Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour

 , ,
, ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Satellitosis in Neurological Disease
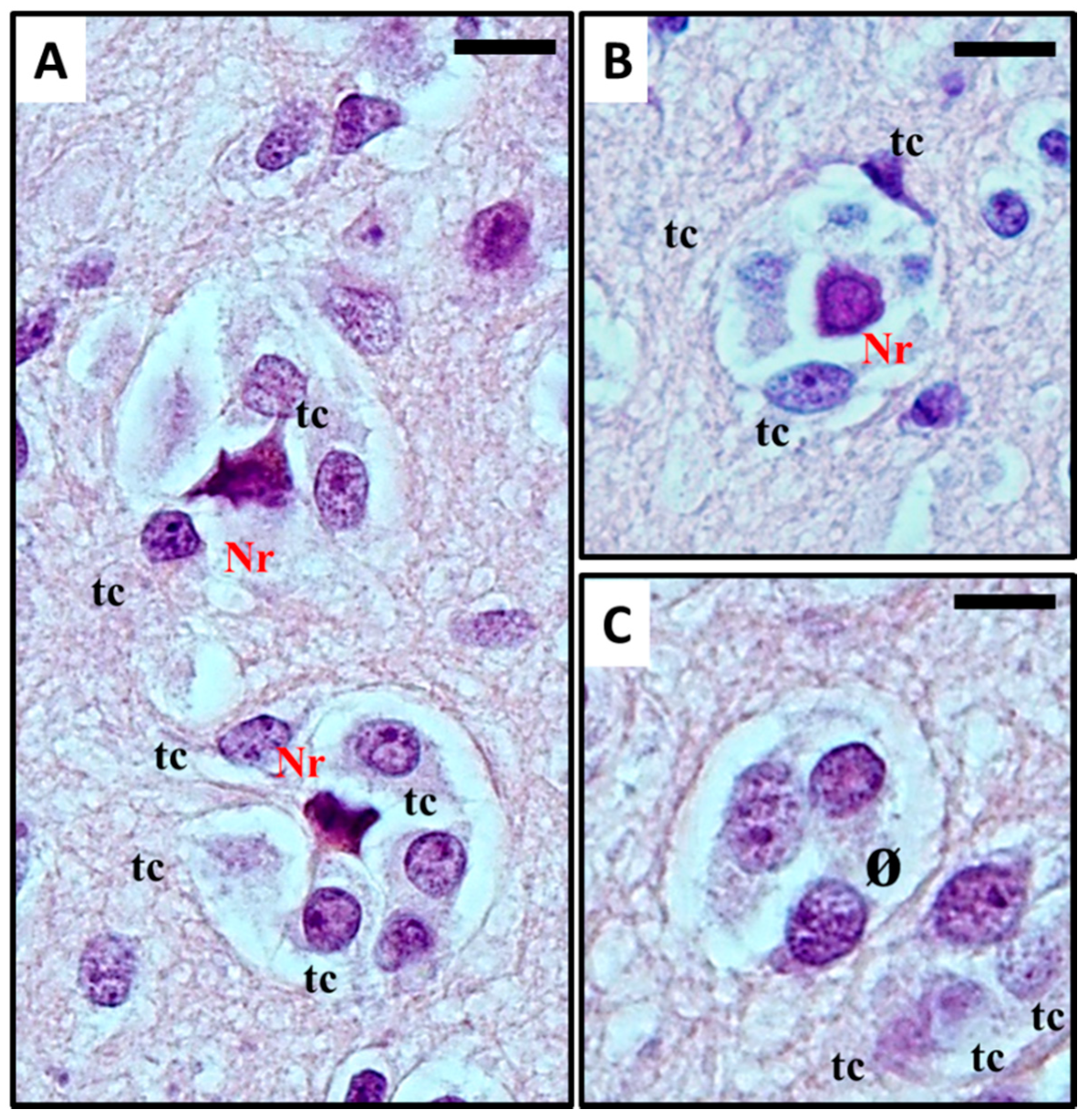
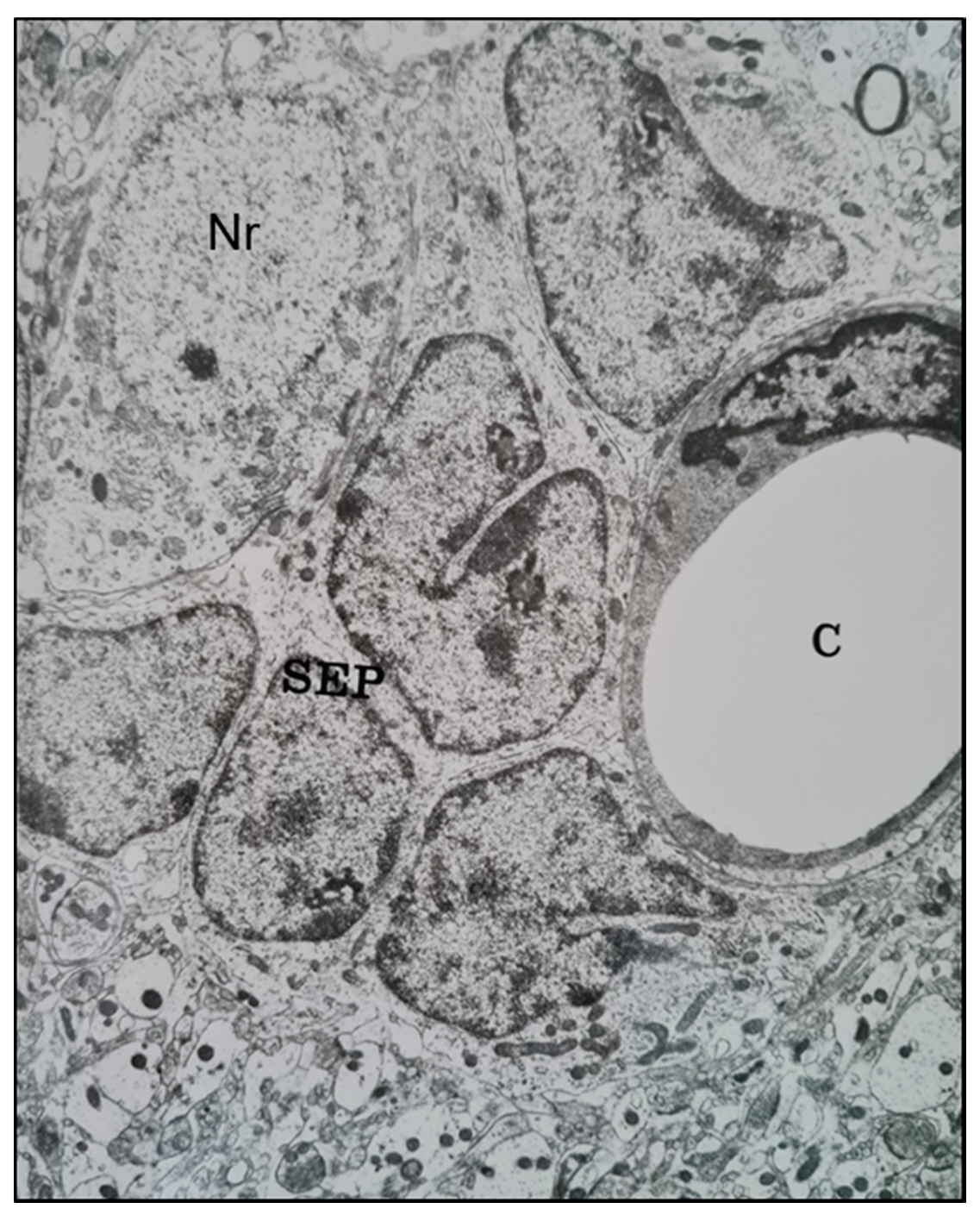
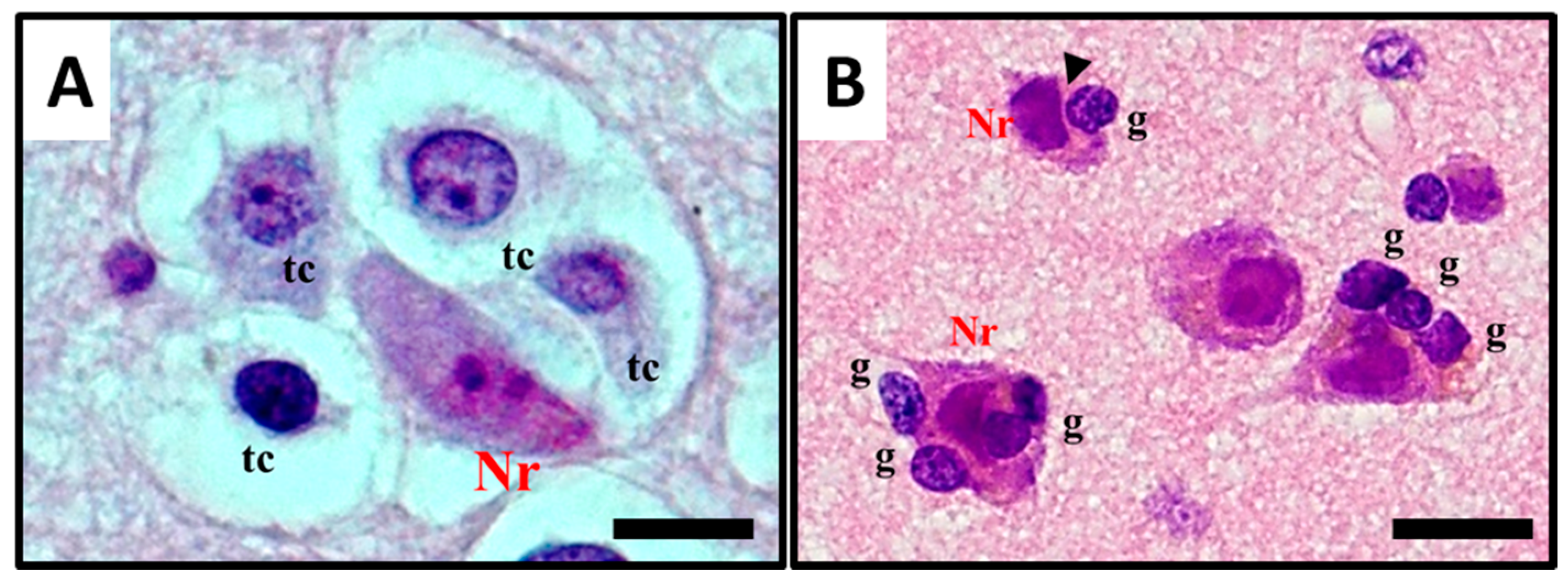
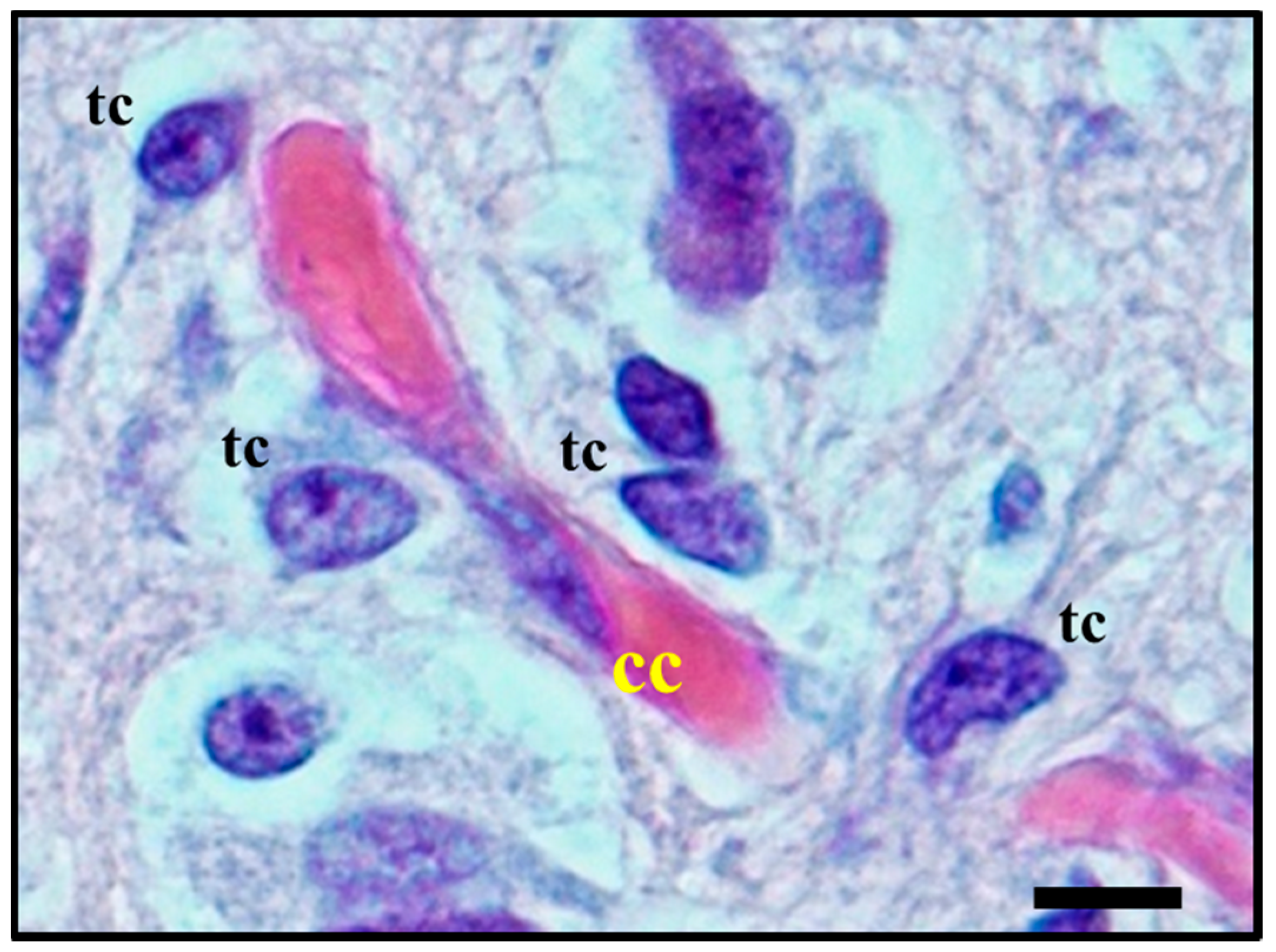
2.1. Histological Characterisation of Perineuronal Satellitosis in Brain Tumours
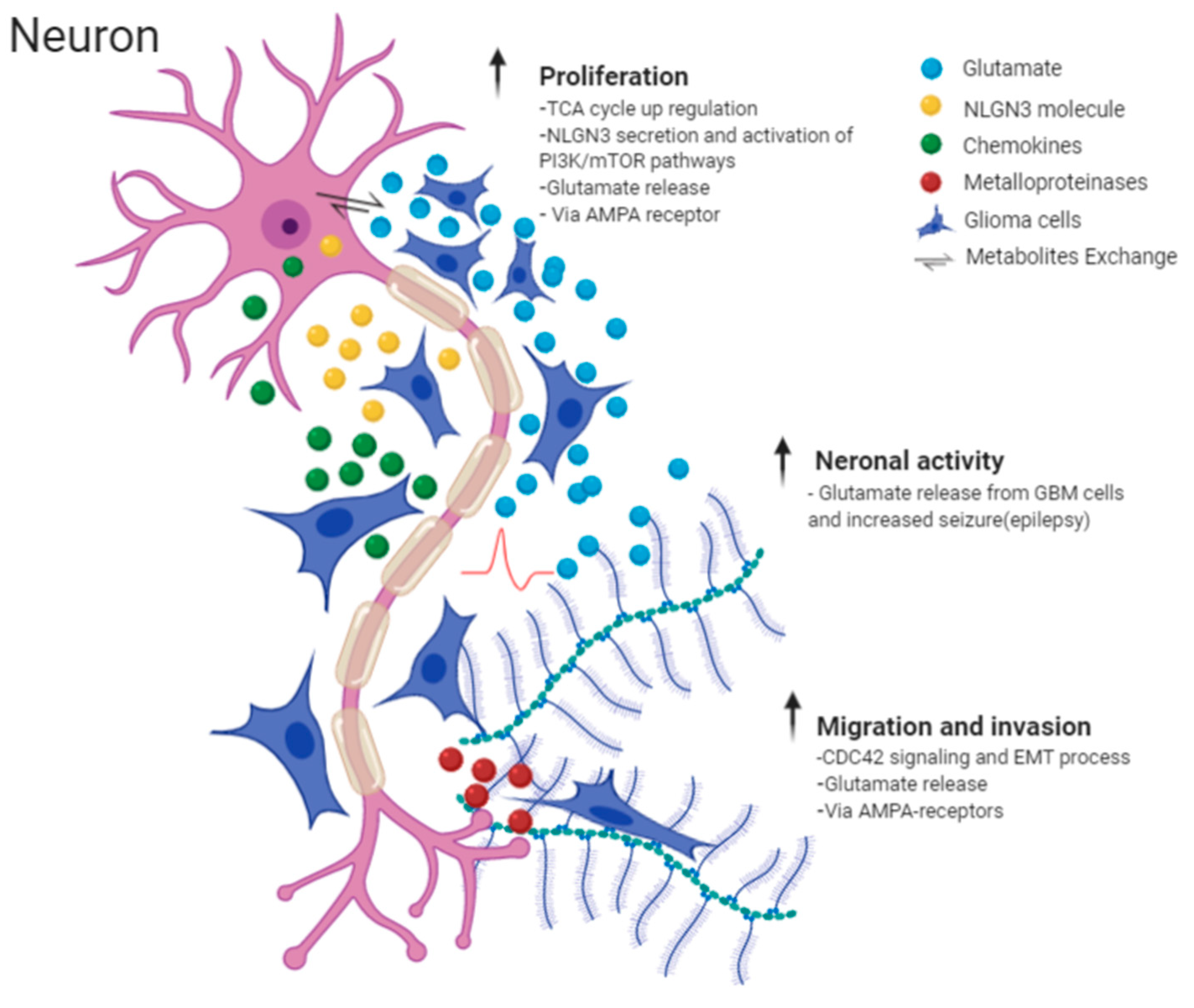
2.2. Molecular Mechanisms Involved in Perineuronal Satellitosis Phenomena
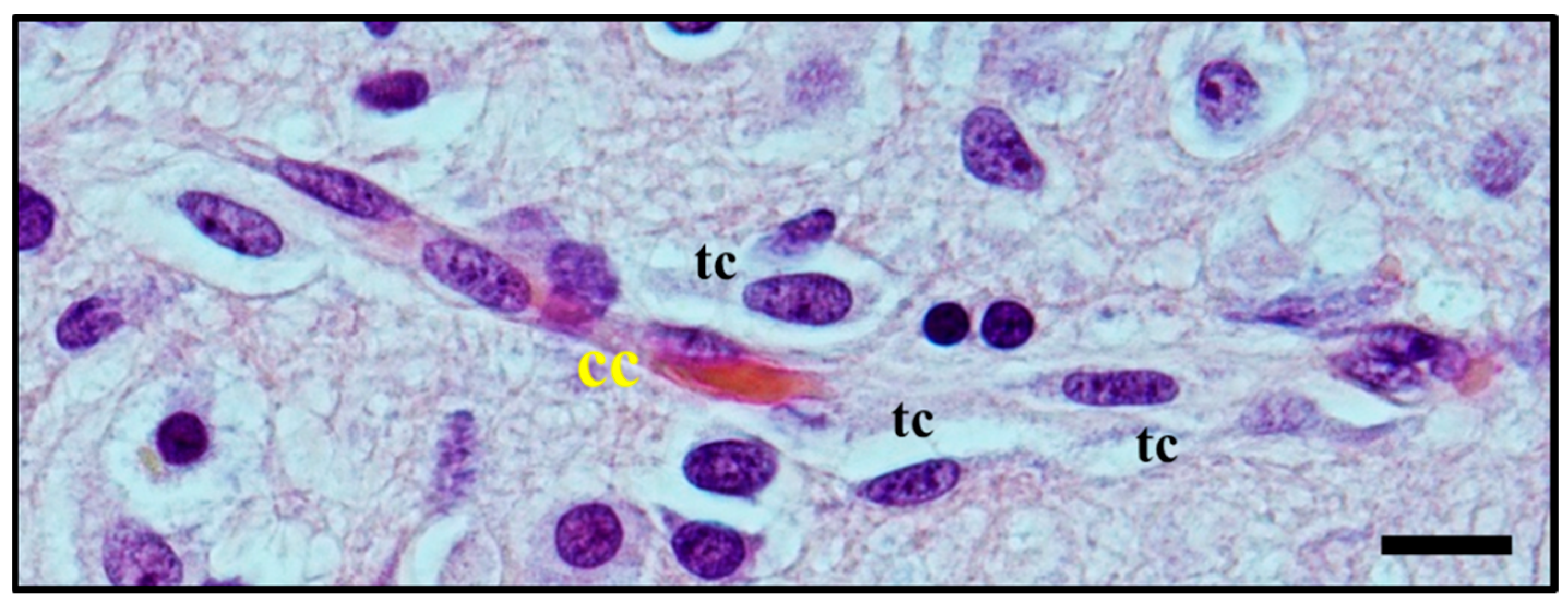
2.3. Histological Characterisation of Perivascular Satellitosis in Brain Tumours
2.4. Molecular Mechanisms Involved in Perivascular Satellitosis Phenomenon
3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Brownson, R.H. Perineuronal satelite cells in the motor cortex of aging brains. J. Neuropathol. Exp. Neurol. 1956. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.K.; Zhou, S.-S.; Russell, M.J.; Geddes, J.; Ellis, W.; Cotman, C.W. Perineuronal satellitosis in the human hippocampal formation. Hippocampus 1993. [Google Scholar] [CrossRef] [PubMed]
- Brain, W.R.; Greenfield, J.G. Late infantile metachromatic leuco-encephalopathy, with primary degeneration of the interfascicular oligodendroglia. Brain 1950. [Google Scholar] [CrossRef] [PubMed]
- Riese, W. The Cerebral Cortex in the Very Old Human Brain1. J. Neuropathol. Exp. Neurol. 1946, 5, 160–164. [Google Scholar] [CrossRef]
- Yokota, O.; Tsuchiya, K.; Hayashi, M.; Kakita, A.; Ohwada, K.; Ishizu, H.; Takahashi, H.; Akiyama, H. Glial clusters and perineuronal glial satellitosis in the basal ganglia of neurofibromatosis type 1. Acta Neuropathol. 2008, 116, 57–66. [Google Scholar] [CrossRef]
- Johnsen, S.D. Book Review: Clinical Neuropathology. Text and Color Atlas. By Catherine Haberland. Demos Publishing, New York, NY, 2007. J. Child Neurol. 2008. [Google Scholar] [CrossRef]
- Pilkington, G.J.; Lantos, P.L. The development of experimental brain tumours a sequential light and electron microscope study of the subependymal plate - II. Microtumours. Acta Neuropathol. 1979. [Google Scholar] [CrossRef]
- Scherer, H.J. Structural development in gliomas. Am. J. Cancer 1938. [Google Scholar] [CrossRef]
- Scherer, H.J. The forms of growth in gliomas and their practical significance. Brain 1940. [Google Scholar] [CrossRef]
