Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells


Simple Summary
Abstract
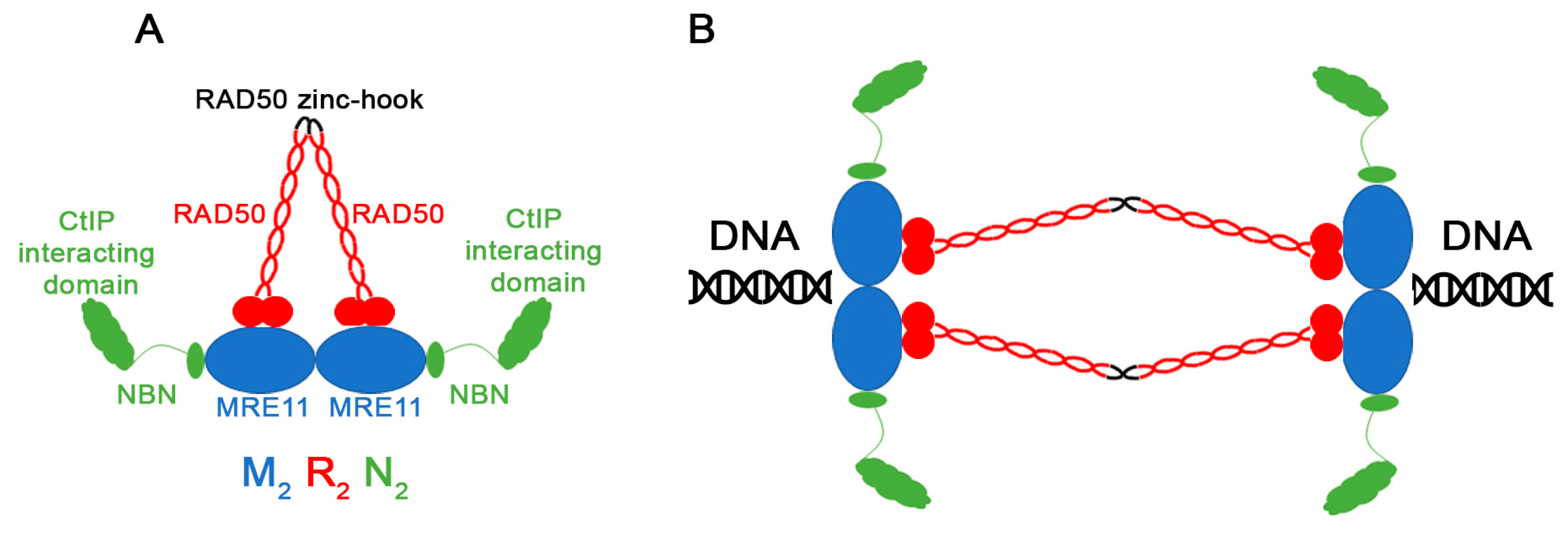
1. Introduction
2. Materials and Methods
2.1. COSMIC Data
2.2. Evolutionary Analyses
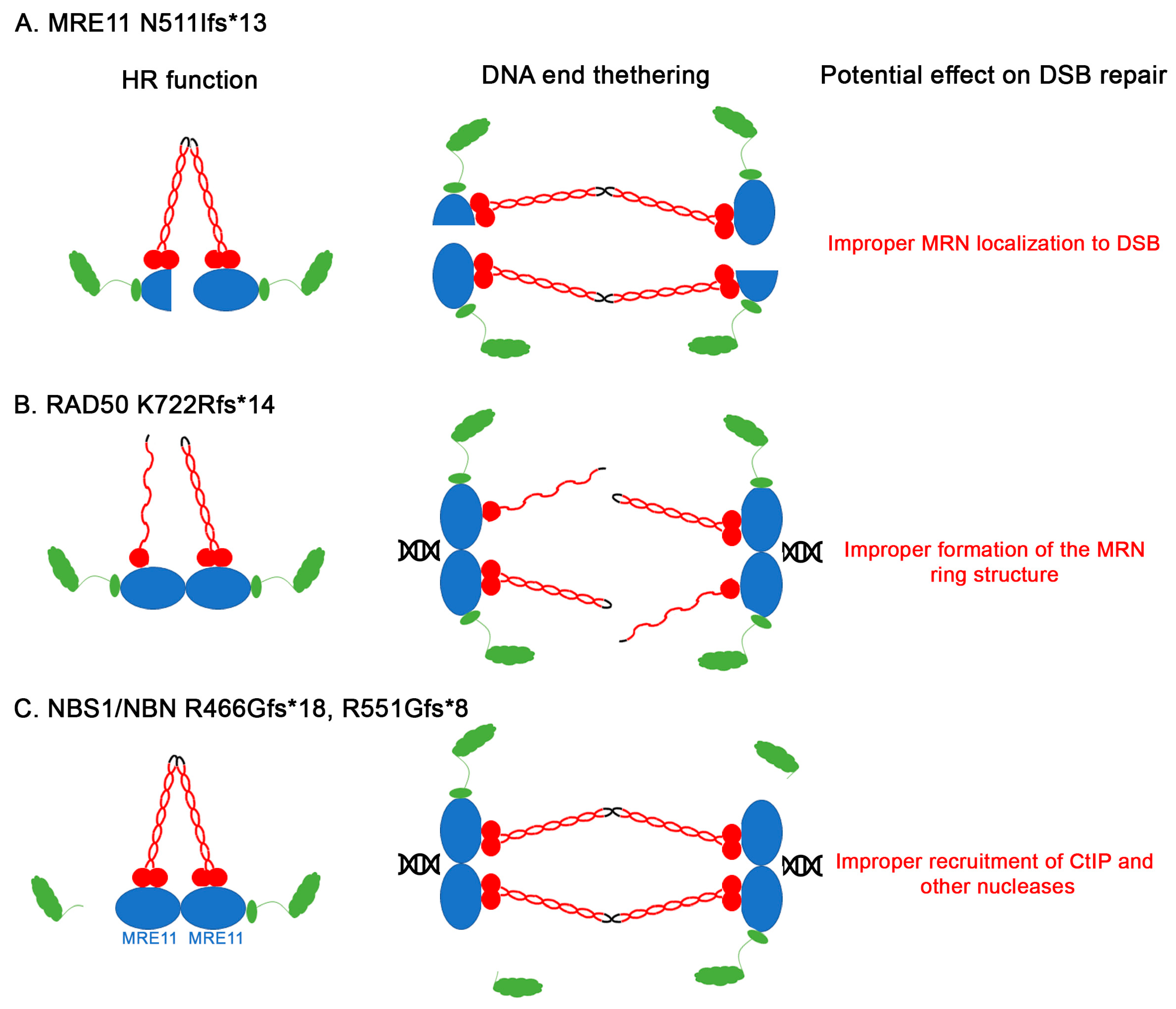
3. Results and Discussion
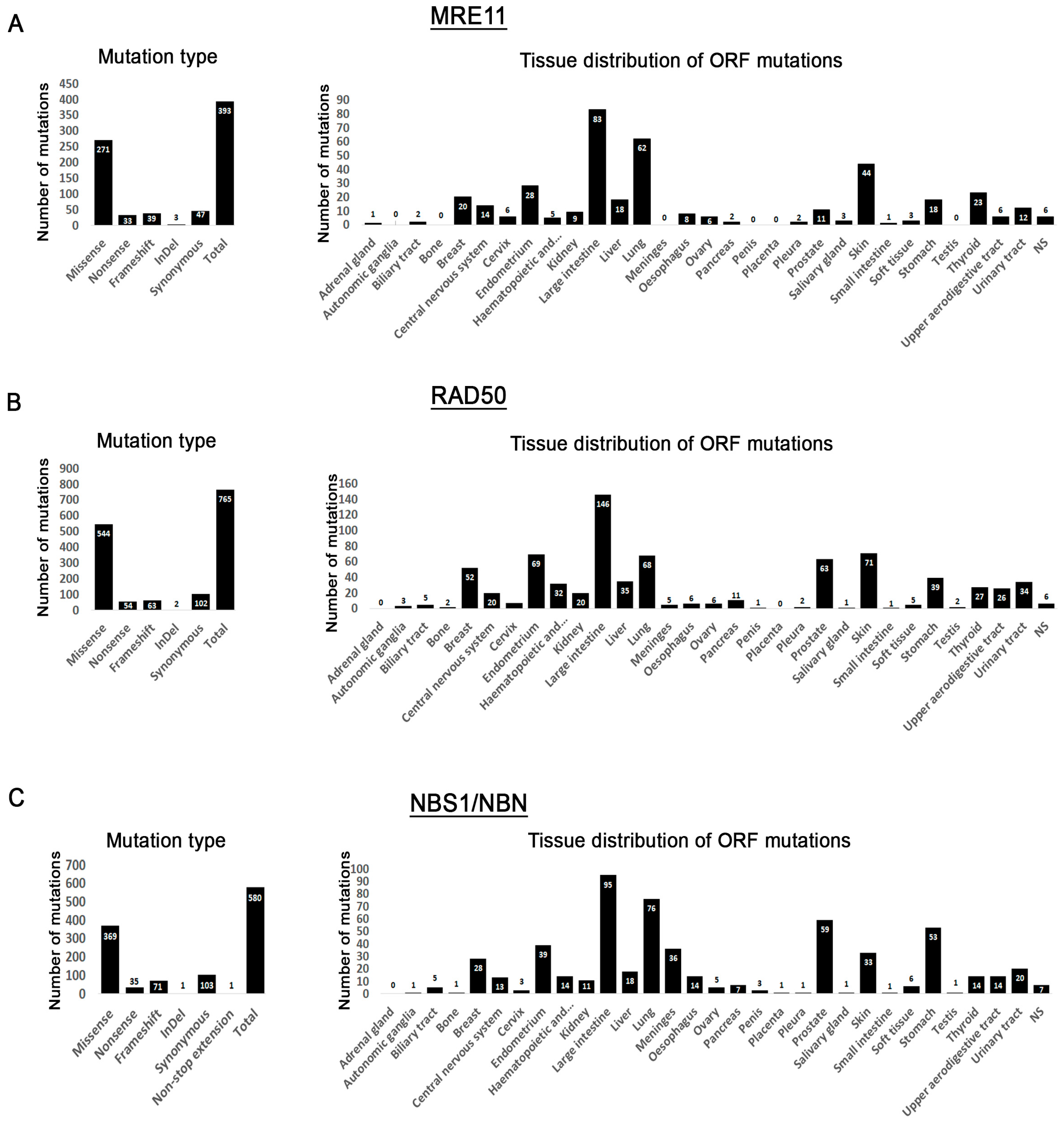
3.1. MRN Mutation Spectrum in Cancer Cells
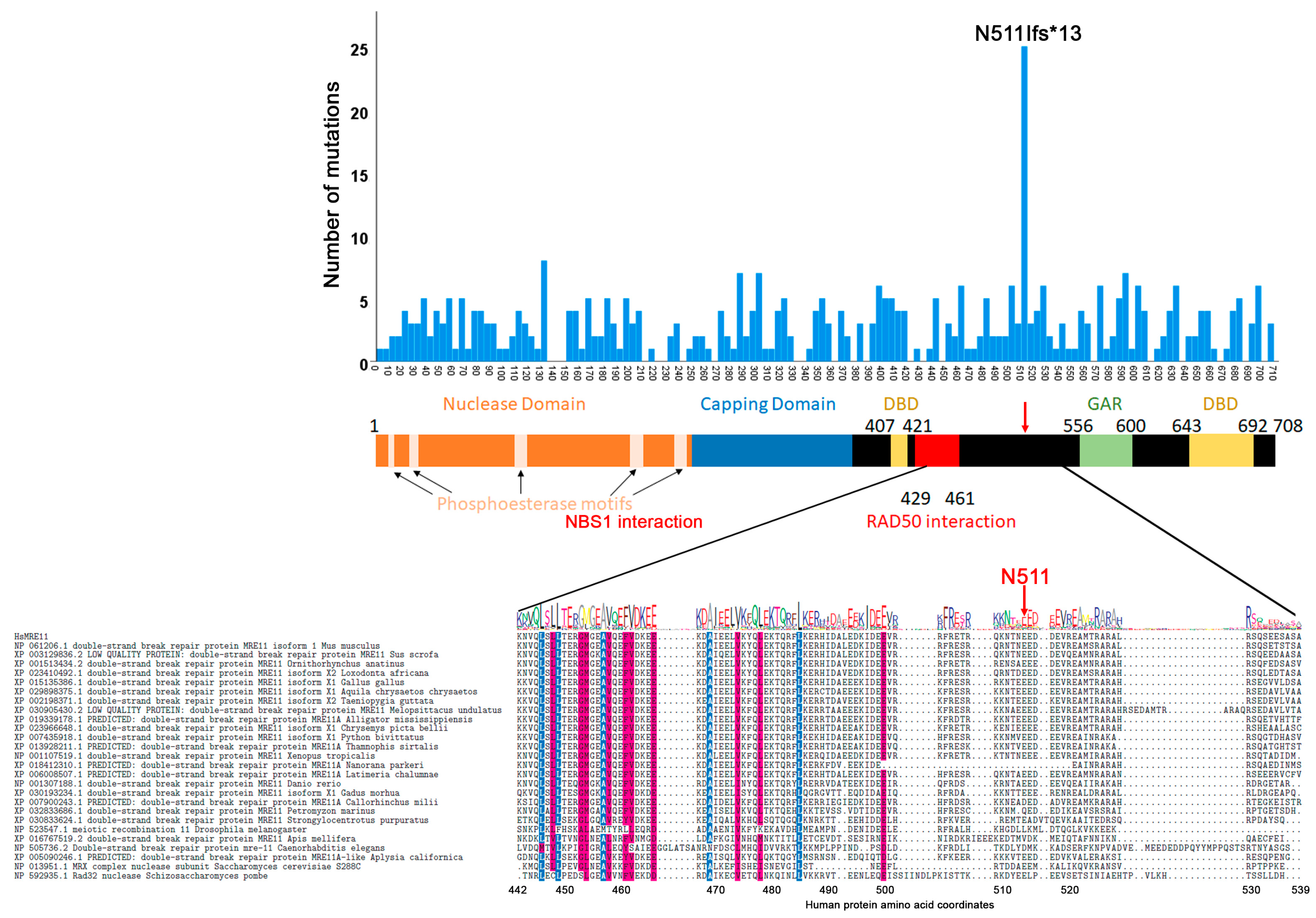
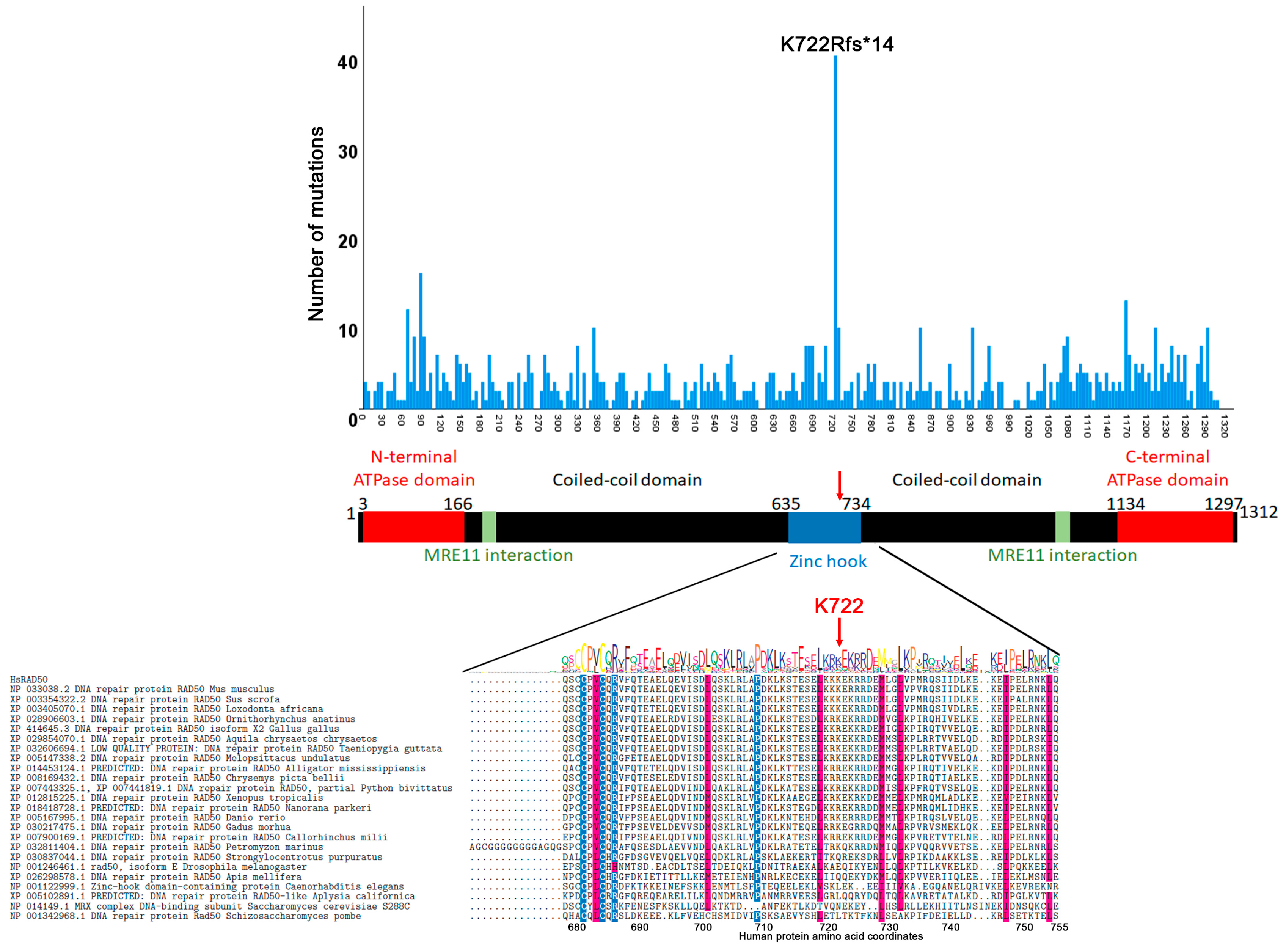
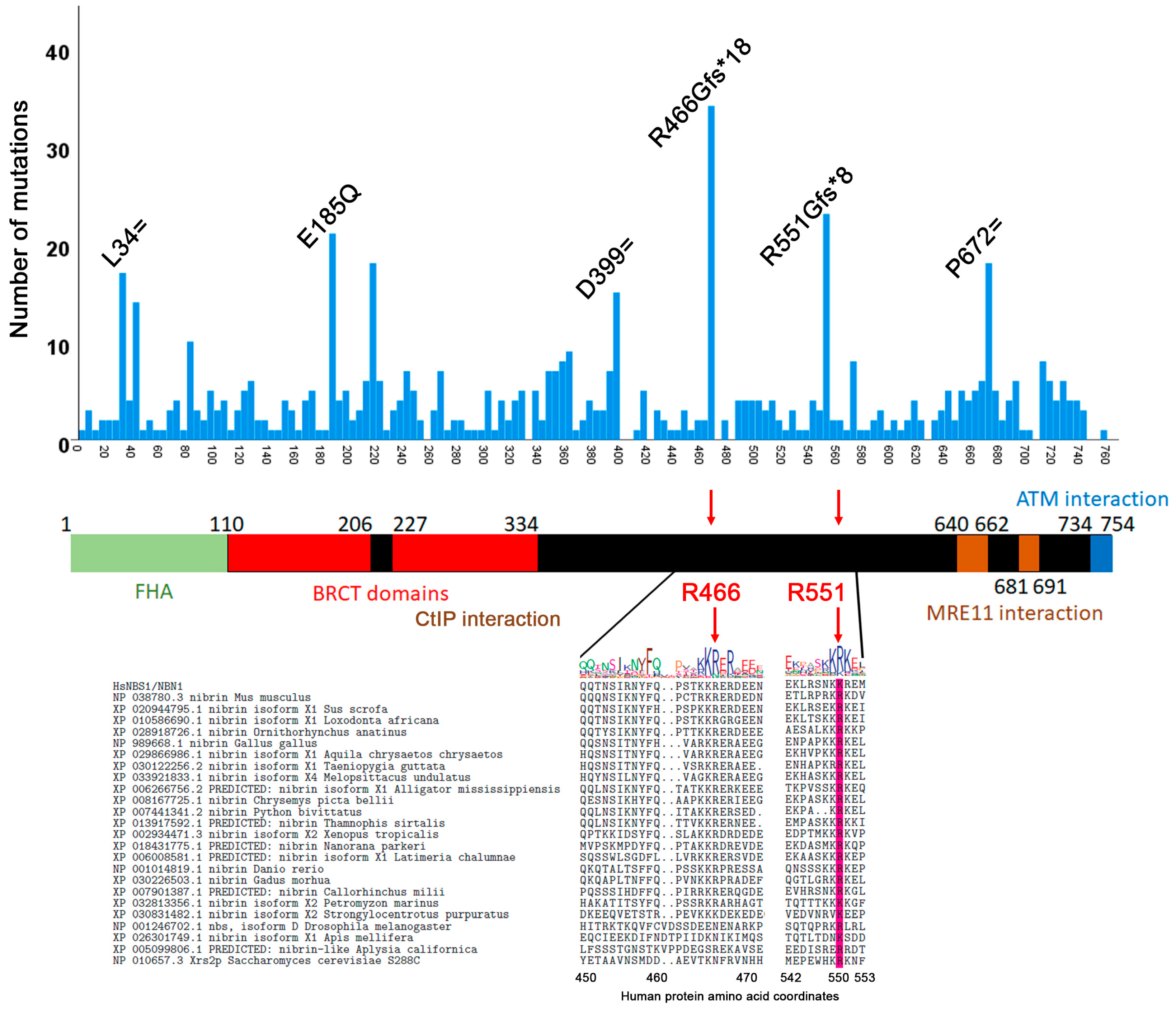
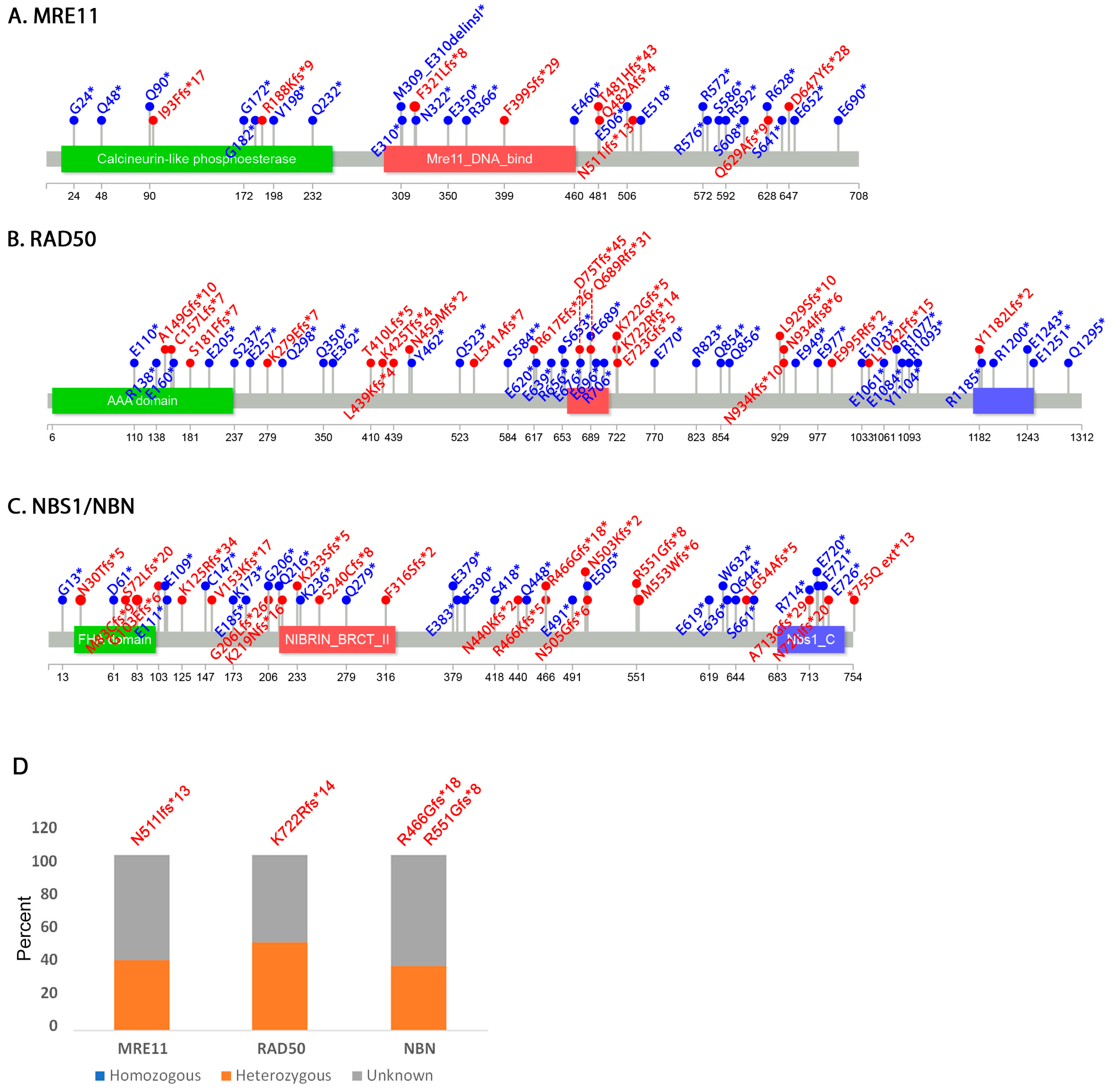
3.2. Mutation Distribution in MRE11, RAD50 and NBS1/NBN
3.3. Other Non-Sense and Frameshift Mutations
3.4. Correlation between Evolutionary Conservation and Point Mutation Spectrum
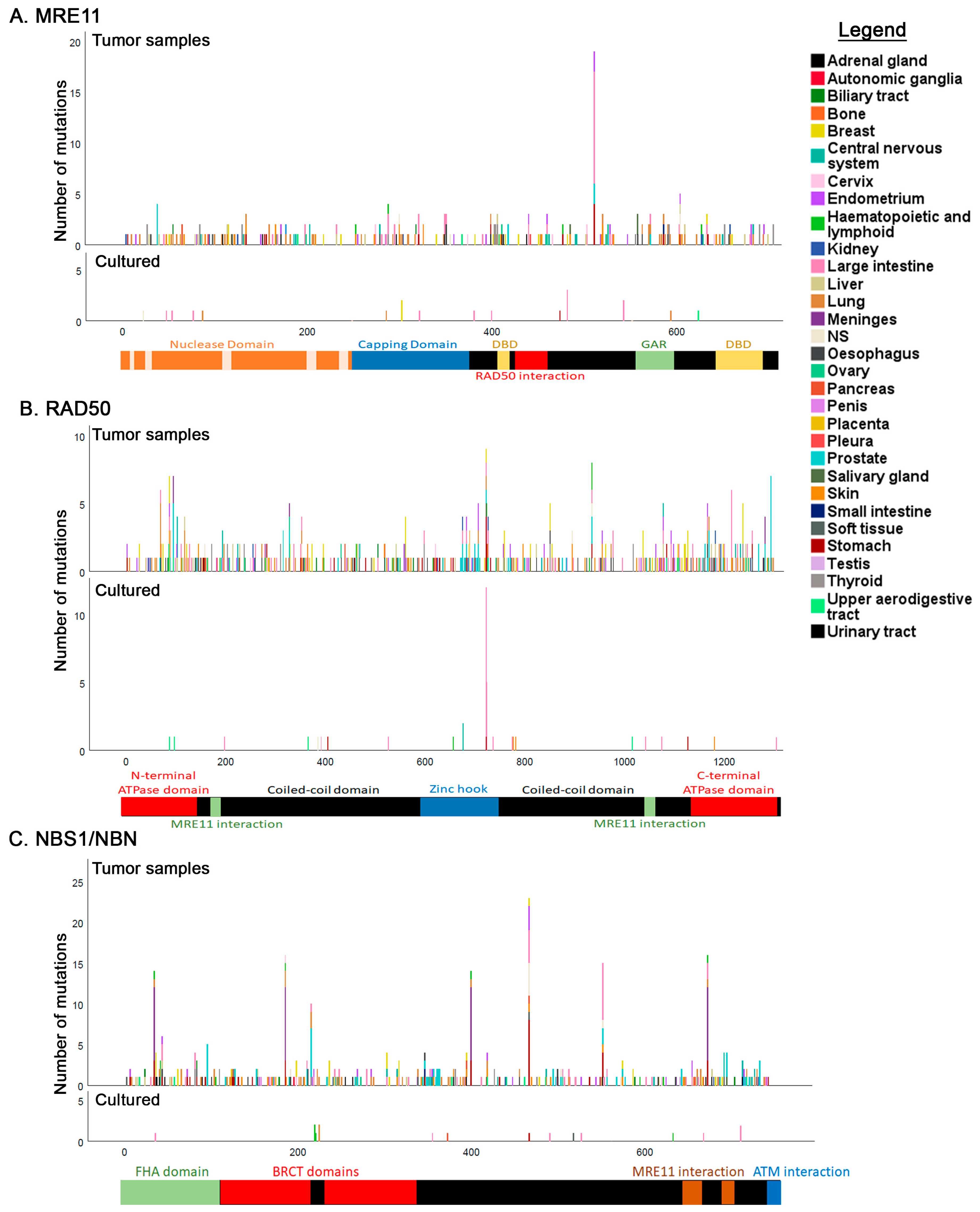
3.5. Tissue Distribution of Mutations
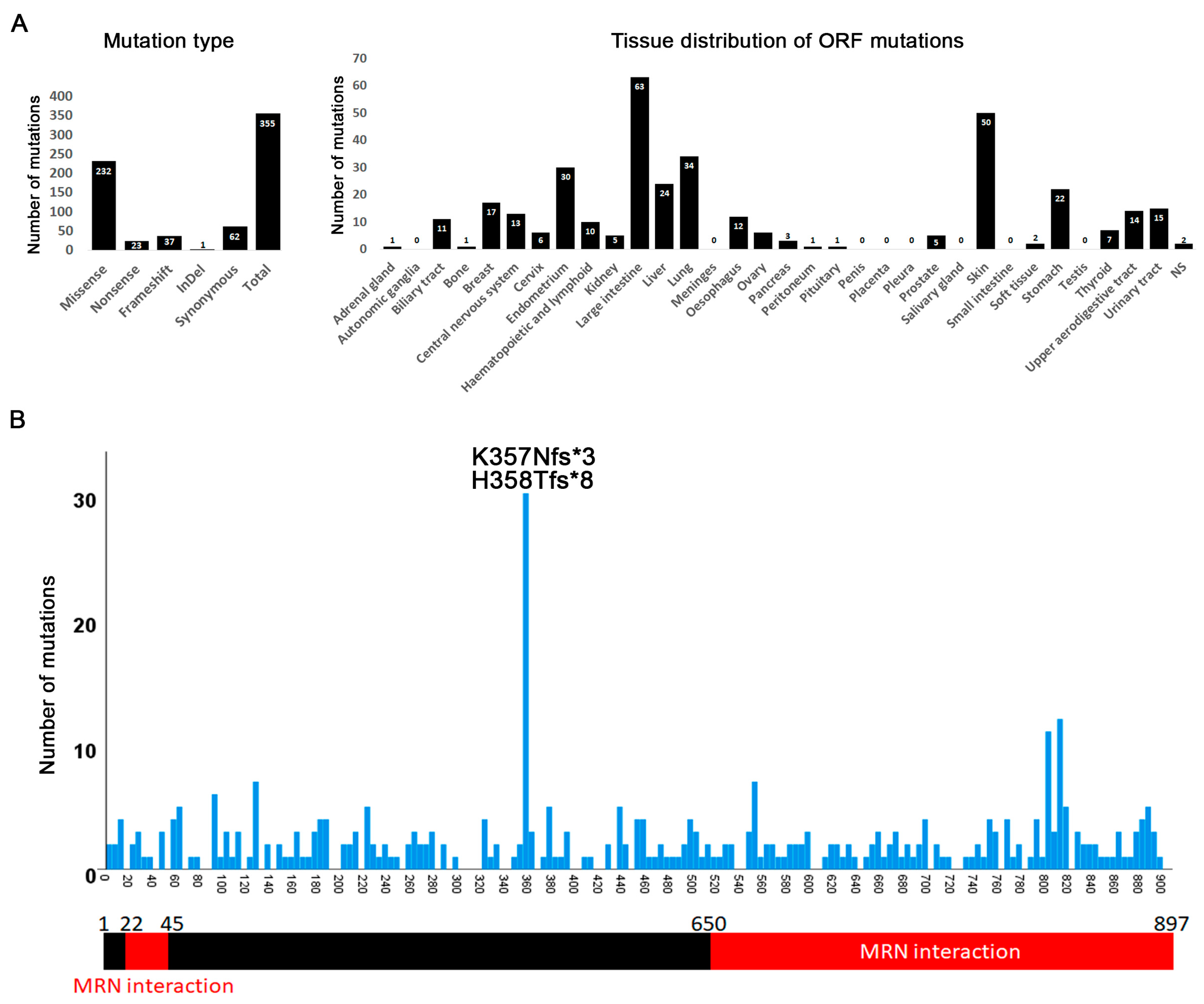
3.6. Mutations in CtIP/RBBP8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Langerak, P.; Russell, P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Origins of the machinery of recombination and sex. Heredity 2002, 88, 125–141. [Google Scholar] [CrossRef]
- Van den Bosch, M.; Bree, R.T.; Lowndes, N.F. The MRN complex: Coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003, 4, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Marsella, A.; Cassani, C.; Casari, E.; Tisi, R.; Longhese, M.P. Structure-function relationships of the Mre11 protein in the control of DNA end bridging and processing. Curr. Genet. 2019, 65, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Karcher, A.; Shin, D.; Fairley, C.; Tainer, J.A.; Carney, J.P. Mre11 and Rad50 from Pyrococcus furiosus: Cloning and biochemical characterization reveal an evolutionarily conserved multiprotein machine. J. Bacteriol. 2000, 182, 6036–6041. [Google Scholar] [CrossRef] [PubMed]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanova, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef]
- Tauchi, H. Positional cloning and functional analysis of the gene responsible for Nijmegen breakage syndrome, NBS1. J. Radiat. Res. 2000, 41, 9–17. [Google Scholar] [CrossRef][Green Version]
- Ivanov, E.L.; Korolev, V.G.; Fabre, F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics 1992, 132, 651–664. [Google Scholar] [PubMed]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef] [PubMed]
- Ajimura, M.; Leem, S.H.; Ogawa, H. Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics 1993, 133, 51–66. [Google Scholar] [PubMed]
- Johzuka, K.; Ogawa, H. Interaction of Mre11 and Rad50: Two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics 1995, 139, 1521–1532. [Google Scholar]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Yuan, S.S.; Lee, E.Y.; Sung, P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Karcher, A.; Craig, L.; Woo, T.T.; Carney, J.P.; Tainer, J.A. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 2001, 105, 473–485. [Google Scholar] [CrossRef]
