Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients

 , , ,
, , , 
 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
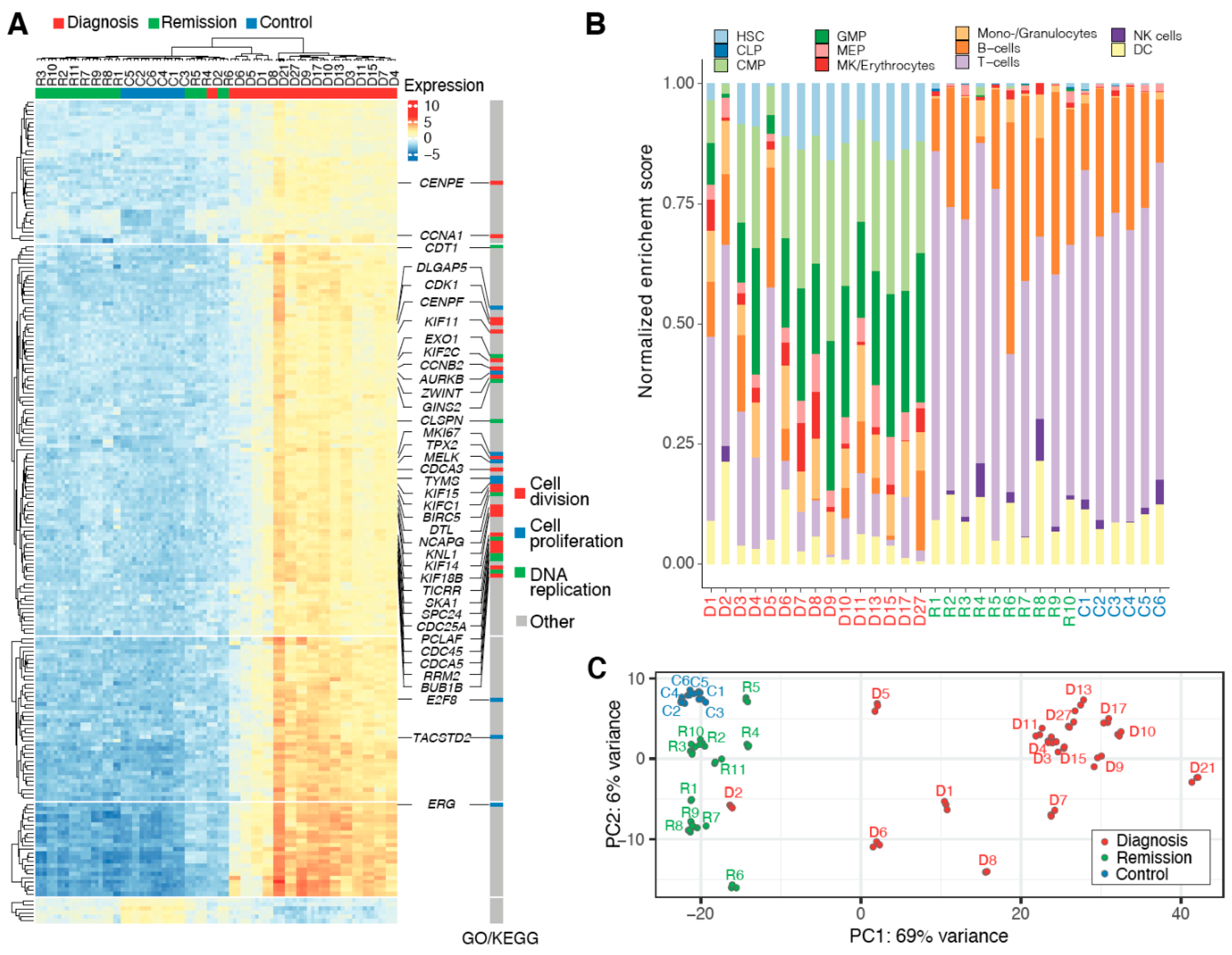
2.1. Heterogeneous CML Transcriptomes Converge at Remission
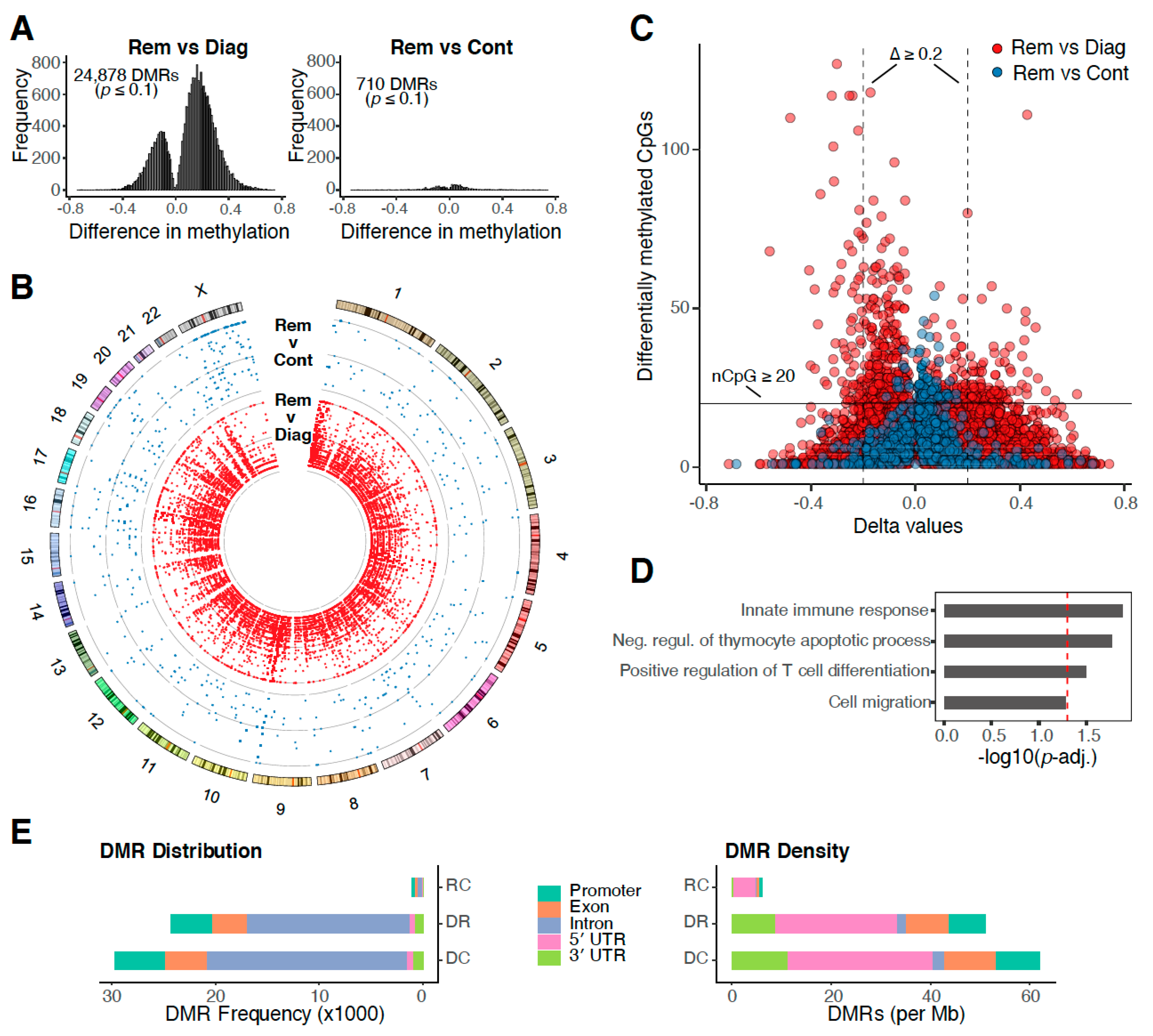
2.2. DNA Methylation Profiles Return to Normal at Remission
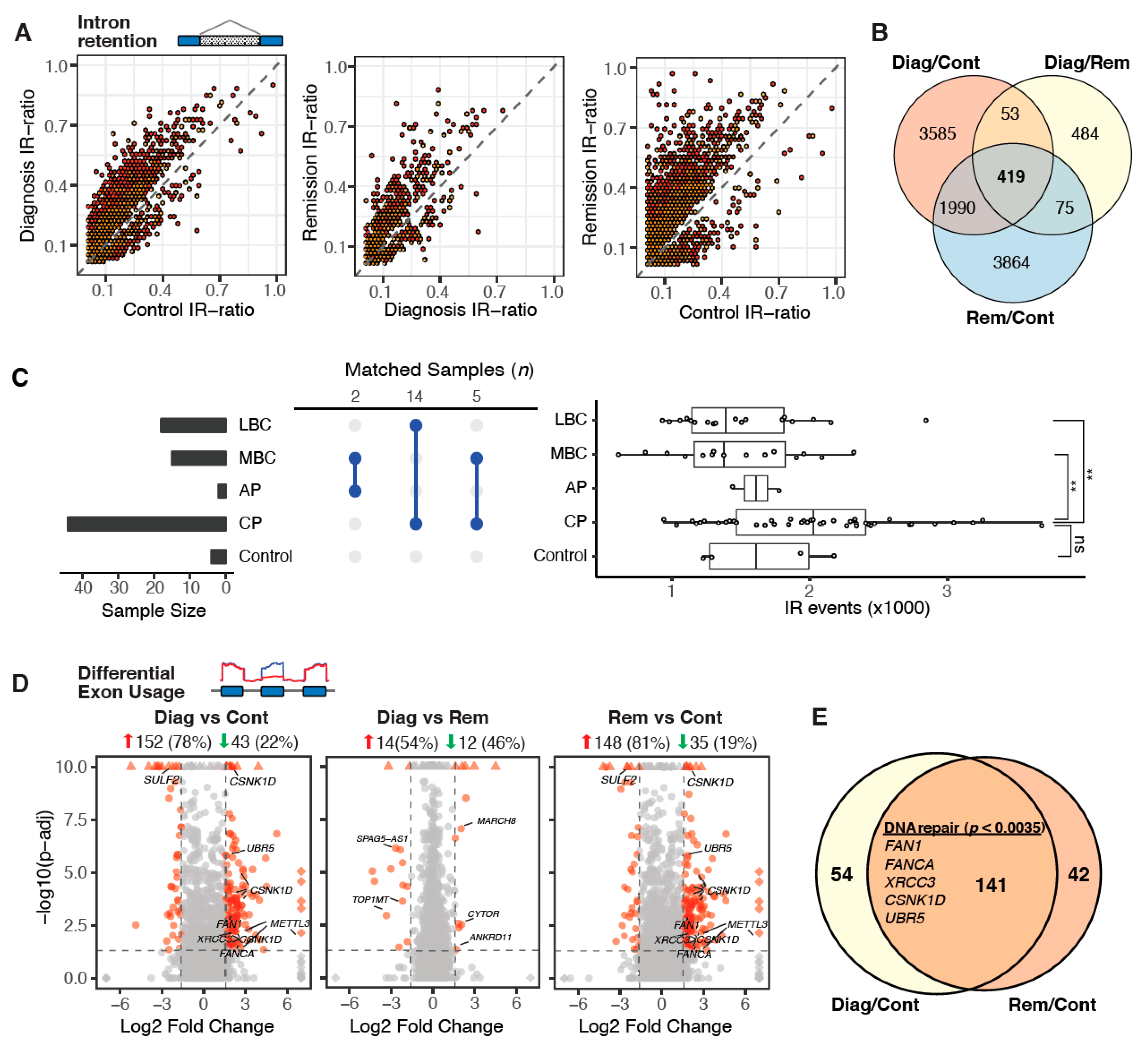
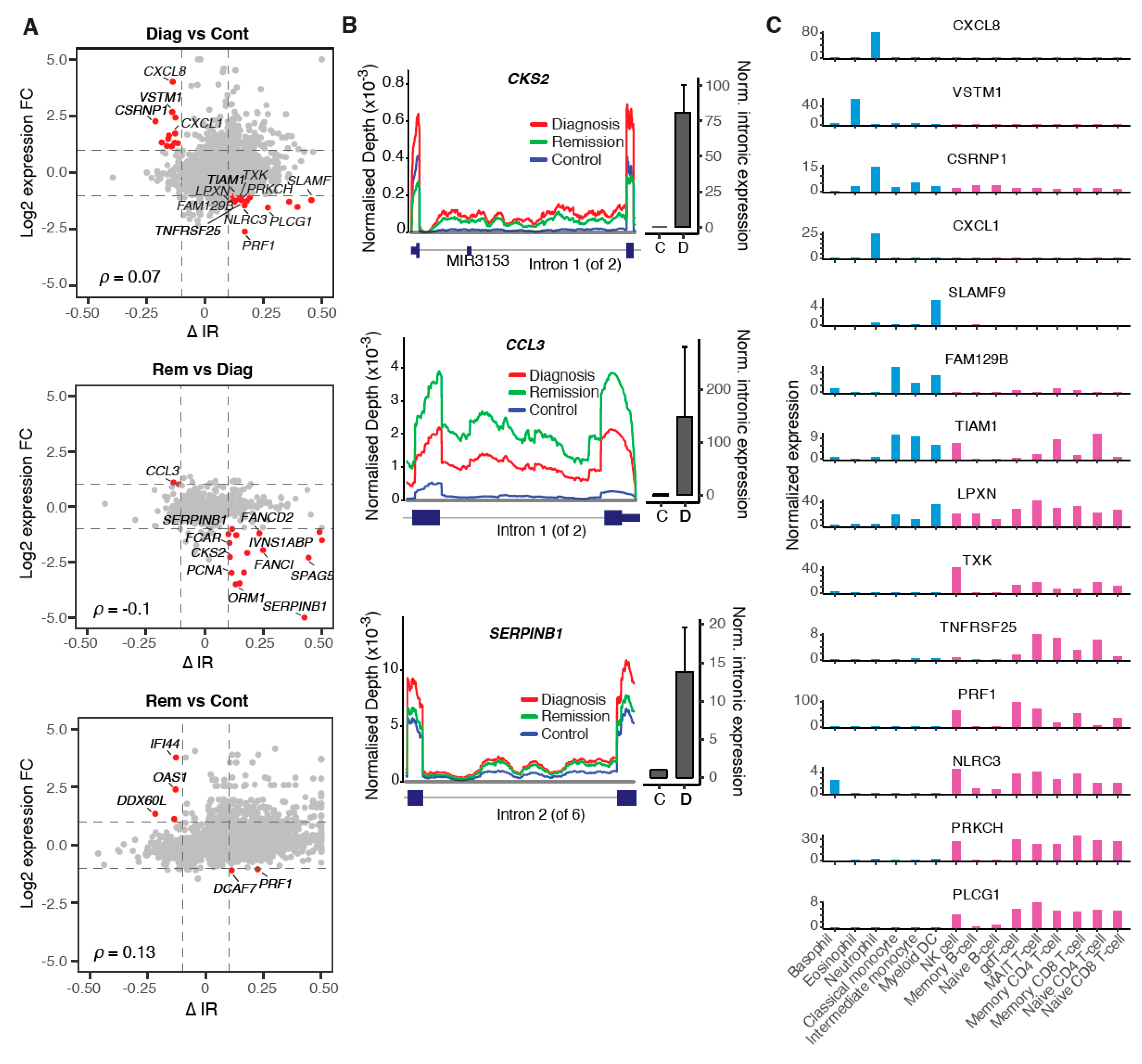
2.3. Aberrant Alternative Splicing Distinguishes Remission Samples from Healthy Controls
2.4. Modulation of IR Levels Supports Lineage-Specific Gene Expression in CML
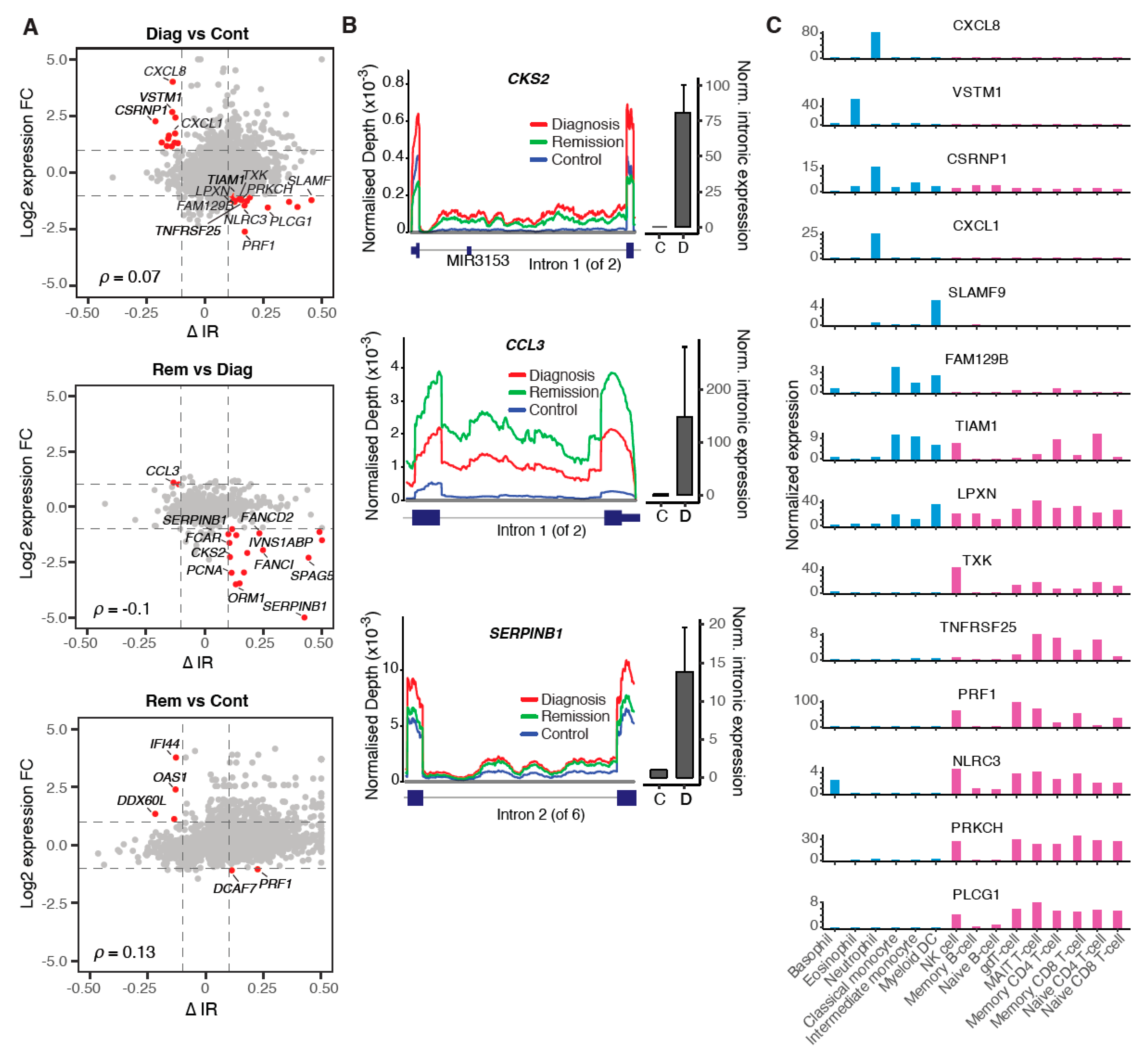
2.5. Multiple Regulatory Mechanisms Modulate Intron Retention in CML
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. RNA Isolation and mRNA-seq
4.3. mRNA Sequencing Data Analysis
4.4. IR Validation
4.5. Whole Genome Bisulfite Sequencing (WGBS)
4.6. WGBS Data Analysis
4.7. ChIP-seq Data Analysis
4.8. Statistical Analyses
4.9. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peng, C.; Li, S. Chronic Myeloid Leukemia (CML) Mouse Model in Translational Research. Methods Mol. Biol. 2016, 1438, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Silver, R.T.; Druker, B.J.; Goldman, J.M.; Gambacorti-Passerini, C.; Guilhot, F.; Schiffer, C.A.; Fischer, T.; Deininger, M.W.; Lennard, A.L.; et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: Results of a phase 2 study. Blood 2002, 99, 1928–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Investig. 