- Scherer, H.J. p. I’étude du Cancer; Bulletin de la Société Chimique de France: Paris, France, 1937; Volume 26. [Google Scholar]
- Lantos, P.L.; Pilkington, G.J. Neuronal changes in experimental gliomas. Neuropathol. Appl. Neurobiol. 1980, 6, 255–266. [Google Scholar] [CrossRef]
- Lantos, P.L.; Pilkington, G.J. The development of experimental brain tumours a sequential light and electron microscope study of the subependymal plate—I. Early lesions (abnormal cell clusters). Acta Neuropathol. 1979. [Google Scholar] [CrossRef] [PubMed]
- Lantos, P.L.; Pilkington, G.J. Cell degeneration and necrosis in experimental gliomas. Br. J. Exp. Pathol. 1978, 59, 85–92. [Google Scholar] [PubMed]
- Dandy, W.E. Removal of right cerebral hemisphere for certain tumors with hemiplegia: Preliminary report. J. Am. Med. Assoc. 1928. [Google Scholar] [CrossRef]
- Wesseling, P.; Kros, J.M.; Jeuken, J.W.M. The pathological diagnosis of diffuse gliomas: Towards a smart synthesis of microscopic and molecular information in a multidisciplinary context. Diagn. Histopathol. 2011, 17, 486–494. [Google Scholar] [CrossRef]
- Saul, R.A.; Sturner, R.A.; Burger, P.C. Hyperplasia of the Myenteric Plexus: Its Association With Early Infantile Megacolon and Neurofibromatosis. Am. J. Dis. Child. 1982. [Google Scholar] [CrossRef]
- Garman, R.H. Histology of the Central Nervous System. Toxicol. Pathol. 2011, 39, 22–35. [Google Scholar] [CrossRef]
- Yamamoto, J.; Kitagawa, T.; Akiba, D.; Nishizawa, S. 5-aminolevulinic acid-induced fluorescence in cerebellar primary central nervous system lymphoma: A case report and literature review. Turk. Neurosurg. 2015. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A state of the science review. Neuro. Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Jeuken, J.W.M.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef]
- Sant, M.; Minicozzi, P.; Lagorio, S.; Børge Johannesen, T.; Marcos-Gragera, R.; Francisci, S.; Oberaigner, W.; Hackl, M.; Van Eycken, E.; Verstreken, M.; et al. Survival of European patients with central nervous system tumors. Int. J. Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 8; ISBN 9780994438126. [Google Scholar]
- Klink, B.; Miletic, H.; Stieber, D.; Huszthy, P.C.; Valenzuela, J.A.C.; Balss, J.; Wang, J.; Schubert, M.; Sakariassen, P.Ø.; Sundstrøm, T.; et al. A Novel, Diffusely Infiltrative Xenograft Model of Human Anaplastic Oligodendroglioma with Mutations in FUBP1, CIC, and IDH1. PLoS ONE 2013, 8, e59773. [Google Scholar] [CrossRef]