- Lammens, K.; Bemeleit, D.J.; Mockel, C.; Clausing, E.; Schele, A.; Hartung, S.; Schiller, C.B.; Lucas, M.; Angermuller, C.; Soding, J.; et al. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 2011, 145, 54–66. [Google Scholar] [CrossRef]
- Das, D.; Moiani, D.; Axelrod, H.L.; Miller, M.D.; McMullan, D.; Jin, K.K.; Abdubek, P.; Astakhova, T.; Burra, P.; Carlton, D.; et al. Crystal structure of the first eubacterial Mre11 nuclease reveals novel features that may discriminate substrates during DNA repair. J. Mol. Biol. 2010, 397, 647–663. [Google Scholar] [CrossRef]
- Lim, H.S.; Kim, J.S.; Park, Y.B.; Gwon, G.H.; Cho, Y. Crystal structure of the Mre11-Rad50-ATPgammaS complex: Understanding the interplay between Mre11 and Rad50. Genes Dev. 2011, 25, 1091–1104. [Google Scholar] [CrossRef]
- Mockel, C.; Lammens, K.; Schele, A.; Hopfner, K.P. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 2012, 40, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal structure of human Mre11: Understanding tumorigenic mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.U.; Lammens, K.; Hopfner, K.P. Structure of the catalytic domain of Mre11 from Chaetomium thermophilum. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 752–757. [Google Scholar] [CrossRef]
- Tisi, R.; Vertemara, J.; Zampella, G.; Longhese, M.P. Functional and structural insights into the MRX/MRN complex, a key player in recognition and repair of DNA double-strand breaks. Comput. Struct. Biotechnol. J. 2020, 18, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; van der Linden, E.; Sanchez, H.; Wyman, C. RAD50, an SMC family member with multiple roles in DNA break repair: How does ATP affect function? Chromosome Res. 2009, 17, 277–288. [Google Scholar] [CrossRef]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef]
- Seifert, F.U.; Lammens, K.; Stoehr, G.; Kessler, B.; Hopfner, K.P. Structural mechanism of ATP-dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J. 2016, 35, 759–772. [Google Scholar] [CrossRef]
- de Jager, M.; Trujillo, K.M.; Sung, P.; Hopfner, K.P.; Carney, J.P.; Tainer, J.A.; Connelly, J.C.; Leach, D.R.; Kanaar, R.; Wyman, C. Differential arrangements of conserved building blocks among homologs of the Rad50/Mre11 DNA repair protein complex. J. Mol. Biol. 2004, 339, 937–949. [Google Scholar] [CrossRef]
- Park, Y.B.; Hohl, M.; Padjasek, M.; Jeong, E.; Jin, K.S.; Krezel, A.; Petrini, J.H.; Cho, Y. Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol. 2017, 24, 248–257. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Craig, L.; Moncalian, G.; Zinkel, R.A.; Usui, T.; Owen, B.A.; Karcher, A.; Henderson, B.; Bodmer, J.L.; McMurray, C.T.; et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 2002, 418, 562–566. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, J. N terminus of CtIP is critical for homologous recombination-mediated double-strand break repair. J. Biol. Chem. 2009, 284, 31746–31752. [Google Scholar] [CrossRef]
