.; Oki, Y.; Advani, R.H.; Winter, J.N.; Bello, C.M.; Spitzer, G.; Palanca-Wessels, M.C.; Kennedy, D.A.; Levine, P.; et al. Brentuximab vedotin demonstrates objective responses in a phase 2 study of relapsed/refractory DLBCL with variable CD30 expression. Blood 2015, 125, 1394–1402. [Google Scholar] [CrossRef]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 2352–2354. [Google Scholar] [CrossRef]
- Bose, P.; Gandhi, V.V.; Konopleva, M.Y. Pathways and mechanisms of venetoclax resistance. Leuk. Lymphoma 2017, 58, 2026–2039. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Meti, N.; Esfahani, K.; Johnson, N.A. The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma. Cancers 2018, 10, 204. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leuk. 2004, 18, 676–684. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- O’Mahony, D.; Morris, J.C.; Quinn, C.; Gao, W.; Wilson, W.H.; Gause, B.; Pittaluga, S.; Neelapu, S.; Brown, M.; Fleisher, T.A.; et al. A Pilot Study of CTLA-4 Blockade after Cancer Vaccine Failure in Patients with Advanced Malignancy. Clin. Cancer Res. 2007, 13, 958–964. [Google Scholar] [CrossRef]
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I Safety and Pharmacokinetic Study of CT-011, a Humanized Antibody Interacting with PD-1, in Patients with Advanced Hematologic Malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Hurvitz, S.A.; Koenig, P.A.; LaPlant, B.R.; Kabat, B.F.; Fernando, D.; Habermann, T.M.; Inwards, D.J.; Verma, M.; Yamada, R.; et al. Phase I Study of Ipilimumab, an Anti-CTLA-4 Monoclonal Antibody, in Patients with Relapsed and Refractory B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2009, 15, 6446–6453. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Laplant, B.; Witzig, T.E.; Johnston, P.B.; Colgan, J.P.; Rech, K.L.; Leis, J.F.; Feldman, A.L.; He, R.; Nowakowski, G.S.; et al. PD-1 Blockade with Pembrolizumab in Relapsed Low Grade Non-Hodgkin Lymphoma. Blood 2017, 130, 4055. [Google Scholar]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Ansell, S.M.; Minnema, M.C.; Johnson, P.; Timmerman, J.M.; Armand, P.; Shipp, M.A.; Rodig, S.J.; Ligon, A.H.; Roemer, M.G.; Reddy, N.; et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J. Clin. Oncol. 2019, 37, 481–489. [Google Scholar] [CrossRef]
- Armand, P.; Rodig, S.; Melnichenko, V.; Thieblemont, C.; Bouabdallah, K.; Tumyan, G.; Özcan, M.; Portino, S.; Fogliatto, L.; Caballero, M.D.; et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 3291–3299. [Google Scholar] [CrossRef]
- Ansell, S.; Gutierrez, M.E.; Shipp, M.A.; Gladstone, D.; Moskowitz, A.; Borello, I.; Popa-McKiver, M.; Farsaci, B.; Zhu, M.L.; Lesokhin, A.M.; et al. A Phase 1 Study of Nivolumab in Combination with Ipilimumab for Relapsed or Refractory Hematologic Malignancies (CheckMate 039). Blood 2016, 128, 183. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Westin, J.; Fowler, N.; Fanale, M.A.; Samaniego, F.; Oki, Y.; Obi, C.; Cao, J.; Cheng, X.; Ma, M.C.J.; et al. Response rates with pembrolizumab in combination with rituximab in patients with relapsed follicular lymphoma: Interim results of an on open-label, phase II study. J. Clin. Oncol. 2017, 35, 7519. [Google Scholar] [CrossRef]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.B.; Fanale, M.A.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef]
- Witzig, T.E.; Vukov, A.M.; Habermann, T.M.; Geyer, S.; Kurtin, P.J.; Friedenberg, W.R.; White, W.L.; Chalchal, H.I.; Flynn, P.J.; Fitch, T.R.; et al. Rituximab Therapy for Patients With Newly Diagnosed, Advanced-Stage, Follicular Grade I Non-Hodgkin’s Lymphoma: A Phase II Trial in the North Central Cancer Treatment Group. J. Clin. Oncol. 2005, 23, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Burris, H.A., 3rd; Morrissey, L.H.; Litchy, S.; Scullin, D.C., Jr.; Bearden, J.D., 3rd; Richards, P.; Greco, F.A. Rituximab monoclonal antibody as initial systemic therapy for patients with low-grade non-Hodgkin lymphoma. Blood 2000, 95, 3052–3056. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.A.; Grillo-Lopez, A.J.; White, C.A.; McLaughlin, P.; Czuczman, M.S.; Link, B.K.; Maloney, D.G.; Weaver, R.L.; Rosenberg, J.; Levy, R. Rituximab Anti-CD20 Monoclonal Antibody Therapy in Non-Hodgkin’s Lymphoma: Safety and Efficacy of Re-Treatment. J. Clin. Oncol. 2000, 18, 3135–3143. [Google Scholar] [CrossRef]
- Palomba, M.; Till, B.; Park, S.; Morschhauser, F.; Cartron, G.; Marks, R.; Penuel, E.; Chitra, S.; Kuhn, M.; Popplewell, L. A phase Ib study evaluating the safety and clinical activity of atezolizumab combined with obinutuzumab in patients with relapsed or refractory non-hodgkin lymphoma (NHL). Hematol. Oncol. 2017, 35, 137–138. [Google Scholar] [CrossRef]
- Younes, A.; John, B.M.; Diefenbach, C.S.; Ferrari, S.; Kahn, C.; Sharman, J.P.; Tani, M.; Ujjani, C.S.; Vitolo, U.; Yuen, S.; et al. Safety and Efficacy of Atezolizumab in Combination with Obinutuzumab and Bendamustine in Patients with Previously Untreated Follicular Lymphoma: An Interim Analysis. Blood 2017, 130, 481. [Google Scholar]
- Younes, A.; Burke, J.M.; Cheson, B.; Diefenbach, C.; Ferrari, S.; Hahn, U.; Hawkes, E.; Khan, C.; Lossos, I.S.; Musuraka, G.; et al. Safety and Efficacy of Atezolizumab in Combination with Rituximab Plus CHOP in Previously Untreated Patients with Diffuse Large B-Cell Lymphoma (DLBCL): Primary Analysis of a Phase I/II Study. Blood 2018, 132, 2969. [Google Scholar] [CrossRef]
- Bond, D.A.; Yildiz, V.; Wei, L.; Alinari, L.; William, B.M.; Brammer, J.E.; Christian, B.A.; Blum, K.A.; Maddocks, K.J. A Phase I Study of Nivolumab and Lenalidomide in Relapsed/ Refractory B Cell Lymphoma. Blood 2019, 134, 4091. [Google Scholar] [CrossRef]
- Casulo, C.; Santoro, A.; Ando, K.; Le Gouill, S.; Ruan, J.; Radford, J.; Arcaini, L.; Pinto, A.; Bouabdallah, R.; Izutsu, K.; et al. Durvalumab (Anti PD-L1) As Monotherapy or in Combination Therapy for Relapsed/Refractory (r/r) Diffuse Large B-Cell Lymphoma (DLBCL) and Follicular Lymphoma (FL): A Subgroup Analysis from the Phase 1/2 Fusion NHL-001 Global Multicenter Trial. Blood 2019, 134, 5320. [Google Scholar] [CrossRef]