- Blumbergs, P.C. Surgical Pathology of the Nervous System and its Coverings. Pathology 2003. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008. [Google Scholar] [CrossRef]
- Baumert, B.G.; Rutten, I.; Dehing-Oberije, C.; Twijnstra, A.; Dirx, M.J.M.; Debougnoux-Huppertz, R.M.T.L.; Lambin, P.; Kubat, B. A pathology-based substrate for target definition in radiosurgery of brain metastases. Int. J. Radiat. Oncol. Biol. Phys. 2006. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.; Monje, M. Neuronal Activity in Ontogeny and Oncology. Trends Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019. [Google Scholar] [CrossRef]
- Oliveira, A.I.; Anjo, S.I.; Vieira De Castro, J.; Serra, S.C.; Salgado, A.J.; Manadas, B.; Costa, B.M. Crosstalk between glial and glioblastoma cells triggers the “go-or-grow” phenotype of tumor cells. Cell Commun. Signal. 2017. [Google Scholar] [CrossRef]
- Venkatesh, V.S.; Lou, E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2019, 9, 96. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Howarth, C.; Sibson, N.R. The role of astrocytes in CNS tumours: Pre-clinical models and novel imaging approaches. Front. Cell. Neurosci. 2013, 7, 40. [Google Scholar]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J. Astrocyte Regulation of Synaptic Behavior. Annu. Rev. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: A review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Bélanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281. [Google Scholar]
- Mangia, S.; Simpson, I.A.; Vannucci, S.J.; Carruthers, A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009, 109, 55–62. [Google Scholar] [CrossRef]
- Pei, Z.; Lee, K.C.; Khan, A.; Erisnor, G.; Wang, H.Y. Pathway analysis of glutamate-mediated, calcium-related signaling in glioma progression. Biochem. Pharmacol. 2020, 176, 113814. [Google Scholar] [CrossRef]