2010, 120, 2254–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, A.S.; Melo, J.V. The impact of gene profiling in chronic myeloid leukaemia. Best Pract. Res. Clin. Haematol. 2009, 22, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Giles, F.; Zhang, X.; Wang, X.; Zhang, Z.; Lee, T.S.; Yeh, C.H.; Albitar, M. Three novel alternative splicing mutations in BCR-ABL1 detected in CML patients with resistance to kinase inhibitors. Int. J. Lab. Hematol. 2011, 33, 326–331. [Google Scholar] [CrossRef]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Dvinge, H.; Bradley, R.K. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, U.; Pinello, N.; Jia, F.; Alasmari, S.; Ritchie, W.; Keightley, M.C.; Shini, S.; Lieschke, G.J.; Wong, J.J.; Rasko, J.E.J. Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol. 2017, 18, 216. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.J.; Au, A.Y.; Ritchie, W.; Rasko, J.E. Intron retention in mRNA: No longer nonsense: Known and putative roles of intron retention in normal and disease biology. Bioessays 2016, 38, 41–49. [Google Scholar] [CrossRef]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Topakian, T.; Altenberger, C.; Cerny-Reiterer, S.; Herndlhofer, S.; Ziegler, B.; Datlinger, P.; Byrgazov, K.; Bock, C.; Mannhalter, C.; et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016, 30, 