- Khouri, I.F.; Curbelo, I.F.; Bassett, R.L.; Allison, J.P.; Gulbis, A.M.; Seliger, B.; Turturro, F.; Jabbour, E.J.; Milton, D.R.; Vence, L.M. Ipilimumab plus Lenalidomide after Allogeneic and Autologous Stem Cell Transplantation for Patients with Lymphoid Malignancies. Clin. Cancer Res. 2017, 24, 1011–1018. [Google Scholar] [CrossRef]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.-J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, N.L.; Costello, B.A.; LaPlant, B.R.; Ansell, S.M.; Kuruvilla, J.G.; Reeder, C.B.; Thye, L.S.; Anderson, D.M.; Krysiak, K.; Ramirez, C.; et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: A phase 2 consortium trial. Blood 2018, 131, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Goy, A.; Mehta, A.; Ramchandren, R.; Pagel, J.M.; Svoboda, J.; Guan, S.; Hill, J.S.; Kwei, K.; Liu, E.A.; et al. Safety and activity of ibrutinib in combination with durvalumab in patients with relapsed or refractory follicular lymphoma or diffuse large B-cell lymphoma. Am. J. Hematol. 2020, 95, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Santoro, A.; Gritti, G.; Brice, P.; Barr, P.M.; Kuruvilla, J.; Cunningham, D.; Kline, J.; Johnson, N.A.; Mehta-Shah, N.; et al. Nivolumab Combined With Brentuximab Vedotin for Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma: Efficacy and Safety From the Phase II CheckMate 436 Study. J. Clin. Oncol. 2019, 37, 3081–3089. [Google Scholar] [CrossRef]
- Frigault, M.J.; Armand, P.; Redd, R.; Jeter, E.; Merryman, R.W.; Coleman, K.C.; Herrera, A.F.; Dahi, P.; Nieto, Y.; LaCasce, A.; et al. PD-1 blockade for diffuse large B-cell lymphoma after autologous stem cell transplantation. Blood Adv. 2020, 4, 122–126. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Coyle, L.; Morley, N.J.; Rambaldi, A.; Mason, K.D.; Verhoef, G.; Furness, C.L.; Zhang, A.; Jung, A.S.; Cohan, D.; Franklin, J.L. Open-Label, phase 2 study of blinatumomab as second salvage therapy in adults with relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Leuk. Lymphoma 2020, 61, 2103–2112. [Google Scholar] [CrossRef]
- Poh, C.; Frankel, P.; Ruel, C.; Abedi, M.; Schwab, E.; Costello, C.L.; Zain, J.; Budde, L.E.; William, B.M.; Foss, F.M.; et al. Blinatumomab/Lenalidomide in Relapsed/Refractory Non-Hodgkin’s Lymphoma: A Phase I California Cancer Consortium Study of Safety, Efficacy and Immune Correlative Analysis. Blood 2019, 134, 760. [Google Scholar] [CrossRef]
- Katz, D.A.; Chu, M.P.; David, K.A.; Thieblemont, C.; Morley, N.J.; Khan, S.S.; Chen, Y.; Kalabus, J.; Morris, J.; Anderson, A.; et al. Open-Label, Phase 2 Study of Blinatumomab after First-Line Rituximab-Chemotherapy in Adults with Newly Diagnosed, High-Risk Diffuse Large B-Cell Lymphoma. Blood 2019, 134, 4077. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bartlett, N.L.; Assouline, S.; Yoon, S.-S.; Bosch, F.; Sehn, L.H.; Cheah, C.Y.; Shadman, M.; Gregory, G.P.; Ku, M.; et al. Mosunetuzumab Induces Complete Remissions in Poor Prognosis Non-Hodgkin Lymphoma Patients, Including Those Who Are Resistant to or Relapsing After Chimeric Antigen Receptor T-Cell (CAR-T) Therapies, and Is Active in Treatment through Multiple Lines. Blood 2019, 134, 6. [Google Scholar] [CrossRef]
- Hutchings, M.; Lugtenburg, P.; Mous, R.; Clausen, M.R.; Chamuleau, M.; Linton, K.; Rule, S.; Lopez, J.S.; Oliveri, R.S.; Demarco, D.; et al. Epcoritamab (GEN3013; DuoBody-CD3×CD20) to induce complete response in patients with relapsed/refractory B-cell non-Hodgkin lymphoma (B-NHL): Complete dose escalation data and efficacy results from a phase I/II trial. J. Clin. Oncol. 2020, 38, 8009. [Google Scholar] [CrossRef]
- Bannerji, R.; Advani, R.H.; Brown, J.R.; Arnason, J.E.; Barnes, J.A.; Allan, J.N.; Ansell, S.M.; O’Brien, S.M.; Chavez, J.C.; Adriaens, L.; et al. Safety and Preliminary Clinical Activity of REGN1979, an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Patients with B-NHL Previously Treated with CD20-Directed Antibody Therapy. Blood 2017, 130, 1550. [Google Scholar]
- Bannerji, M.R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.; Ansell, S.M.; O’Brien, S.M.; Duell, J.; Martin, F.P.; Joyce, R.M.; et al. Odronextamab (REGN1979), a Human CD20 x CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-Hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Morschhauser, F.; Carlo-Stella, C.; Offner, F.; Salles, G.A.; Hutchings, M.; Iacoboni, G.; Sureda, A.; Crump, M.; Martinez-Lopez, J.; Thomas, D.; et al. Dual CD20-Targeted Therapy With Concurrent CD20-TCB and Obinutuzumab Shows Highly Promising Clinical Activity and Manageable Safety in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma: Preliminary Results From a Phase Ib Trial. Blood 2019, 134, 1584. [Google Scholar] [CrossRef]
- Wong, R.; Pepper, C.; Brennan, P.; Nagorsen, D.; Man, S.; Fegan, C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematol. 2013, 98, 1930–1938. [Google Scholar] [CrossRef]
- Robinson, H.R.; Qi, J.; Cook, E.M.; Nichols, C.; Dadashian, E.L.; Underbayev, C.; Herman, S.E.M.; Saba, N.S.; Keyvanfar, K.; Sun, C.; et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood 2018, 132, 521–532. [Google Scholar] [CrossRef]
- Martens, A.W.J.; Janssen, S.R.; Derks, I.A.M.; Iii, H.C.A.; Izhak, L.; Van Kampen, R.; Tonino, S.H.; Eldering, E.; Van Der Windt, G.J.W.; Kater, A.P. CD3xCD19 DART molecule treatment induces non-apoptotic killing and is efficient against high-risk chemotherapy and venetoclax-resistant chronic lymphocytic leukemia cells. J. Immunother. Cancer 2020, 8, e000218. [Google Scholar] [CrossRef]
- Circosta, P.; Elia, A.R.; Landra, I.; Machiorlatti, R.; Todaro, M.; Aliberti, S.; Brusa, D.; Deaglio, S.; Chiaretti, S.; Bruna, R.; et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. OncoImmunology 2018, 7, e1341032. [Google Scholar] [CrossRef] [PubMed]
- Gohil, S.H.; Evans, R.; Harasser, M.; El-Kholy, M.; Paredes-Moscosso, S.; Della Peruta, M.; Nathwani, A.C. Ibrutinib enhances the efficacy of ROR1 bispecific T cell engager mediated cytotoxicity in chronic lymphocytic leukaemia. Br. J. Haematol. 2019, 186, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.B.; Rivière, I.; Sénéchal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Halton, E.; Lamanna, N.; et al. Autologous CD19-Targeted CAR T Cells in Patients with Residual CLL following Initial Purine Analog-Based Therapy. Mol. Ther. 2018, 26, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.A.; Casulo, C.; Munshi, P.N.; Maloney, D.G.; De Vos, S.; et al. Interim analysis of ZUMA-5: A phase II study of axicabtagene ciloleucel (axi-cel) in patients (pts) with relapsed/refractory indolent non-Hodgkin lymphoma (R/R iNHL). J. Clin. Oncol. 2020, 38, 8008. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; Steinmetz, R.N.; Riddell, S.R.; et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood 2019, 134, 636–640. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor–Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Abramson, M.J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, D.M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, M.C.; et al. Pivotal Safety and Efficacy Results from Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (liso-cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood 2019, 134, 241. [Google Scholar] [CrossRef]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther. Oncol. 