- Civita, P.; Franceschi, S.; Aretini, P.; Ortenzi, V.; Menicagli, M.; Lessi, F.; Pasqualetti, F.; Giuseppe Naccarato, A.; Maria Mazzanti, C. Laser capture microdissection and RNA-seq analysis: High sensitivity approaches to explain histopathological heterogeneity in human glioblastoma FFPE archived tissues. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Okura, H.; Golbourn, B.J.; Shahzad, U.; Agnihotri, S.; Sabha, N.; Krieger, J.R.; Figueiredo, C.A.; Chalil, A.; Landon-Brace, N.; Riemenschneider, A.; et al. A role for activated Cdc42 in glioblastoma multiforme invasion. Oncotarget 2016. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.I. Aberrant signaling pathways in Glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Veeravalli, K.K.; Rao, J.S. MMP-9 and uPAR regulated glioma cell migration. Cell Adhes. Migr. 2012, 6, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Esencay, M.; Mendez, O.; Yee, H.; Smirnova, I.; Huang, Y.; Chiriboga, L.; Lukyanov, E.; Liu, M.; Newcomb, E.W. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1α/CXCR4 expression in glioblastomas: One plausible explanation of Scherer’s structures. Am. J. Pathol. 2008, 173, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Esencay, M.; Sarfraz, Y.; Zagzag, D. CXCR7 is induced by hypoxia and mediates glioma cell migration towards SDF-1α. BMC Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Foo, K.; Blumenthal, L.; Man, H.Y. Regulation of neuronal bioenergy homeostasis by glutamate. Neurochem. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011. [Google Scholar] [CrossRef]
- Maus, A.; Peters, G.J. Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007. [Google Scholar] [CrossRef]
- Sontheimer, H. A role for glutamate in growth and invasion of primary brain tumors. J. Neurochem. 2008. [Google Scholar] [CrossRef]
- Takano, T.; Lin, J.H.C.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001. [Google Scholar] [CrossRef] [PubMed]
- Savaskan, N.E.; Heckel, A.; Hahnen, E.; Engelhorn, T.; Doerfler, A.; Ganslandt, O.; Nimsky, C.; Buchfelder, M.; Eyüpoglu, I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Engelhorn, T.; Savaskan, N.E.; Schwarz, M.A.; Kreutzer, J.; Meyer, E.P.; Hahnen, E.; Ganslandt, O.; Dörfler, A.; Nimsky, C.; Buchfelder, M.; et al. Cellular characterization of the peritumoral edema zone in malignant brain tumors. Cancer Sci. 2009. [Google Scholar] [CrossRef] [PubMed]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993. [Google Scholar] [CrossRef]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Buckingham, S.C.; Sontheimer, H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 2012. [Google Scholar] [CrossRef]