1861–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.C.H.; Rasko, J.E.J.; Wong, J.J. We skip to work: Alternative splicing in normal and malignant myelopoiesis. Leukemia 2018, 32, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Nikolaou, C.; Althammer, S.; Sammeth, M.; Beato, M.; Valcarcel, J.; Guigo, R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009, 16, 996–1001. [Google Scholar] [CrossRef]
- Wong, J.J.; Gao, D.; Nguyen, T.V.; Kwok, C.T.; van Geldermalsen, M.; Middleton, R.; Pinello, N.; Thoeng, A.; Nagarajah, R.; Holst, J.; et al. Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat. Commun. 2017, 8, 15134. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Vanichkina, D.P.; Schmitz, U.; Wong, J.J.; Rasko, J.E.J. Challenges in defining the role of intron retention in normal biology and disease. Semin. Cell Dev. Biol. 2018, 75, 40–49. [Google Scholar] [CrossRef]
- Wong, J.J.; Ritchie, W.; Ebner, O.A.; Selbach, M.; Wong, J.W.; Huang, Y.; Gao, D.; Pinello, N.; Gonzalez, M.; Baidya, K.; et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.R.; Ritchie, W.; Wong, J.J.; Schmitz, U.; Middleton, R.; An, X.; Mohandas, N.; Rasko, J.E.; Blobel, G.A. A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood 2016, 127, e24–e34. [Google Scholar] [CrossRef] [Green Version]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; De Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Gstoettenbauer, A.; Colinge, J.; Kaupe, I.; Bilban, M.; Burkard, T.R.; Valent, P.; Superti-Furga, G. The