2019, 15, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, T.; Soumerai, J.D.; Dorritie, K.A.; Stephens, D.M.; Riedell, P.A.; Arnason, J.E.; Kipps, T.J.; Gillenwater, H.H.; Gong, L.; Dubovsky, J.A.; et al. Rapid Undetectable MRD (uMRD) Responses in Patients with Relapsed/Refractory (R/R) Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL) Treated with Lisocabtagene Maraleucel (liso-cel), a CD19-Directed CAR T Cell Product: Updated Results from Transcend CLL 004, a Phase 1/2 Study Including Patients with High-Risk Disease Previously Treated with Ibrutinib. Blood 2019, 134, 503. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Mcgettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Popplewell, L.L.; Wagner, J.R.; Naranjo, A.; Blanchard, M.S.; Mott, M.R.; Norris, A.P.; Wong, C.W.; Urak, R.Z.; Chang, W.-C.; et al. Phase 1 studies of central memory–derived CD19 CAR T–cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016, 127, 2980–2990. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Gauthier, J.; Hirayama, A.V.; Purushe, J.; Hay, K.A.; Lymp, J.; Li, D.H.; Yeung, C.C.S.; Sheih, A.; Pender, B.S.; Hawkins, R.M.; et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020, 135, 1650–1660. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Anandappa, A.J.; Sun, J.; Kim, J.; Leet, D.E.; Bozym, D.J.; Chen, C.; Williams, L.; Shukla, S.A.; Zhang, W.; et al. A cloning and expression system to probe T-cell receptor specificity and assess functional avidity to neoantigens. Blood 2018, 132, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nat. Cell Biol. 2017, 543, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.D.; Badovinac, V.P. Defining Memory CD8 T Cell. Front. Immunol. 2018, 9, 2692. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Kubota, M.; Yudanin, N.; Turner, D.; Camp, P.; Thome, J.J.C.; Bickham, K.L.; Lerner, H.; Goldstein, M.; Sykes, M.; et al. Distribution and Compartmentalization of Human Circulating and Tissue-Resident Memory T Cell Subsets. Immunity 2013, 38, 187–197. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Pourgheysari, B.; Bruton, R.; Parry, H.; Billingham, L.; Fegan, C.; Murray, J.; Moss, P. The number of cytomegalovirus-specific CD4+ T cells is markedly expanded in patients with B-cell chronic lymphocytic leukemia and determines the total CD4+ T-cell repertoire. Blood 2010, 116, 2968–2974. [Google Scholar] [CrossRef]
- D’Arena, G.; Laurenti, L.; Minervini, M.M.; Deaglio, S.; Bonello, L.; De Martino, L.; De Padua, L.; Savino, L.; Tarnani, M.; De Feo, V.; et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk. Res. 2011, 35, 363–368. [Google Scholar] [CrossRef]
- Tonino, S.H.; Van De Berg, P.J.; La Yong, S.; Berge, I.J.T.; Kersten, M.J.; Van Lier, R.A.W.; Van Oers, M.H.; Kater, A.P. Expansion of effector T cells associated with decreased PD-1 expression in patients with indolent B cell lymphomas and chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 1785–1794. [Google Scholar] [CrossRef]
- Mackus, W.J.M.; Frakking, F.N.J.; Grummels, A.; Gamadia, L.E.; De Bree, G.J.; Hamann, D.; Van Lier, R.A.; Van Oers, M.H.J. Expansion of CMV-specific CD8+CD45RA+CD27- T cells in B-cell chronic lymphocytic leukemia. Blood 2003, 102, 1057–1063. [Google Scholar] [CrossRef]
- Nunes, C.; Wong, R.; Mason, M.; Fegan, C.; Man, S.; Pepper, C. Expansion of a CD8+PD-1+ Replicative Senescence Phenotype in Early Stage CLL Patients Is Associated with Inverted CD4:CD8 Ratios and Disease Progression. Clin. Cancer Res. 2011, 18, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.P.; Contesti, J.; Huergo-Zapico, L.; Lopez-Larrea, C.; Fernández-Guizán, A.; Acebes-Huerta, A.; Gonzalez-Huerta, A.J.; Gonzalez, E.; Fernandez-Alvarez, C.; Gonzalez, S. Prognostic significance of CD8 and CD4 T cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2010, 51, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Weiss, L.; Gassner, F.; Rubenzer, G.; Holler, C.; Greil, R. Difference in the relative distribution of CD4+ T-cell subsets in B-CLL with mutated and unmutated immunoglobulin (Ig) VH genes: Implication for the course of disease. J. Immunother. 2009, 32, 302–309. [Google Scholar] [CrossRef]
- De Weerdt, I.; Hofland, T.; De Boer, R.; Dobber, J.A.; Dubois, J.; Van Nieuwenhuize, D.; Mobasher, M.; De Boer, F.; Hoogendoorn, M.; Velders, G.A.; et al. Distinct immune composition in lymph node and peripheral blood of CLL patients is reshaped during venetoclax treatment. Blood Adv. 2019, 3, 2642–2652. [Google Scholar] [CrossRef]
- Palma, M.; Gentilcore, G.; Heimersson, K.; Mozaffari, F.; Näsman-Glaser, B.; Young, E.; Rosenquist, R.; Hansson, L.; Österborg, A.; Mellstedt, H. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 2017, 102, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Pfeifer, D.; Bartholomé, K.; Follo, M.; Timmer, J.; Fisch, P.; Veelken, H. Definition and characterization of the systemic T-cell dysregulation in untreated indolent B-cell lymphoma and very early CLL. Blood 2011, 117, 3836–3846. [Google Scholar] [CrossRef]
- Rossmann, E.D.; Lewin, N.; Jeddi-Tehrani, M.; Österborg, A.; Mellstedt, H. Intracellular T cell cytokines in patients with B cell chronic lymphocytic leukaemia (B-CLL). Eur. J. Haematol. 2002, 68, 299–306. [Google Scholar] [CrossRef]
- Görgün, G.; Holderried, T.A.W.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Podhorecka, M.; Dmoszynska, A.; Rolinski, J.; Wasik, E. T type 1/type 2 subsets balance in B-cell chronic lymphocytic leukemia—The three-color flow cytometry analysis. Leuk. Res. 2002, 26, 657–660. [Google Scholar] [CrossRef]
- Pangault, C.; Amé-Thomas, P.; Ruminy, P.; Rossille, D.; Caron, G.; Baia, M.; De Vos, J.; Roussel, M.; Monvoisin, C.; Lamy, T.; et al. Follicular lymphoma cell niche: Identification of a preeminent IL-4-dependent TFH–B cell axis. Leukemia 2010, 24, 2080–2089. [Google Scholar] [CrossRef]
- Hilchey, S.P.; Rosenberg, A.F.; Hyrien, O.; Secor-Socha, S.; Cochran, M.; Brady, M.T.; Wang, J.-C.E.; Sanz, I.; Burack, W.R.; Quataert, S.A.; et al. Follicular lymphoma tumor–infiltrating T-helper (TH) cells have the same polyfunctional potential as normal nodal TH cells despite skewed differentiation. Blood 2011, 118, 3591–3602. [Google Scholar] [CrossRef]
- Glas, A.M.; Knoops, L.; Delahaye, L.; Kersten, M.J.; Kibbelaar, R.E.; Wessels, L.A.; Van Laar, R.; Van Krieken, J.H.J.M.; Baars, J.W.; Raemaekers, J.; et al. Gene-Expression and Immunohistochemical Study of Specific T-Cell Subsets and Accessory Cell Types in the Transformation and Prognosis of Follicular Lymphoma. J. Clin. Oncol. 2007, 25, 390–398. [Google Scholar] [CrossRef]
- Edinger, J.T.; Kant, J.A.; Swerdlow, S.H. Cutaneous Marginal Zone Lymphomas Have Distinctive Features and Include 2 Subsets. Am. J. Surg. Pathol. 2010, 34, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Koulis, A.; Diss, T.; Isaacson, P.G.; Dogan, A. Characterization of tumor-infiltrating T lymphocytes in B-cell lymphomas of mucosa-associated lymphoid tissue. Am. J. Pathol. 1997, 151, 1353–1360. [Google Scholar] [PubMed]