- Pallud, J.; Capelle, L.; Huberfeld, G. Tumoral epileptogenicity: How does it happen’. Epilepsia 2013. [Google Scholar] [CrossRef]
- Pallud, J.; Audureau, E.; Blonski, M.; Sanai, N.; Bauchet, L.; Fontaine, D.; Mandonnet, E.; Dezamis, E.; Psimaras, D.; Guyotat, J.; et al. Epileptic seizures in diffuse low-grade gliomas in adults. Brain 2014. [Google Scholar] [CrossRef]
- Patt, S.; Steenbeck, J.; Hochstetter, A.; Kraft, R.; Huonker, R.; Haueisen, J.; Haberland, N.; Ebmeier, K.; Hliscs, R.; Fiehler, J.; et al. Source localization and possible causes of interictal epileptic activity in tumor-associated epilepsy. Neurobiol. Dis. 2000. [Google Scholar] [CrossRef]
- Senner, V.; Köhling, R.; Püttmann-Cyrus, S.; Straub, H.; Paulus, W.; Speckmann, E.J. A new neurophysiological/neuropathological ex vivo model localizes the origin of glioma-associated epileptogenesis in the invasion area. Acta Neuropathol. 2004, 107, 1–7. [Google Scholar] [CrossRef]
- Köhling, R.; Senner, V.; Paulus, W.; Speckmann, E.J. Epileptiform activity preferentially arises outside tumor invasion zone in glioma xenotransplants. Neurobiol. Dis. 2006. [Google Scholar] [CrossRef] [PubMed]
- Moots, P.L.; Maciunas, R.J.; Eisert, D.R.; Parker, R.A.; Laporte, K.; Khalil, B.A. The Course of Seizure Disorders in Patients with Malignant Gliomas. Arch. Neurol. 1995. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Kuner, T.; Wick, W.; Winkler, F. Synaptic input to brain tumors: Clinical implications. Neuro. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019. [Google Scholar] [CrossRef]
- Secretion, N.-; Venkatesh, H.S.; Johung, T.B.; Mallick, P.; Monje, M.; Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A. Neuronal Activity Promotes Glioma Growth through Article Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar]
- Lytton, D.G.; Resuhr, L.M. Galen on abnormal swellings. J. Hist. Med. Allied Sci. 1978. [Google Scholar] [CrossRef]
- Lenzi, P.; Bocci, G.; Natale, G. John Hunter and the origin of the term “angiogenesis”. Angiogenesis 2016. [Google Scholar] [CrossRef]
- Virchow, R. Die krankhaften Geschwülste; Verlag von August Hirschwald: Berlin, Germany, 1978. [Google Scholar]
- Thiersch, C. Der Epithelialkrebs, Namentlich der Haut: Eine Anatomisch-Klinische Untersuchung; Verlag von Wilhelm Engelmann: Leipzig, Germany, 1865; Volume 1. [Google Scholar]