- Lloyd, J.; Chapman, J.R.; Clapperton, J.A.; Haire, L.F.; Hartsuiker, E.; Li, J.; Carr, A.M.; Jackson, S.P.; Smerdon, S.J. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 2009, 139, 100–111. [Google Scholar] [CrossRef]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Boswell, Z.K.; Rahman, S.; Canny, M.D.; Latham, M.P. A dynamic allosteric pathway underlies Rad50 ABC ATPase function in DNA repair. Sci. Rep. 2018, 8, 1639. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Lee, J.H.; Paull, T.T. Rad50 ATPase activity is regulated by DNA ends and requires coordination of both active sites. Nucleic Acids Res. 2017, 45, 5255–5268. [Google Scholar] [CrossRef]
- Kinoshita, E.; van Rossum-Fikkert, S.; Sanchez, H.; Kertokalio, A.; Wyman, C. Human RAD50 makes a functional DNA-binding complex. Biochimie 2015, 113, 47–53. [Google Scholar] [CrossRef]
- Williams, G.J.; Williams, R.S.; Williams, J.S.; Moncalian, G.; Arvai, A.S.; Limbo, O.; Guenther, G.; SilDas, S.; Hammel, M.; Russell, P.; et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol. 2011, 18, 423–431. [Google Scholar] [CrossRef]
- Paull, T.T.; Deshpande, R.A. The Mre11/Rad50/Nbs1 complex: Recent insights into catalytic activities and ATP-driven conformational changes. Exp. Cell Res. 2014, 329, 139–147. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Paudyal, S.C.; You, Z. Sharpening the ends for repair: Mechanisms and regulation of DNA resection. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 647–657. [Google Scholar] [CrossRef]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef]
- Gobbini, E.; Cassani, C.; Villa, M.; Bonetti, D.; Longhese, M.P. Functions and regulation of the MRX complex at DNA double-strand breaks. Microb. Cell 2016, 3, 329–337. [Google Scholar] [CrossRef]
- Bonetti, D.; Colombo, C.V.; Clerici, M.; Longhese, M.P. Processing of DNA Ends in the Maintenance of Genome Stability. Front. Genet. 2018, 9, 390. [Google Scholar] [CrossRef]
- Runge, K.W.; Li, Y. A curious new role for MRN in Schizosaccharomyces pombe non-homologous end-joining. Curr. Genet. 2018, 64, 359–364. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.; Lobrich, M. The pendulum of the Ku-Ku clock. DNA Repair 2018, 71, 164–171. [Google Scholar] [CrossRef]
- Lafrance-Vanasse, J.; Williams, G.J.; Tainer, J.A. Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog. Biophys. Mol. Biol. 2015, 117, 182–193. [Google Scholar] [CrossRef]
- Langerak, P.; Mejia-Ramirez, E.; Limbo, O.; Russell, P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011, 7, e1002271. [Google Scholar] [CrossRef]
- Teixeira-Silva, A.; Ait Saada, A.; Hardy, J.; Iraqui, I.; Nocente, M.C.; Freon, K.; Lambert, S.A.E. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef]
- Bhaskara, V.; Dupre, A.; Lengsfeld, B.; Hopkins, B.B.; Chan, A.; Lee, J.H.; Zhang, X.; Gautier, J.; Zakian, V.; Paull, T.T. Rad50 adenylate kinase activity regulates DNA tethering by Mre11/Rad50 complexes. Mol. Cell 2007, 25, 647–661. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Williams, G.J.; Limbo, O.; Williams, R.S.; Kuhnlein, J.; Lee, J.H.; Classen, S.; Guenther, G.; Russell, P.; Tainer, J.A.; et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2016, 35, 791. [Google Scholar] [CrossRef]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef]