- Riedel, S.; Kraft, M.; Kucharzik, T.; Pauels, H.G.; Tiemann, M.; Steinbuchel, A.; Domschke, W.; Lugering, N. CD4 + Th1-cells Predominate in Low-grade B-Cell Lymphoma of Gastric Mucosa-associated Lymphoid Tissue (MALT type). Scand. J. Gastroenterol. 2001, 36, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Fabert, C.; Soubeyran, I.; Velasco, V.; Parrens, M.; Jeannet, R.; Lereclus, E.; Gachard, N.; Feuillard, J.; Faumont, N. Inflamed phenotype of splenic marginal zone B-cell lymphomas with expression of PD-L1 by intratumoral monocytes/macrophages and dendritic cells. Cell. Mol. Immunol. 2019, 16, 621–624. [Google Scholar] [CrossRef]
- Song, D.G.; Ye, Q.; Carpenito, C.; Poussin, M.; Wang, L.P.; Ji, C.; Figini, M.; June, C.H.; Coukos, G.; Powell, D.J., Jr. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011, 71, 4617–4627. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef]
- Andorsky, D.J.; Yamada, R.E.; Said, J.; Pinkus, G.S.; Betting, D.J.; Timmerman, J.M. Programmed Death Ligand 1 Is Expressed by Non-Hodgkin Lymphomas and Inhibits the Activity of Tumor-Associated T Cells. Clin. Cancer Res. 2011, 17, 4232–4244. [Google Scholar] [CrossRef]
- Li, L.; Zhang, J.; Chen, J.; Xu-Monette, Z.Y.; Miao, Y.; Xiao, M.; Young, K.H.; Wang, S.; Medeiros, L.J.; Wang, M.; et al. B-cell receptor–mediated NFATc1 activation induces IL-10/STAT3/PD-L1 signaling in diffuse large B-cell lymphoma. Blood 2018, 132, 1805–1817. [Google Scholar] [CrossRef]
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201. [Google Scholar] [CrossRef]
- Kwon, D.; Kim, S.; Kim, P.-J.; Go, H.; Nam, S.J.; Paik, J.H.; Kim, Y.A.; Kim, T.M.; Heo, D.S.; Kim, C.W.; et al. Clinicopathological analysis of programmed cell death 1 and programmed cell death ligand 1 expression in the tumour microenvironments of diffuse large B cell lymphomas. Histopathology 2015, 68, 1079–1089. [Google Scholar] [CrossRef]
- Chen, B.J.; Dashnamoorthy, R.; Galera, P.; Makarenko, V.; Chang, H.; Ghosh, S.; Evens, A.M. The immune checkpoint molecules PD-1, PD-L1, TIM-3 and LAG-3 in diffuse large B-cell lymphoma. Oncotarget 2019, 10, 2030–2040. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, J.; Tumuluru, S.; Bao, R.; Leukam, M.; Venkataraman, G.; Phillip, J.; Fitzpatrick, C.; McElherne, J.; MacNabb, B.W.; Orlowski, R.; et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T-cell–inflamed phenotype. Blood 2019, 133, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, H.; Xiao, T.; Liu, J.-Z.; Liu, G.; Wang, J.-X.; Li, G.-Y.; Wang, L.-X. Prognostic value of PD-1 and TIM-3 on CD3+ T cells from diffuse large B-cell lymphoma. Biomed. Pharmacother. 2015, 75, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, R.; Miao, Y.; Tran, T.; Adams, L.; Roscoe, N.; Xu, B.; Manyam, G.C.; Tan, X.; Zhang, H.; et al. PD-1/PD-L1 expression and interaction by automated quantitative immunofluorescent analysis show adverse prognostic impact in patients with diffuse large B-cell lymphoma having T-cell infiltration: A study from the International DLBCL Consortium Program. Mod. Pathol. 2019, 32, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhang, L.; Chen, L.; Liu, G.; Feng, Z.; Gao, L. Tim-3 expression is increased on peripheral T cells from diffuse large B cell lymphoma. Tumor Biol. 2014, 35, 7951–7956. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, S.E.; Beiske, K.; Blaker, Y.N.; Førsund, M.S.; Holte, H.; Østenstad, B.; Kimby, E.; Köksal, H.; Wälchli, S.; Bai, B.; et al. TIGIT and PD-1 Mark Intratumoral T Cells with Reduced Effector Function in B-cell Non-Hodgkin Lymphoma. Cancer Immunol. Res. 2019, 7, 355–362. [Google Scholar] [CrossRef]
- Laurent, C.; Charmpi, K.; Gravelle, P.; Tosolini, M.; Franchet, C.; Ysebaert, L.; Brousset, P.; Bidaut, A.; Ycart, B.; Fournié, J.-J. Several immune escape patterns in non-Hodgkin’s lymphomas. OncoImmunology 2015, 4, e1026530. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Xiao, M.; Au, Q.; Padmanabhan, R.; Xu, B.; Hoe, N.; Rodríguez-Perales, S.; Torres-Ruiz, R.; Manyam, G.C.; Visco, C.; et al. Immune Profiling and Quantitative Analysis Decipher the Clinical Role of Immune-Checkpoint Expression in the Tumor Immune Microenvironment of DLBCL. Cancer Immunol. Res. 2019, 7, 644–657. [Google Scholar] [CrossRef]
- Motta, M.; Rassenti, L.; Shelvin, B.J.; Lerner, S.; Kipps, T.J.; Keating, M.J.; Wierda, W.G. Increased expression of CD152 (CTLA-4) by normal T lymphocytes in untreated patients with B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1788–1793. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Gassner, F.J.; Ratswohl, C.; Zaborsky, N.; Rebhandl, S.; Schubert, M.; Steiner, M.; Gutjahr, J.C.; Pleyer, L.; Egle, A.; et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. OncoImmunology 2017, 7, e1371399. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Niki, T.; Hirashima, M.; Novak, A.J.; Witzig, T.E.; Ansell, S.M. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Xiu, B.; Novak, A.J.; Ansell, S.M. PD-1 expression defines two distinct T-cell sub-populations in follicular lymphoma that differentially impact patient survival. Blood Cancer J. 2015, 5, e281. [Google Scholar] [CrossRef] [PubMed]
- Gravelle, P.; Do, C.; Franchet, C.; Mueller, S.; Oberic, L.; Ysebaert, L.; LaRocca, L.M.; Hohaus, S.; Calmels, M.-N.; Frenois, F.-X.; et al. Impaired functional responses in follicular lymphoma CD8+TIM-3+ T lymphocytes following TCR engagement. OncoImmunology 2016, 5, e1224044. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.-P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1+ T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef]
- Josefsson, S.E.; Huse, K.; Kolstad, A.; Beiske, K.; Pende, D.; Steen, C.B.; Inderberg, E.M.; Lingjærde, O.C.; Østenstad, B.; Smeland, E.B.; et al. T Cells Expressing Checkpoint Receptor TIGIT Are Enriched in Follicular Lymphoma Tumors and Characterized by Reversible Suppression of T-cell Receptor Signaling. Clin. Cancer Res. 2018, 24, 870–881. [Google Scholar] [CrossRef]
- Myklebust, J.H.; Irish, J.M.; Brody, J.; Czerwinski, D.K.; Houot, R.; Kohrt, H.E.; Timmerman, J.; Said, J.; Green, M.R.; Delabie, J.; et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood 2013, 121, 1367–1376. [Google Scholar] [CrossRef]
- Hilchey, S.P.; De, A.; Rimsza, L.M.; Bankert, R.B.; Bernstein, S.H. Follicular Lymphoma Intratumoral CD4+CD25+GITR+Regulatory T Cells Potently Suppress CD3/CD28-Costimulated Autologous and Allogeneic CD8+CD25−and CD4+CD25−T Cells. J. Immunol. 2007, 178, 4051–4061. [Google Scholar] [CrossRef]