chemokine interleukin-8 and the surface activation protein CD69 are markers for Bcr-Abl activity in chronic myeloid leukemia. Mol. Oncol. 2008, 2, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, S.N.; Magee, J.A. PRKCH regulates hematopoietic stem cell function and predicts poor prognosis in acute myeloid leukemia. Exp. Hematol 2017, 53, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Plaschka, C.; Lariviere, L.; Wenzeck, L.; Seizl, M.; Hemann, M.; Tegunov, D.; Petrotchenko, E.; Borchers, C.; Baumeister, W.; Herzog, F. Architecture of the RNA polymerase II–Mediator core initiation complex. Nature 2015, 518, 376. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.; Gao, D.; Thomas, A.; Singh, B.; Au, A.; Wong, J.J.; Bomane, A.; Cosson, B.; Eyras, E.; Rasko, J.E.; et al. IRFinder: Assessing the impact of intron retention on mammalian gene expression. Genome Biol. 2017, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lu, Y.; Tian, W. Epigenetic features are significantly associated with alternative splicing. BMC Genom. 2012, 13, 123. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Zheng, L.; Park, J.W.; Lv, R.; Chen, H.; Jiao, F.; Xu, W.; Mu, S.; Wen, H.; Qiu, J.; et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol. Cell 2014, 56, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Jena, N.; Deng, M.; Sicinska, E.; Sicinski, P.; Daley, G.Q. Critical role for cyclin D2 in BCR/ABL-induced proliferation of hematopoietic cells. Cancer Res. 2002, 62, 535–541. [Google Scholar] [PubMed]