- Goldmann, E. The Growth of Malignant Disease in Man and the Lower Animals, with Special Reference to the Vascular System. J. R. Soc. Med. 1908, 1, 1–13. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Diksin, M.; Smith, S.J.; Rahman, R. The molecular and phenotypic basis of the glioma invasive perivascular niche. Int. J. Mol. Sci. 2017, 18, 2342. [Google Scholar] [CrossRef] [PubMed]
- Bolteus, A.J.; Berens, M.E.; Pilkington, G.J. Migration and invasion in brain neoplasms. Curr. Neurol. Neurosci. Rep. 2001, 1, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Kienast, Y.; Fuhrmann, M.; Von Baumgarten, L.; Burgold, S.; Mitteregger, G.; Kretzschmar, H.; Herms, J. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia 2009. [Google Scholar] [CrossRef] [PubMed]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Duda, D.G.; Emblem, K.E.; Taylor, J.W.; Gerstner, E.R.; Loeffler, J.S.; Batchelor, T.T.; Jain, R.K. Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with Glioblastoma. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014. [Google Scholar] [CrossRef]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. Elife 2016. [Google Scholar] [CrossRef] [PubMed]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Thurston, G.; Davis, S.; Wiegand, S.J.; Holash, J.; Rudge, J.S.; Yancopoulos, G.D. Complementary and coordinated roles of the VEGFs and angiopoietins during normal and pathologic vascular formation. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2002. [Google Scholar]
- Küsters, B.; Leenders, W.P.J.; Wesseling, P.; Smits, D.; Verrijp, K.; Ruiter, D.J.; Peters, J.P.W.; Van der Kogel, A.J.; De Waal, R.M.W. Vascular endothelial growth factor-A165 induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res. 2002, 62, 341–345. [Google Scholar]
- Montana, V.; Sontheimer, H. Bradykinin promotes the Chemotactic invasion of primary brain tumors. J. Neurosci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Ploegmakers, K.J.; Grevers, F.; Verbovšek, U.; Silvestre-Roig, C.; Aronica, E.; Tigchelaar, W.; Turnšek, T.L.; Molenaar, R.J.; Van Noorden, C.J.F. CD133+ and Nestin+ Glioma Stem-Like Cells Reside Around CD31+ Arterioles in Niches that Express SDF-1α, CXCR4, Osteopontin and Cathepsin K. J. Histochem. Cytochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Wormer, J.R.; Kakar, H.; Breznik, B.; van der Swaan, B.; Hulsbos, R.; Tigchelaar, W.; Tonar, Z.; Khurshed, M.; Molenaar, R.J.; et al. Periarteriolar Glioblastoma Stem Cell Niches Express Bone Marrow Hematopoietic Stem Cell Niche Proteins. J. Histochem. Cytochem. 2018. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016. [Google Scholar] [CrossRef]