- Xerri, L.; Chetaille, B.; Serriari, N.; Attias, C.; Guillaume, Y.; Arnoulet, C.; Olive, D. Programmed death 1 is a marker of angioimmunoblastic T-cell lymphoma and B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Hum. Pathol. 2008, 39, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Harrington, B.K.; Wheeler, E.; Hornbuckle, K.; Shana’Ah, A.Y.; Youssef, Y.; Smith, L.; Ii, Q.H.; Klamer, B.; Zhang, X.; Long, M.; et al. Modulation of immune checkpoint molecule expression in mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qian, J.; Lu, Y.; Li, H.; Bao, H.; He, D.; Liu, Z.; Zheng, Y.; He, J.; Li, Y.; et al. Immune evasion of mantle cell lymphoma: Expression of B7-H1 leads to inhibited T-cell response to and killing of tumor cells. Haematol. 2013, 98, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Vranic, S.; Ghosh, N.; Kimbrough, J.; Bilalovic, N.; Bender, R.; Arguello, D.; Veloso, Y.; Dizdarevic, A.; Gatalica, Z. PD-L1 Status in Refractory Lymphomas. PLoS ONE 2016, 11, e0166266. [Google Scholar] [CrossRef]
- Goyal, A.; Moore, J.B.; Gimbel, D.; Carter, J.B.; Kroshinsky, D.; Ferry, J.A.; Harris, N.L.; Duncan, L.M. PD-1, S-100 and CD1a expression in pseudolymphomatous folliculitis, primary cutaneous marginal zone B-cell lymphoma (MALT lymphoma) and cutaneous lymphoid hyperplasia. J. Cutan. Pathol. 2014, 42, 6–15. [Google Scholar] [CrossRef]
- Muenst, S.; Hoeller, S.; Willi, N.; Dirnhofer, S.; Tzankov, A. Diagnostic and Prognostic Utility of PD-1 In B Cell Lymphomas. Dis. Markers 2010, 29, 47–53. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef]
- Twa, D.D.W.; Chan, F.C.; Ben-Neriah, S.; Woolcock, B.W.; Mottok, A.; Tan, K.L.; Slack, G.W.; Gunawardana, J.; Lim, R.S.; McPherson, A.W.; et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 2014, 123, 2062–2065. [Google Scholar] [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.-H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgun, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.-D.; Siska, P.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8+ T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Clear, A.J.; Kelly, G.; Fatah, R.; Matthews, J.; MacDougall, F.; Lister, T.A.; Lee, A.M.; Calaminici, M.; Gribben, J.G. Follicular lymphoma cells induce T-cell immunologic synapse dysfunction that can be repaired with lenalidomide: Implications for the tumor microenvironment and immunotherapy. Blood 2009, 114, 4713–4720. [Google Scholar] [CrossRef] [PubMed]
- Kiaii, S.; Clear, A.J.; Ramsay, A.G.; Davies, D.; Sangaralingam, A.; Lee, A.; Calaminici, M.; Neuberg, D.S.; Gribben, J.G. Follicular Lymphoma Cells Induce Changes in T-Cell Gene Expression and Function: Potential Impact on Survival and Risk of Transformation. J. Clin. Oncol. 2013, 31, 2654–2661. [Google Scholar] [CrossRef]
- Laurent, C.; Müller, S.; Do, C.; Al-Saati, T.; Allart, S.; LaRocca, L.M.; Hohaus, S.; Duchez, S.; Quillet-Mary, A.; Laurent, G.; et al. Distribution, function, and prognostic value of cytotoxic T lymphocytes in follicular lymphoma: A 3-D tissue-imaging study. Blood 2011, 118, 5371–5379. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Amedei, A.; Manghetti, M.; Costa, F.; Baldari, C.T.; Quazi, A.S.; Telford, J.L.; Romagnani, S.; Del Prete, G. Impaired T-cell regulation of B-cell growth in Helicobacter pylori–related gastric low-grade MALT lymphoma. Gastroenterol. 1999, 117, 1105–1112. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Rathmell, J.C. Metabolic pathways in T cell fate and function. Trends Immunol. 2012, 33, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef]
- Ricci, J.-E.; Chiche, J. Metabolic Reprogramming of Non-Hodgkin’s B-Cell Lymphomas and Potential Therapeutic Strategies. Front. Oncol. 2018, 8, 556. [Google Scholar] [CrossRef]
- Tosolini, M.; Algans, C.; Pont, F.; Ycart, B.; Fournié, J.-J. Large-scale microarray profiling reveals four stages of immune escape in non-Hodgkin lymphomas. OncoImmunology 2016, 5, e1188246. [Google Scholar] [CrossRef]
- Siska, P.; Van Der Windt, G.J.W.; Kishton, R.J.; Cohen, S.; Eisner, W.; Maciver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Yokote, T.; Akioka, T.; Hiraoka, N.; Nishiwaki, U.; Miyoshi, T.; Iwaki, K.; Takayama, A.; Masuda, Y.; Hatooka, J.; et al. Infiltration of effector regulatory T cells predicts poor prognosis of diffuse large B-cell lymphoma, not otherwise specified. Blood Adv. 2017, 1, 486–493. [Google Scholar] [CrossRef] [PubMed]
- El-Dien, M.M.S.; Abdou, A.G.; Asaad, N.Y.; El-Wahed, M.M.A.; Kora, M.A.E.-H.M. Intratumoral FOXP3+ Regulatory T Cells in Diffuse Large B-Cell Lymphoma. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Cha, Z.; Gu, H.; Zang, Y.; Wang, Z.; Li, J.; Huang, W.; Qin, A.; Zhu, L.; Tu, X.; Cheng, N.; et al. The prevalence and function of CD4 + CXCR5 + Foxp3 + follicular regulatory T cells in diffuse large B cell lymphoma. Int. Immunopharmacol. 2018, 61, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Novak, A.J.; Stenson, M.J.; Witzig, T.E.; Ansell, S.M. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood 2006, 107, 3639–3646. [Google Scholar] [CrossRef]
- Tzankov, A.; Meier, C.; Hirschmann, P.; Went, P.; Pileri, S.A.; Dirnhofer, S. Correlation of high numbers of intratumoral FOXP3+ regulatory T cells with improved survival in germinal center-like diffuse large B-cell lymphoma, follicular lymphoma and classical Hodgkin’s lymphoma. Haematol. 2008, 93, 193–200. [Google Scholar] [CrossRef]
- Lee, A.M.; Clear, A.J.; Calaminici, M.; Davies, A.J.; Jordan, S.; MacDougall, F.; Matthews, J.; Norton, A.J.; Gribben, J.G.; Lister, T.A.; et al. Number of CD4+ Cells and Location of Forkhead Box Protein P3–Positive Cells in Diagnostic Follicular Lymphoma Tissue Microarrays Correlates With Outcome. J. Clin. Oncol. 2006, 24, 5052–5059. [Google Scholar] [CrossRef]
- Carreras, J.; Lopez-Guillermo, A.; Fox, B.C.; Colomo, L.; Martinez, A.; Roncador, G.; Montserrat, E.; Campo, E.; Banham, A.H. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood 2006, 108, 2957–2964. [Google Scholar] [CrossRef]
- Wahlin, B.E.; Aggarwal, M.; Montes-Moreno, S.; Gonzalez, L.F.; Roncador, G.; Sanchez-Verde, L.; Christensson, B.; Sander, B.; Kimby, E. A Unifying Microenvironment Model in Follicular Lymphoma: Outcome Is Predicted by Programmed Death-1-Positive, Regulatory, Cytotoxic, and Helper T Cells and Macrophages. Clin. Cancer Res. 2010, 16, 637–650. [Google Scholar] [CrossRef]