- Green, I.D.; Pinello, N.; Song, R.; Lee, Q.; Halstead, J.M.; Kwok, C.T.; Wong, A.C.H.; Nair, S.S.; Clark, S.J.; Roediger, B.; et al. Macrophage development and activation involve coordinated intron retention in key inflammatory regulators. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Yang, W.; Han, M.; Zhang, Y.; Shen, T.; Nie, H.; Zhou, Z.; Dai, Y.; Yang, Y.; Liu, P.; et al. Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res. 2016, 44, 6817–6829. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, S.; Guigo, R. Dynamic changes in intron retention are tightly associated with regulation of splicing factors and proliferative activity during B-cell development. Nucleic Acids Res. 2020, 48, 1327–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seggewiss, R.; Loré, K.; Greiner, E.; Magnusson, M.K.; Price, D.A.; Douek, D.C.; Dunbar, C.E.; Wiestner, A. Imatinib inhibits T-cell receptor–mediated T-cell proliferation and activation in a dose-dependent manner. Blood 2005, 105, 2473–2479. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Schmitt, A.; Giannopoulos, K.; Chen, B.; Rojewski, M.; Döhner, H.; Bunjes, D.; Schmitt, M. Imatinib impairs the proliferation and function of CD4+ CD25+ regulatory T cells in a dose-dependent manner. Int. J. Oncol. 2007, 31, 1133–1139. [Google Scholar]