- Reginato, G.; Cejka, P. The MRE11 complex: A versatile toolkit for the repair of broken DNA. DNA Repair 2020, 91–92, 102869. [Google Scholar] [CrossRef]
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 2013, 12, 791–799. [Google Scholar] [CrossRef]
- Dzikiewicz-Krawczyk, A. The importance of making ends meet: Mutations in genes and altered expression of proteins of the MRN complex and cancer. Mutat. Res. 2008, 659, 262–273. [Google Scholar] [CrossRef]
- Yi, K.; Ju, Y.S. Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med. 2018, 50, 98. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Prim. 2019, 5, 64. [Google Scholar] [CrossRef]
- Rahman, S.; Canny, M.D.; Buschmann, T.A.; Latham, M.P. A Survey of Reported Disease-Related Mutations in the MRE11-RAD50-NBS1 Complex. Cells 2020, 9, 1678. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Bodenhofer, U.; Bonatesta, E.; Horejs-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Bridgham, J.T.; Carroll, S.M.; Thornton, J.W. Evolution of hormone-receptor complexity by molecular exploitation. Science 2006, 312, 97–101. [Google Scholar] [CrossRef]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017, 33, 283–297. [Google Scholar] [CrossRef]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair 2010, 9, 1299–1306. [Google Scholar] [CrossRef]
- Dery, U.; Coulombe, Y.; Rodrigue, A.; Stasiak, A.; Richard, S.; Masson, J.Y. A glycine-arginine domain in control of the human MRE11 DNA repair protein. Mol. Cell Biol. 2008, 28, 3058–3069. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Kim, T.M.; An, C.H.; Rhee, J.K.; Jung, S.H.; Lee, S.H.; Baek, I.P.; Kim, M.S.; Lee, S.H.; Chung, Y.J. Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget 2015, 6, 27725–27735. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Karcher, A.; Shin, D.S.; Craig, L.; Arthur, L.M.; Carney, J.P.; Tainer, J.A. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 2000, 101, 789–800. [Google Scholar] [CrossRef]
- Tatebe, H.; Lim, C.T.; Konno, H.; Shiozaki, K.; Shinohara, A.; Uchihashi, T.; Furukohri, A. Rad50 zinc hook functions as a constitutive dimerization module interchangeable with SMC hinge. Nat. Commun. 2020, 11, 370. [Google Scholar] [CrossRef]
- Soh, Y.M.; Basquin, J.; Gruber, S. A rod conformation of the Pyrococcus furiosus Rad50 coiled coil. Proteins 2020. [Google Scholar] [CrossRef]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef]