- Lim, H.W.; Hillsamer, P.; Banham, A.H.; Kim, C.H.; Banham, A.H. Cutting Edge: Direct Suppression of B Cells by CD4+CD25+ Regulatory T Cells. J. Immunol. 2005, 175, 4180–4183. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Kochanek, M.; Darabi, K.; Popov, A.; Jensen, M.; Endl, E.; Knolle, P.A.; Thomas, R.K.; Von Bergwelt-Baildon, M.; Debey, S.; et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005, 106, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Jak, M.; Mous, R.; Remmerswaal, E.B.M.; Spijker, R.; Jaspers, A.; Yagüe, A.; Eldering, E.; Van Lier, R.A.W.; Van Oers, M.H.J. Enhanced formation and survival of CD4+CD25hiFoxp3+T-cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Piper, K.P.; Karanth, M.; McLarnon, A.; Kalk, E.; Khan, N.; Murray, J.; Pratt, G.; Moss, P.A.H. Chronic lymphocytic leukaemia cells drive the global CD4+ T cell repertoire towards a regulatory phenotype and leads to the accumulation of CD4+ forkhead box P3+ T cells. Clin. Exp. Immunol. 2011, 166, 154–163. [Google Scholar] [CrossRef]
- D’Arena, G.; D’Auria, F.; Simeon, V.; Laurenti, L.; Deaglio, S.; Mansueto, G.; Del Principe, M.I.; Statuto, T.; Pietrantuono, G.; Guariglia, R.; et al. A shorter time to the first treatment may be predicted by the absolute number of regulatory T-cells in patients with Rai stage 0 chronic lymphocytic leukemia. Am. J. Hematol. 2012, 87, 628–631. [Google Scholar] [CrossRef]
- Craig, V.J.; Cogliatti, S.B.; Arnold, I.; Gerke, C.; Balandat, J.-E.; Wundisch, T.; Müller, A. B-cell receptor signaling and CD40 ligand-independent T cell help cooperate in Helicobacter-induced MALT lymphomagenesis. Leukemia 2010, 24, 1186–1196. [Google Scholar] [CrossRef]
- Wickenden, K.; Nawaz, N.; Mamand, S.; Kotecha, D.; Wilson, A.L.; Wagner, S.D.; Ahearne, M.J. PD1hi cells associate with clusters of proliferating B-cells in marginal zone lymphoma. Diagn. Pathol. 2018, 13, 74. [Google Scholar] [CrossRef]
- Cha, Z.; Qian, G.; Zang, Y.; Gu, H.; Huang, Y.; Zhu, L.; Li, J.; Liu, Y.; Tu, X.; Song, H.; et al. Circulating CXCR5+CD4+ T cells assist in the survival and growth of primary diffuse large B cell lymphoma cells through interleukin 10 pathway. Exp. Cell Res. 2017, 350, 154–160. [Google Scholar] [CrossRef]
- Taga, K.; Tosato, G. IL-10 inhibits human T cell proliferation and IL-2 production. J. Immunol. 1992, 148, 1143–1148. [Google Scholar]
- Miles, B.; Connick, E. Control of the Germinal Center by Follicular Regulatory T Cells During Infection. Front. Immunol. 2018, 9, 2704. [Google Scholar] [CrossRef]
- Pascutti, M.F.; Jak, M.; Tromp, J.M.; Derks, I.A.M.; Remmerswaal, E.B.M.; Thijssen, R.; Van Attekum, M.H.A.; Van Bochove, G.G.; Luijks, D.M.; Pals, S.T.; et al. IL-21 and CD40L signals from autologous T cells can induce antigen-independent proliferation of CLL cells. Blood 2013, 122, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Amé-Thomas, P.; Le Priol, J.; Yssel, H.; Caron, G.; Pangault, C.; Jean, R.; Martin, N.; Marafioti, T.; Gaulard, P.; Lamy, T.; et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: Role in the survival of malignant B cells. Leukemia 2011, 26, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Rawal, S.; Chu, F.; Zhang, M.; Park, H.J.; Nattamai, D.; Kannan, S.; Sharma, R.; Delgado, D.; Chou, T.; Lin, H.Y.; et al. Cross Talk between Follicular Th Cells and Tumor Cells in Human Follicular Lymphoma Promotes Immune Evasion in the Tumor Microenvironment. J. Immunol. 2013, 190, 6681–6693. [Google Scholar] [CrossRef] [PubMed]
- Amé-Thomas, P.; Hoeller, S.; Artchounin, C.; Misiak, J.; Braza, M.S.; Jean, R.; Le Priol, J.; Monvoisin, C.; Martin, N.; Gaulard, P.; et al. CD10 delineates a subset of human IL-4 producing follicular helper T cells involved in the survival of follicular lymphoma B cells. Blood 2015, 125, 2381–2385. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Jr, W.G.K. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors I karos and A iolos via modulation of the E 3 ubiquitin ligase complex CRL 4 CRBN. Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef]
- Dredge, K.; Marriott, J.B.; Todryk, S.M.; Muller, G.W.; Chen, R.; Stirling, D.I.; Dalgleish, A.G. Protective Antitumor Immunity Induced by a Costimulatory Thalidomide Analog in Conjunction with Whole Tumor Cell Vaccination Is Mediated by Increased Th1-Type Immunity. J. Immunol. 2002, 168, 4914–4919. [Google Scholar] [CrossRef]
- Galustian, C.; Meyer, B.; Labarthe, M.-C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2008, 58, 1033–1045. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Evans, R.; Kiaii, S.; Svensson, L.; Hogg, N.; Gribben, J.G. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by alter-ing Rho GTPase signaling that is reversible with lenalidomide. Blood 2013, 121, 2704–2714. [Google Scholar] [CrossRef]
- Chiu, H.; Trisal, P.; Bjorklund, C.; Carrancio, S.; Toraño, E.G.; Guarinos, C.; Papazoglou, D.; Hagner, P.R.; Beldi-Ferchiou, A.; Tarte, K.; et al. Combination lenalidomide-rituximab immunotherapy activates anti-tumour immunity and induces tumour cell death by complementary mechanisms of action in follicular lymphoma. Br. J. Haematol. 2019, 185, 240–253. [Google Scholar] [CrossRef]
- Ioannou, N.; Hagner, P.R.; Stokes, M.; Gandhi, A.K.; Apollonio, B.; Fanous, M.; Papazoglou, D.; Sutton, L.A.; Rosenquist, R.; Amini, R.-M.; et al. Triggering interferon signaling in T cells with avadomide sensitizes CLL to anti-PD-L1/PD-1 immunotherapy. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Zabaleta, A.; Alignani, D.; Lasa, M.; Maiso, P.; Jelinek, T.; Segura, V.; Delgado, J.A.; Rodriguez-Otero, P.; Prosper, F.; et al. New Insights into the Mechanism of Action (MoA) of First-in-Class IgG-Based Bcma T-Cell Bispecific Antibody (TCB) for the Treatment of Multiple Myeloma (MM). Blood 2016, 128, 2096. [Google Scholar] [CrossRef]
- Hofland, T.; De Weerdt, I.; Ter Burg, H.; De Boer, R.; Tannheimer, S.; Tonino, S.H.; Kater, A.P.; Eldering, E. Dissection of the Effects of JAK and BTK Inhibitors on the Functionality of Healthy and Malignant Lymphocytes. J. Immunol. 2019, 203, 2100–2109. [Google Scholar] [CrossRef]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjønnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Taskén, K. The PI3K p110δ Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef]
- Hanna, B.S.; Roessner, P.M.; Scheffold, A.; Jebaraj, B.M.C.; Demerdash, Y.; Öztürk, S.; Lichter, P.; Stilgenbauer, S.; Seiffert, M. PI3Kδ inhibition modulates regulatory and effector T-cell differentiation and function in chronic lymphocytic leukemia. Leukemia 2018, 33, 1427–1438. [Google Scholar] [CrossRef]
- Petersen, C.T.; Hassan, M.; Morris, A.B.; Jeffery, J.; Lee, K.; Jagirdar, N.; Staton, A.D.; Raikar, S.S.; Spencer, H.T.; Sulchek, T.; et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv. 2018, 2, 210–223. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef]
- Mamontov, P.; Eberwine, R.A.; Perrigoue, J.; Das, A.; Friedman, J.R.; Mora, J.R. A negative role for the interleukin-2-inducible T-cell kinase (ITK) in human Foxp3+ TREG differentiation. PLoS ONE 2019, 14, e0215963. [Google Scholar] [CrossRef]
- Hu, J.; August, A. Naive and innate memory phenotype CD4+ T cells have different requirements for active Itk for their development. J. Immunol. 2008, 180, 6544–6552. [Google Scholar] [CrossRef]