- Jabouille, A.; Delugin, M.; Pineau, R.; Dubrac, A.; Soulet, F.; Lhomond, S.; Pallares-Lupon, N.; Prats, H.; Bikfalvi, A.; Chevet, E.; et al. Glioblastoma invasion and cooption depend on IRE1α endoribonuclease activity. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science (80-) 1999. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Bolin, S.; Phillips, J.J.; Persson, A.I. Signals that regulate the oncogenic fate of neural stem cells and progenitors. Exp. Neurol. 2014, 260, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013. [Google Scholar] [CrossRef] [PubMed]
- Infanger, D.W.; Cho, Y.J.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Munn, L.L.; Jain, R.K. Vascular regulation of antitumor immunity. Science (80-) 2019, 365, 544–545. [Google Scholar] [CrossRef]
- Martens, T.; Laabs, Y.; Günther, H.S.; Kemming, D.; Zhu, Z.; Witte, L.; Hagel, C.; Westphal, M.; Lamszus, K. Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor-2. Clin. Cancer Res. 2008. [Google Scholar] [CrossRef]
- Foray, C.; Barca, C.; Backhaus, P.; Schelhaas, S.; Winkeler, A.; Viel, T.; Schäfers, M.; Grauer, O.; Jacobs, A.H.; Zinnhardt, B. Multimodal Molecular Imaging of the Tumour Microenvironment. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Zlochower, A.; Chow, D.S.; Chang, P.; Khatri, D.; Boockvar, J.A.; Filippi, C.G. Deep learning AI applications in the imaging of glioma. Top. Magn. Reson. Imaging 2020, 29. [Google Scholar] [CrossRef]
- Benzakoun, J.; Robert, C.; Legrand, L.; Pallud, J.; Meder, J.F.; Oppenheim, C.; Dhermain, F.; Edjlali, M. Anatomical and functional MR imaging to define tumoral boundaries and characterize lesions in neuro-oncology. Cancer/Radiotherapie 2020, 24, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.I.; Song, H.; Ming, G. li Applications of Human Brain Organoids to Clinical Problems. Dev. Dyn. 2019, 248, 53–64. [Google Scholar]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.S.; Kim, J.S.; Yeo, H.C.; Bae, C.M.; Han, H.J.; Baek, K.; Chang, W.; Lim, K.S.; Yun, S.P.; Shin, I.S.; et al. A simple metastatic brain cancer model using human embryonic stem cell-derived cerebral organoids. FASEB J. 2020, 1–12. [Google Scholar] [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Models | Molecular Mechanism Proposed | References |