- Liu, J.; McCleland, M.; Stawiski, E.W.; Gnad, F.; Mayba, O.; Haverty, P.M.; Durinck, S.; Chen, Y.J.; Klijn, C.; Jhunjhunwala, S.; et al. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat. Commun. 2014, 5, 3830. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.; Hohl, M.; Fleming, J.C.; Petrini, J.H. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat. Struct. Mol. Biol. 2005, 12, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Tauchi, H.; Nakamura, A.; Kondo, N.; Sakamoto, S.; Endo, S.; Smeets, D.; Solder, B.; Belohradsky, B.H.; Der Kaloustian, V.M.; et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat. Genet. 1998, 19, 179–181. [Google Scholar] [CrossRef]
- Ueno, M.; Nakazaki, T.; Akamatsu, Y.; Watanabe, K.; Tomita, K.; Lindsay, H.D.; Shinagawa, H.; Iwasaki, H. Molecular characterization of the Schizosaccharomyces pombe nbs1+ gene involved in DNA repair and telomere maintenance. Mol. Cell. Biol. 2003, 23, 6553–6563. [Google Scholar] [CrossRef]
- Komatsu, K. NBS1 and multiple regulations of DNA damage response. J. Radiat. Res. 2016, 57 (Suppl. 1), i11–i17. [Google Scholar] [CrossRef]
- Almawi, A.W.; Matthews, L.A.; Guarne, A. FHA domains: Phosphopeptide binding and beyond. Prog. Biophys. Mol. Biol. 2017, 127, 105–110. [Google Scholar] [CrossRef]
- Wu, Q.; Jubb, H.; Blundell, T.L. Phosphopeptide interactions with BRCA1 BRCT domains: More than just a motif. Prog. Biophys. Mol. Biol. 2015, 117, 143–148. [Google Scholar] [CrossRef]
- Tauchi, H.; Matsuura, S.; Kobayashi, J.; Sakamoto, S.; Komatsu, K. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene 2002, 21, 8967–8980. [Google Scholar] [CrossRef]
- Yachida, S.; Wood, L.D.; Suzuki, M.; Takai, E.; Totoki, Y.; Kato, M.; Luchini, C.; Arai, Y.; Nakamura, H.; Hama, N.; et al. Genomic Sequencing Identifies ELF3 as a Driver of Ampullary Carcinoma. Cancer Cell 2016, 29, 229–240. [Google Scholar] [CrossRef]
- Moelans, C.B.; de Ligt, J.; van der Groep, P.; Prins, P.; Besselink, N.J.M.; Hoogstraat, M.; Ter Hoeve, N.D.; Lacle, M.M.; Kornegoor, R.; van der Pol, C.C.; et al. The molecular genetic make-up of male breast cancer. Endocr. Relat. Cancer 2019, 26, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Taylor-Weiner, A.; Lelic, N.; Jones, R.T.; Kim, J.C.; Francis, J.M.; Abedalthagafi, M.; Borges, L.F.; Coumans, J.V.; Curry, W.T.; et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2014, 2, 167. [Google Scholar] [CrossRef] [PubMed]
- Chong, I.Y.; Cunningham, D.; Barber, L.J.; Campbell, J.; Chen, L.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Guettler, S.; Garcia-Murillas, I.; et al. The genomic landscape of oesophagogastric junctional adenocarcinoma. J. Pathol. 2013, 231, 301–310. [Google Scholar] [CrossRef]
- Lim, B.; Mun, J.; Kim, J.H.; Kim, C.W.; Roh, S.A.; Cho, D.H.; Kim, Y.S.; Kim, S.Y.; Kim, J.C. Genome-wide mutation profiles of colorectal tumors and associated liver metastases at the exome and transcriptome levels. Oncotarget 2015, 6, 22179–22190. [Google Scholar] [CrossRef]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Kang, H.; Tan, M.; Bishop, J.A.; Jones, S.; Sausen, M.; Ha, P.K.; Agrawal, N. Whole-Exome Sequencing of Salivary Gland Mucoepidermoid Carcinoma. Clin. Cancer Res. 2017, 23, 283–288. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, C.; Li, X.; Luo, W.; Roy, B.; Xiong, T.; Zhang, X.; Yang, H.; Wang, J.; Ye, Z.; et al. The landscape of somatic mutation in sporadic Chinese colorectal cancer. Oncotarget 2018, 9, 27412–27422. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wei, Q.; Bondy, M.L.; Li, D.; Brewster, A.; Shete, S.; Yu, T.K.; Sahin, A.; Meric-Bernstam, F.; Hunt, K.K.; et al. Polymorphisms and haplotypes of the NBS1 gene are associated with risk of sporadic breast cancer in non-Hispanic white women ≤55 years. Carcinogenesis 2006, 27, 2209–2216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lan, Q.; Shen, M.; Berndt, S.I.; Bonner, M.R.; He, X.; Yeager, M.; Welch, R.; Keohavong, P.; Donahue, M.; Hainaut, P.; et al. Smoky coal exposure, NBS1 polymorphisms, p53 protein accumulation, and lung cancer risk in Xuan Wei, China. Lung Cancer 2005, 49, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Ryk, C.; Kumar, R.; Thirumaran, R.K.; Hou, S.M. Polymorphisms in the DNA repair genes XRCC1, APEX1, XRCC3 and NBS1, and the risk for lung cancer in never- and ever-smokers. Lung Cancer 2006, 54, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of microarray data using Z score transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef]
- Xiao, Y.; Weaver, D.T. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997, 25, 2985–2991. [Google Scholar] [CrossRef]
- Luo, G.; Yao, M.S.; Bender, C.F.; Mills, M.; Bladl, A.R.; Bradley, A.; Petrini, J.H. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. USA 1999, 96, 7376–7381. [Google Scholar] [CrossRef]
- Dumon-Jones, V.; Frappart, P.O.; Tong, W.M.; Sajithlal, G.; Hulla, W.; Schmid, G.; Herceg, Z.; Digweed, M.; Wang, Z.Q. Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res. 2003, 63, 7263–7269. [Google Scholar]
- Delia, D.; Piane, M.; Buscemi, G.; Savio, C.; Palmeri, S.; Lulli, P.; Carlessi, L.; Fontanella, E.; Chessa, L. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum. Mol. Genet. 2004, 13, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, M.S.; Brody, J.R.; Elghalbzouri-Maghrani, E.; Zdzienicka, M.Z.; Kern, S.E. Does tumorigenesis select for or against mutations of the DNA repair-associated genes BRCA2 and MRE11?: Considerations from somatic mutations in microsatellite unstable (MSI) gastrointestinal cancers. BMC Genet. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Oba, D.; Hayashi, M.; Minamitani, M.; Hamano, S.; Uchisaka, N.; Kikuchi, A.; Kishimoto, H.; Takagi, M.; Morio, T.; Mizutani, S. Autopsy study of cerebellar degeneration in siblings with ataxia-telangiectasia-like disorder. Acta Neuropathol. 