- Kapnick, S.M.; Stinchcombe, J.C.; Griffiths, G.M.; Schwartzberg, P.L. Inducible T Cell Kinase Regulates the Acquisition of Cytolytic Capacity and Degranulation in CD8(+) CTLs. J. Immunol. 2017, 198, 2699–2711. [Google Scholar] [CrossRef]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef]
- Parry, H.M.; Mirajkar, N.; Cutmore, N.; Zuo, J.; Long, H.; Kwok, M.; Oldrieve, C.; Hudson, C.; Stankovic, T.; Paneesha, S.; et al. Long-Term Ibrutinib Therapy Reverses CD8(+) T Cell Exhaustion in B Cell Chronic Lymphocytic Leukaemia. Front. Immunol. 2019, 10, 2832. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Sivina, M.; Robins, H.; Yusko, E.; Vignali, M.; O’Brien, S.; Keating, M.J.; Ferrajoli, A.; Estrov, Z.; Jain, N.; et al. Ibrutinib Therapy Increases T Cell Repertoire Diversity in Patients with Chronic Lymphocytic Leukemia. J. Immunol. 2017, 198, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.S.; Yazdanparast, H.; Demerdash, Y.; Roessner, P.M.; Schulz, R.; Lichter, P.; Stilgenbauer, S.; Seiffert, M. Combining ibrutinib and checkpoint blockade improves CD8+ T-cell function and control of chronic lymphocytic leukemia in Em-TCL1 mice. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Fraietta, J.A.; Qayyum, S.; Zhang, Q.; Maus, M.V.; Liu, X.; Nunez-Cruz, S.; Klichinsky, M.; et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clin Cancer Res. 2016, 22, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Kohrt, H.E.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef]
- Stephens, D.M.; Byrd, J.C. How I manage ibrutinib intolerance and complications in patients with chronic lymphocytic leukemia. Blood 2019, 133, 1298–1307. [Google Scholar] [CrossRef]
- Witzig, T.E.; Nowakowski, G.S.; Habermann, T.M.; Goy, A.; Hernandez-Ilizaliturri, F.J.; Chiappella, A.; Vitolo, U.; Fowler, N.; Czuczman, M.S. A comprehensive review of lenalidomide therapy for B-cell non-Hodgkin lymphoma. Ann. Oncol. 2015, 26, 1667–1677. [Google Scholar] [CrossRef]
- Vitolo, U.; Trněný, M.; Belada, D.; Carella, A.M.; Chua, N.; Abrisqueta, P.; Demeter, J.; Flinn, I.W.; Hong, X.; Kim, W.S.; et al. Obinutuzumab or Rituximab Plus CHOP in Patients with Previously Untreated Diffuse Large B-Cell Lymphoma: Final Results from an Open-Label, Randomized Phase 3 Study (GOYA). Blood 2016, 128, 470. [Google Scholar] [CrossRef]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Huckelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in Expansion Potential of Naive Chimeric Antigen Receptor T Cells from Healthy Donors and Untreated Chronic Lymphocytic Leukemia Patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.; Ubelhart, R.; Schubert, M.L.; Fan, F.; He, B.; Hoffmann, J.M.; Wang, L.; Wang, S.; Gong, W.; Neuber, B.; et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int. J. Cancer 2019, 145, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, S.; Falcone, L.; Camisa, B.; De Girardi, F.; Biondi, M.; Giglio, F.; Ciceri, F.; Bonini, C.; Bondanza, A.; Casucci, M. Next-Generation Manufacturing Protocols Enriching TSCM CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front. Immunol. 2020, 11, 1217. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther. Oncol. 2020, 18, 272–281. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]

{kind=link}
| Study | Treatment | Phase | Disease | N | CR | ORR | Trial ID (NCT) |
|---|---|---|---|---|---|---|---|
| O’Mahony et al., 2007 [20] | Ipilimumab | II | FL | 2 | 0/2 (0%) | 1/2 (50%) | N/A |
| MCL | 2 | 0/2 (0%) | 1/2 (50%) | ||||
| Berger et al., 2008 [21] | Pidilzumab | I | CLL | 3 | 0/3 (0%) | 0/3 (0%) | N/A |
| DLBCL | 2 | 0/3 (0%) | 0/2 (0%) | ||||
| FL | 1 | 1/1 (100%) | 1/1 (100%) | ||||
| Ansell et al., 2009 [23] | Ipilimumab | I | FL | 14 | 0/14 (0%) | 1/14 (7%) | NCT00089076 |
| DLBCL | 3 | 1/3 (33%) | 1/3 (33%) | ||||
| MCL | 1 | 0/1 (0%) | 0/1 (0%) | ||||
| Bashey et al., 2009 [22] | Ipilimumab | I | MCL | 1 | 0 | 1/1 (100%) | NCT00060372 |
| CLL | 2 | 0 | 0/2 (0%) | ||||
| Westin et al., 2014 [31] | Pidilizumab and rituximab | II | FL | 29 | 15/29 (52%) | 19/29 (66%) | NCT00904722 |
| Ansell et al., 2016 [29] | Nivolumab and ipilimumab | Ib | B-NHL | 15 | 0/15 (0%) | 3/15 (20%) | NCT01592370 |
| Lesokhin et al., 2016 [26] | Nivolumab | Ib | FL | 10 | 1/10 (10%) | 4/10 (40%) | NCT01592370 |
| MCL | 4 | 0/4 (0%) | 0/4 (0%) | ||||
| MZL | 1 | 0/1 (0%) | 0/1 (0%) | ||||
| DLBCL | 11 | 2/11 (18%) | 4/11 (36%) | ||||
| Ding et al., 2017 [25] | Pembrolizumab | II | CLL | 16 | 0/16 (0%) | 0/16 (0%) | NCT02332980 |
| CLL with RT | 9 | 1/9 (11%) | 4/9 (44%) | ||||
| Ding et al., 2017 [24] | Pembrolizumab | II | FL | 18 | 0/18 (0%) | 2/18 (11%) | NCT02332980 |
| MZL | 2 | 0/2 (0%) | 0/2 (0%) | ||||
| Nastoupil et al., 2017 [30] | Pembrolizumab and rituximab | II | FL | 25 | 12/25 (48%) | 16/25 (64%) | NCT02446457 |
| Younes et al., 2017 [36] | Atezolizumab, Obinutuzumab and Bendamustin | Ib/II | FL | 15 | 10/15 (67%) | 12/15 (80%) | NCT02596971 |
| Ansell et al., 2019 [27] | Nivolumab | II | auto-HCT failed DLBCL | 87 | 3/87 (3%) | 9/87 (10%) | NCT02038933 |
| auto-HCT ineligible DLBCL | 34 | 0/34 (0%) | 1/34 (3%) | ||||
| Khouri et al., 2018 [40] | Ipilimumab and lenalidomide | II | FL (allo-HSCT) | 2 | 1/2 (50%) | 2/2 (100%) | NCT01919619 |
| MCL (allo-HSCT) | 3 | 2/3 (67%) | 3/3 (100%) | ||||
| DLBCL (allo-HSCT) | 1 | 0/1 (0%) | 1/1 (100%) | ||||
| CLL (allo-HSCT) | 2 | 0/2 (0%) | 0/2 (0%) | ||||
| MCL (auto-HSCT) | 2 | 2/2 (100%) | 2/2 (100%) | ||||
| FL (auto-HSCT) | 1 | 1/1 (100%) | 1/1 (100%) | ||||
| Palomba et al., 2017 [35] | Atezolizumab and obinutuzumab | Ib | FL | 26 | N/A | 57% | NCT02220842 |
| DLBCL | 23 | N/A | 16% | ||||
| Younes et al., 2018 [37] | Atezolizumab, Rituximab and CHOP | I/II | DLBCL | 40 | 31/40 (78%) | 35/40 (88%) | NCT02596971 |
| Armand et al., 2019 [28] | Pembrolizumab Pembrolizumab | Ib II | PMBCL PMBCL | 21 53 | 7/21 (33%) 7/53 (13%) | 48% 45% | NCT01953692 NCT02576990 |
| Bond et al., 2019 [38] | Nivolumab and lenalidomide | I | B-cell lymphoma * | 10 | 1/10 (10%) | 3/10 (30%) | NCT03015896 |
| Casulo et al., 2019 [39] | Durvalumab monotherapy or in combination with lenalidomide ± rituximab or rituximab ± bendamustine | I/II | DLBCL | 38 | 3/38 (8%) | 7/38 (18%) | NCT02733042 |
| FL | 22 | 6/22 (27%) | 13/22 (59%) | ||||
| Younes et al., 2019 [41] | Nivolumab and ibrutinib | I/IIa | CLL/SLL | 36 (30 CLL) | 0/36 (0%) | 22/36 (61%) | NCT02329847 |
| RT | 20 | 2/20 (10%) | 13/20 (65%) | ||||
| FL | 40 | 4/40 (10%) | 13/40 (33%) | ||||
| DLBCL | 45 | 7/45 (16%) | 16/45 (36%) | ||||
| Zinzani et al., 2019 [46] | Nivolumab and brentuximab vedotin | I/II | PMBCL | 30 | 11/30 (37%) | 22/30 (73%) | NCT02581631 |