- De Lavallade, H.; Khoder, A.; Hart, M.; Sarvaria, A.; Sekine, T.; Alsuliman, A.; Mielke, S.; Bazeos, A.; Stringaris, K.; Ali, S. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood J. Am. Soc. Hematol. 2013, 122, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Rohon, P.; Porkka, K.; Mustjoki, S. Immunoprofiling of patients with chronic myeloid leukemia at diagnosis and during tyrosine kinase inhibitor therapy. Eur. J. Haematol. 2010, 85, 387–398. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nakamae, H.; Katayama, T.; Nakane, T.; Koh, H.; Nakamae, M.; Hirose, A.; Hagihara, K.; Terada, Y.; Nakao, Y. Different immunoprofiles in patients with chronic myeloid leukemia treated with imatinib, nilotinib or dasatinib. Leuk. Lymphoma 2012, 53, 1084–1089. [Google Scholar] [CrossRef]
- Reinwald, M.; Boch, T.; Hofmann, W.-K.; Buchheidt, D. Risk of Infectious Complications in Hemato-Oncological Patients Treated with Kinase Inhibitors. Biomark. Insights 2016, 10, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, Y.; Wang, C.; Wu, Y.N.; Xing, Y. SURVIV for survival analysis of mRNA isoform variation. Nat. Commun. 2016, 7, 11548. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Pinello, N.; Nguyen, T.V.; Thoeng, A.; Nagarajah, R.; Holst, J.; Rasko, J.E.; Wong, J.J. DNA methylation/hydroxymethylation regulate gene expression and alternative splicing during terminal granulopoiesis. Epigenomics 2019, 11, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Blum, R.; Bowman, C.; Hu, D.; Shilatifard, A.; Shen, S.; Dynlacht, B.D. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol. Cell 2014, 53, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef] [Green Version]
- Monteuuis, G.; Schmitz, U.; Petrova, V.; Kearney, P.S.; Rasko, J.E.J. Holding on to junk bonds: Intron retention in cancer and therapy. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv 2017, 120295. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Clark, S.J.; Statham, A.; Stirzaker, C.; Molloy, P.L.; Frommer, M. DNA methylation: Bisulphite modification and analysis. Nat. Protoc. 2006, 1, 2353. [Google Scholar] [CrossRef]
- Nair, S.S.; Luu, P.L.; Qu, W.; Maddugoda, M.; Huschtscha, L.; Reddel, R.; Chenevix-Trench, G.; Toso, M.; Kench, J.G.; Horvath, L.G.; et al. Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X Ten. Epigenet. Chromatin 2018, 11, 24. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Remission | Control | |
|---|---|---|---|
| Number of samples | 16 | 11 | 6 |
| Sex (F/M) | 11/5 | 8/3 | 2/4 |
| Median age years (range) | 54 (21–69) | 56 (23–70) | 37 (30–42) |
| Trial (RESIST/ENESTxtnd/PINNACLE/PERSISTEM) | 1/4/4/7 | 1/4/4/2 | |
| Timepoint | Screening (×12)/Day 1 | 6–35 months | |