|---|---|---|
| Human Tissue sample and Animals | (First evidence) Histological visualisation
| [1,2,3,4,5,6,8,9,11,13] |
| Human Tissue sample | Metabolic exchange and migration
| [41] |
| Animals (in vitro, in vivo) | Chemotactic attraction and migration
| [46,47] |
| Animals In vivo, in vitro Patient derived GB cells | Metabolic exchange and spared within brain parenchyma
| [29,51,52,53] |
| Human In vivo; in vitro Patient derived GB cells | Neuronal activity on glioma promotion
| [66,68,69,70] |
| Experimental Models | Molecular Mechanism Proposed | References |
|---|---|---|
| Human, Animals in vivo | First histological description
| [8,12] |
| Animals (in vitro cells, in vivo) | GB cells interact with BBB:
| [80] |
| Animals (in vitro cells, in vivo) | Mechanism of Individual co-option and Collective co-option:
| [80,81] |
| Animals (In vivo cell lines) | Vascular recruitment
| [88,89] |
| Human /Animal (In vivo; in vitro Patient-derived-GB, Tissue sample) | Chemotactic attraction and invasion
| [90,91,92,93,94,95,96] |
| Human/Animals In vivo; in vitro | GB cell/pericyte fusion-hybrids
| [86] |
| Human In vivo cellls; in vitro | Surrounding microvasculature and migration
| [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Civita, P.; Valerio, O.; Naccarato, A.G.; Gumbleton, M.; Pilkington, G.J. Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour. Cancers 2020, 12, 3720. https://doi.org/10.3390/cancers12123720
Civita P, Valerio O, Naccarato AG, Gumbleton M, Pilkington GJ. Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour. Cancers. 2020; 12(12):3720. https://doi.org/10.3390/cancers12123720
Chicago/Turabian StyleCivita, Prospero, Ortenzi Valerio, Antonio Giuseppe Naccarato, Mark Gumbleton, and Geoffrey J. Pilkington. 2020. "Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour" Cancers 12, no. 12: 3720. https://doi.org/10.3390/cancers12123720
APA StyleCivita, P., Valerio, O., Naccarato, A. G., Gumbleton, M., & Pilkington, G. J. (2020). Satellitosis, a Crosstalk between Neurons, Vascular Structures and Neoplastic Cells in Brain Tumours; Early Manifestation of Invasive Behaviour. Cancers, 12(12), 3720. https://doi.org/10.3390/cancers12123720