2010, 119, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Alsbeih, G.; Al-Hadyan, K.; Al-Harbi, N. Assessment of carriers’ frequency of a novel MRE11 mutation responsible for the rare ataxia telangiectasia-like disorder. Genet. Test. 2008, 12, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Varon, R.; Schoch, C.; Reis, A.; Hiddemann, W.C.; Sperling, K.; Schnittger, S. Mutation analysis of the Nijmegen breakage syndrome gene (NBS1) in nineteen patients with acute myeloid leukemia with complex karyotypes. Leuk. Lymphoma 2003, 44, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.; Swiatek-Koscielna, B.; Kaluzna, E.M.; Rembowska, J.; Dzikiewicz-Krawczyk, A.; Zawada, M.; Januszkiewicz-Lewandowska, D. Effect of irradiation on DNA synthesis, NBN gene expression and chromosomal stability in cells with NBN mutations. Arch. Med. Sci 2017, 13, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Ebi, H.; Matsuo, K.; Sugito, N.; Suzuki, M.; Osada, H.; Tajima, K.; Ueda, R.; Takahashi, T. Novel NBS1 heterozygous germ line mutation causing MRE11-binding domain loss predisposes to common types of cancer. Cancer Res. 2007, 67, 11158–11165. [Google Scholar] [CrossRef]
- Mendez, G.; Cilli, D.; Berardinelli, F.; Viganotti, M.; Ascenzi, P.; Tanzarella, C.; Antoccia, A.; di Masi, A. Cleavage of the BRCT tandem domains of nibrin by the 657del5 mutation affects the DNA damage response less than the Arg215Trp mutation. IUBMB Life 2012, 64, 853–861. [Google Scholar] [CrossRef]
- Dzikiewicz-Krawczyk, A.; Mosor, M.; Januszkiewicz, D.; Nowak, J. Impact of heterozygous c.657-661del, p.I171V and p.R215W mutations in NBN on nibrin functions. Mutagenesis 2012, 27, 337–343. [Google Scholar] [CrossRef]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.; Tosetto, M.; Gorman, J.; O’Donoghue, D.; Sheahan, K.; Hyland, J.; Mulcahy, H.; Gibbons, D.; O’Sullivan, J. Effects of radiation on levels of DNA damage in normal non-adjacent mucosa from colorectal cancer cases. J. Gastrointest. Cancer 2013, 44, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Campregher, C.; Schmid, G.; Ferk, F.; Knasmuller, S.; Khare, V.; Kortum, B.; Dammann, K.; Lang, M.; Scharl, T.; Spittler, A.; et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLoS ONE 2012, 7, e50541. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Roelens, B.; Zawadzki, K.A.; Villeneuve, A.M. Interdependent and separable functions of Caenorhabditis elegans MRN-C complex members couple formation and repair of meiotic DSBs. Proc. Natl. Acad. Sci. USA 2018, 115, E4443–E4452. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Williams, R.S. CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair 2017, 56, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; Woo, J.K.; Yip, K.Y.; Seo, J.; Hui, E.P.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef]
- Laurent, C.; Nicolae, A.; Laurent, C.; Le Bras, F.; Haioun, C.; Fataccioli, V.; Amara, N.; Adelaide, J.; Guille, A.; Schiano, J.M.; et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood 2020, 135, 360–370. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Kurman, R.J.; Nakayama, K.; Velculescu, V.E.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; Shih Ie, M. Low-grade serous carcinomas of the ovary contain very few point mutations. J. Pathol. 2012, 226, 413–420. [Google Scholar] [CrossRef]