| Frigault et al., 2020 [47] | Pembrolizumab after auto-SCT | II | DLBCL | 29 | 17/29 (59%) | 17/29 (59%) | NCT02362997 |
| Herrera et al., 2020 [45] | Durvalumab and ibrutinib | Ib/II | FL GCB DLBCL Non-GCB DLBCL | 27 16 16 | 1/27 (4%) 1/16 (6%) 5/16 (31%) | 7/27 (26%) 2/16 (13%) 6/16 (38%) | NCT02401048 |
| Study | Treatment | Phase | Disease | N | CR | ORR | Trial ID (NCT) |
|---|---|---|---|---|---|---|---|
| Goebeler et al., 2016 [48] | Blinatumomab | 1 | FL | 15 * | 6/15 (40%) | 12/15 (80%) | NCT00274742 |
| MCL | 7 * | 3/7 (43%) | 5/7 (71%) | ||||
| DLBCL | 11 * | 4/11 (36%) | 6/11 (55%) | ||||
| Viardot et al., 2016 [49] | Blinatumomab | 2 | DLBCL | 21 | 4/21 (19%) | 9/21 (43%) | NCT01741792 |
| Bannerji et al., 2017 [55] | REGN1979 (CD3xCD20) | 1 | DLBCL | 22 | 0/22 (0%) | 4/22 (18%) | NCT02290951 |
| FL | 11 | 0/11 (0%) | 4/11 (36%) | ||||
| MZL | 2 | 0/2 (0%) | 1/2 (50%) | ||||
| MCL | 2 | 0/3 (0%) | 1/3 (33%) | ||||
| Katz et al., 2019 [52] | Blinatumomab after R-CHOP | 2 | DLBCL | 28 | NA | 25/28 (89%) | NCT03023878 |
| Morschhauser et al. 2019 [57] | Glofitamab and obinutuzumab | - | B-NHL ** (all included) | 21 | 9/21 (43%) | 10/21 (48%) | NCT03075696 |
| B-NHL ** (highest glofitamab dose) | 10 | 8/10 (80%) | 9/10 (90%) | ||||
| Poh et al., 2019 [51] | Blinatumomab and lenalidomide | 1 | B-NHL | 18 | 50% | 83% | NCT02568553 |
| Schuster et al., 2019 [53] | Mosunetuzumab | 1/1b | indolent B-NHL *** | 64 | 27/64 (42%) | 41/64 (64%) | NCT02500407 |
| aggressive B-NHL *** | 119 | 22/119 (18%) | 41/119 (34%) | ||||
| Bannerji et al., 2020 [56] | REGN1979 | 1 | FL (≥5 mg) | 28 | 21/28 (75%) | 26/28 (93%) | NCT02290951 |
| FL (≥80 mg) | 16 | 11/16 (69%) | 15/16 (94%) | ||||
| DLBCL (no prior CAR, ≥5 mg) | 30 | 9/30 (30%) | 14/30 (47%) | ||||
| DLBCL (no prior CAR, ≥80 mg) | 10 | 6/10 (60%) | 6/10 (60%) | ||||
| DLBCL (relapse after CAR, ≥5 mg) | 23 | 5/23 (22%) | 7/23 (30%) | ||||
| DLBCL (relapse after CAR, ≥80 mg) | 21 | 5/21 (24%) | 7/21 (33%) | ||||
| Coyle et al., 2020 [50] | Blinatumomab | 2 | B-NHL **** | 41 | 9/41 (22%) | 15/41 (37%) | NCT02910063 |
| Hutchings et al., 2020 [54] | Subcutaneous epcoritamab (GEN3013; CD3xCD20) | 1/2 | DLBCL/HGBCL | 9 | 4/9 (44%) | 5/9 (56%) | NCT03625037 |
| FL | 6 | 0/6 (0%) | 6/6 (100%) |
| Study | CAR type | Phase | Disease | N | CR | ORR | Trial ID (NCT) |
|---|---|---|---|---|---|---|---|
| First Generation CAR | |||||||
| Till et al., 2008 [66] | anti-CD20 | I | FL | 7 | 2/7 (29%) | 3/7 (43%) | NCT00012207 |
| Second Generation 28z CAR | |||||||
| Brentjens et al., 2011 [69] | anti-CD19 | I/II | CLL | 7 | 0/7 (0%) | 0/7 (0%) | NCT00466531 |
| Kochenderfer et al., 2012 [67] | anti-CD19 | I/II | FL | 3 | 0/3 (0%) | 3/3 (100%) | NCT00924326 |
| CLL | 4 | 1/4 (25%) | 3/4 (75%) | ||||
| Kochenderfer et al., 2013 [68] | anti-CD19 | I | CLL | 4 | 0/4 (0%) | 0/4 (0%) | NCT01087294 |
| DLBCL | 2 | 0/2 (0%) | 0/2 (0%) | ||||
| MCL | 4 | 0/4 (0%) | 2/4 (50%) | ||||
| Kochenderfer et al., 2015 [70] | anti-CD19 | I/II | CLL | 4 | 3/4 (75%) | 4/4 (100%) | NCT00924326 |
| MZL | 1 | 0/1 (0%) | 1/1 (100%) | ||||
| DLBCL | 4 | 2/4 (50%) | 4/4 (50%) | ||||
| Ramos et al., 2016 [72] | anti-k light chain | I | CLL | 2 | 0/2 (0%) | 0/2 (0%) | NCT00881920 |
| DLBCL | 2 | 0/2 (0%) | 0/2 (0%) | ||||
| MCL | 1 | 0/1 (0%) | 0/1 (0%) | ||||
| Wang et al., 2016 [86] | anti-CD19 (1st gen) | I/II | DLBCL | 7 | 2/7 (29%) | 3/7 (43%) | NCT01318317 |
| MCL | 1 | 1/1 (100%) | 1/1 (100%) | ||||
| anti-CD19 (2nd gen) | I | DLBCL | 4 | 3/4 (75%) | 3/4 (75%) | NCT01815749 | |
| MCL | 4 | 3/4 (75%) | 3/4 (75%) | ||||
| Kochenderfer et al., 2017 [71] | anti-CD19 | I/II | DLBCL | 19 | 11/19 (58%) | 15/19 (79%) | NCT00924326 |
| FL | 2 | 2/2 (100%) | 2/2 (100%) | ||||
| MCL | 1 | 1/1 (100%) | 1/1 (100%) | ||||
| Geyer et al., 2018 [73] | anti-CD19 | I | CLL * | 8 | 2/8 (25%) | 2/8 (25%) | NCT01416974 |
| Locke et al., 2018 [74] | anti-CD19 | I/II | DLBCL | 101 | 59/101 (58%) | 84/101 (83%) | NCT02348216 |
| Jacobson et al., 2020 [75] | anti-CD19 | II | FL | 80 | 64/80 (80%) | 76/80 (95%) | NCT03105336 |
| MZL | 7 | 5/7 (71%) | 6/7 (86%) | ||||
| Wang et al., 2020 [65] | anti-CD19 | II | MCL | 60 | 40/60 (67%) | 56/60 (93%) | NCT02601313 |
| Second Generation BBz CAR | |||||||
| Kalos et al., 2011 [87] | anti-CD19 | I | CLL | 3 | 2/3 (67%) | 3/3 (100%) | NCT00295477 |
| II | NCT00622232 | ||||||
| Porter et al., 2015 [78] | anti-CD19 | I | CLL | 14 | 4/14 (29%) | 8/14 (57%) | NCT01029366 |
| Fraietta et al., 2016 [84] | anti-CD19 | II | CLL | 3 | 1/3 (33%) | 3/3 (100%) | NCT01747486 |
| I/II | NCT01105247 | ||||||
| I/II | NCT01217749 | ||||||
| Schuster et al., 2017 [79] | anti-CD19 | I | FL | 14 | 6/14 (42%) | 7/14 (50%) | NCT02030834 |
| DLBCL | 14 | 10/14 (71%) | 11/14 (79%) | ||||
| Fraietta et al., 2018 [76] | anti-CD19 | I | CLL | 41 | 8/41 (20%) | 16/41 (39%) | NCT01029366 |
| II | NCT01747486 | ||||||
| I | NCT02640209 | ||||||
| Abramson et al., 2019 [81] | anti-CD19 | I | DLBCL | 255 | 135/255 (53%) | 186/255 (73%) | NCT02631044 |
| Cao et al., 2019 [85] | anti-CD19 ** | N/A | DLBCL | 10 | 5/10 (50%) | 9/10 (90%) | ChiCTR-ONN-16009862, ChiCTR1800019288 |
| Hirayama el al., 2019 [77] | anti-CD19 | I/II | FL | 8 | 7/8 (88%) | 7/8 (88%) | NCT01865617 |
| FL *** | 13 | 6/13 (46%) | 6/13 (46%) | ||||
| Schuster et al., 2019 [64] | anti-CD19 | II | DLBCL | 93 | 40/93 (43%) | 52/93 (56%) | NCT02445248 |
| Siddiqi et al., 2019 [83] | anti-CD19 | I/II | CLL | 22 | 10/22 (64%) | 18/22 (82%) | NCT03331198 |
| Ying et al., 2019 [82] | anti-CD19 (4-1BB or CD28) | I/II | MZL | 1 | 1/1 (100%) | 1/1 (100%) | NCT03528421 |
| FL | 5 | 5/5 (100%) | 5/5 (100%) | ||||
| DLBCL | 2 | 1/2 (50%) | 1/2 (50%) | ||||
| Gauthier et al., 2020 [88] | anti CD19 | I/II | CLL **** | 19 | N/A | 14/17 (88%) | NCT01865617 |
| CLL | 19 | N/A | 10/19 (56%) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Bruggen, J.A.C.; Martens, A.W.J.; Tonino, S.H.; Kater, A.P. Overcoming the Hurdles of Autologous T-Cell-Based Therapies in B-Cell Non-Hodgkin Lymphoma. Cancers 2020, 12, 3837. https://doi.org/10.3390/cancers12123837
van Bruggen JAC, Martens AWJ, Tonino SH, Kater AP. Overcoming the Hurdles of Autologous T-Cell-Based Therapies in B-Cell Non-Hodgkin Lymphoma. Cancers. 2020; 12(12):3837. https://doi.org/10.3390/cancers12123837
Chicago/Turabian Stylevan Bruggen, Jaco A. C., Anne W. J. Martens, Sanne H. Tonino, and Arnon P. Kater. 2020. "Overcoming the Hurdles of Autologous T-Cell-Based Therapies in B-Cell Non-Hodgkin Lymphoma" Cancers 12, no. 12: 3837. https://doi.org/10.3390/cancers12123837
APA Stylevan Bruggen, J. A. C., Martens, A. W. J., Tonino, S. H., & Kater, A. P. (2020). Overcoming the Hurdles of Autologous T-Cell-Based Therapies in B-Cell Non-Hodgkin Lymphoma. Cancers, 12(12), 3837. https://doi.org/10.3390/cancers12123837