| First-line TKI (imatinib/nilotinib) | 8/8 | ||
| RNA-seq and WGBS (RNA-seq only) | 10 (+6) | 10 (+1) | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitz, U.; Shah, J.S.; Dhungel, B.P.; Monteuuis, G.; Luu, P.-L.; Petrova, V.; Metierre, C.; Nair, S.S.; Bailey, C.G.; Saunders, V.A.; et al. Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients. Cancers 2020, 12, 3738. https://doi.org/10.3390/cancers12123738
Schmitz U, Shah JS, Dhungel BP, Monteuuis G, Luu P-L, Petrova V, Metierre C, Nair SS, Bailey CG, Saunders VA, et al. Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients. Cancers. 2020; 12(12):3738. https://doi.org/10.3390/cancers12123738
Chicago/Turabian StyleSchmitz, Ulf, Jaynish S. Shah, Bijay P. Dhungel, Geoffray Monteuuis, Phuc-Loi Luu, Veronika Petrova, Cynthia Metierre, Shalima S. Nair, Charles G. Bailey, Verity A. Saunders, and et al. 2020. "Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients" Cancers 12, no. 12: 3738. https://doi.org/10.3390/cancers12123738
APA StyleSchmitz, U., Shah, J. S., Dhungel, B. P., Monteuuis, G., Luu, P.-L., Petrova, V., Metierre, C., Nair, S. S., Bailey, C. G., Saunders, V. A., Turhan, A. G., White, D. L., Branford, S., Clark, S. J., Hughes, T. P., Wong, J. J.-L., & Rasko, J. E. J. (2020). Widespread Aberrant Alternative Splicing despite Molecular Remission in Chronic Myeloid Leukaemia Patients. Cancers, 12(12), 3738. https://doi.org/10.3390/cancers12123738