- Wu, R.C.; Veras, E.; Lin, J.; Gerry, E.; Bahadirli-Talbott, A.; Baras, A.; Ayhan, A.; Shih, I.M.; Wang, T.L. Elucidating the pathogenesis of synchronous and metachronous tumors in a woman with endometrioid carcinomas using a whole-exome sequencing approach. Mol. Case Stud. 2017, 3, a001693. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; Kim, M.S.; Baek, I.P.; Park, S.W.; Lee, S.H.; Lee, H.H.; Kim, S.S.; Chung, Y.J.; Lee, S.H. The mutational burdens and evolutionary ages of early gastric cancers are comparable to those of advanced gastric cancers. J. Pathol. 2014, 234, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Volkening, L.; Vatselia, A.; Asgedom, G.; Bastians, H.; Lavin, M.; Schindler, D.; Schambach, A.; Bousset, K.; Dork, T. RAD50 regulates mitotic progression independent of DNA repair functions. FASEB J. 2020, 34, 2812–2820. [Google Scholar] [CrossRef]
- Kuroda, S.; Urata, Y.; Fujiwara, T. Ataxia-telangiectasia mutated and the Mre11-Rad50-NBS1 complex: Promising targets for radiosensitization. Acta Med. Okayama 2012, 66, 83–92. [Google Scholar] [CrossRef]
- Chang, L.; Huang, J.; Wang, K.; Li, J.; Yan, R.; Zhu, L.; Ye, J.; Wu, X.; Zhuang, S.; Li, D.; et al. Targeting Rad50 sensitizes human nasopharyngeal carcinoma cells to radiotherapy. BMC Cancer 2016, 16, 190. [Google Scholar] [CrossRef]
- Tran, H.M.; Shi, G.; Li, G.; Carney, J.P.; O’Malley, B.; Li, D. Mutant Nbs1 enhances cisplatin-induced DNA damage and cytotoxicity in head and neck cancer. Otolaryngol. Head Neck Surg. 2004, 131, 477–484. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Residue Conservation from Alignment Data 1 | Statistical Analysis of Mutations from COSMIC (Chi Square) 2 | |||||
|---|---|---|---|---|---|---|---|
| Residue Type | Number of Residues | Observed | Expected | Chi-Square Value | Chi-Square Sum | p-Value | |
| MRE11 | Identity | 113 | 52 | 43.36 | 1.72 | 2.05 | 0.152206 |
| Non-conserved | 595 | 219 | 227.64 | 0.33 | |||
| RAD50 | Identity | 136 | 81 | 56.58 | 10.54 | 11.76 | 0.000605 |
| Non-conserved | 1176 | 463 | 487.42 | 1.22 | |||
| NBN | Identity | 38 | 30 | 18.45 | 7.23 | 7.61 | 0.005805 |
| Non-conserved | 716 | 339 | 350.55 | 0.38 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McPherson, M.T.; Holub, A.S.; Husbands, A.Y.; Petreaca, R.C. Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells. Cancers 2020, 12, 3794. https://doi.org/10.3390/cancers12123794
McPherson MT, Holub AS, Husbands AY, Petreaca RC. Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells. Cancers. 2020; 12(12):3794. https://doi.org/10.3390/cancers12123794
Chicago/Turabian StyleMcPherson, Matthew T., Ashton S. Holub, Aman Y. Husbands, and Ruben C. Petreaca. 2020. "Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells" Cancers 12, no. 12: 3794. https://doi.org/10.3390/cancers12123794
APA StyleMcPherson, M. T., Holub, A. S., Husbands, A. Y., & Petreaca, R. C. (2020). Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells. Cancers, 12(12), 3794. https://doi.org/10.3390/cancers12123794