Proper Read Filtering Method to Adequately Analyze Whole-Transcriptome Sequencing and RNA Based Immune Repertoire Sequencing Data for Tumor Milieu Research


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
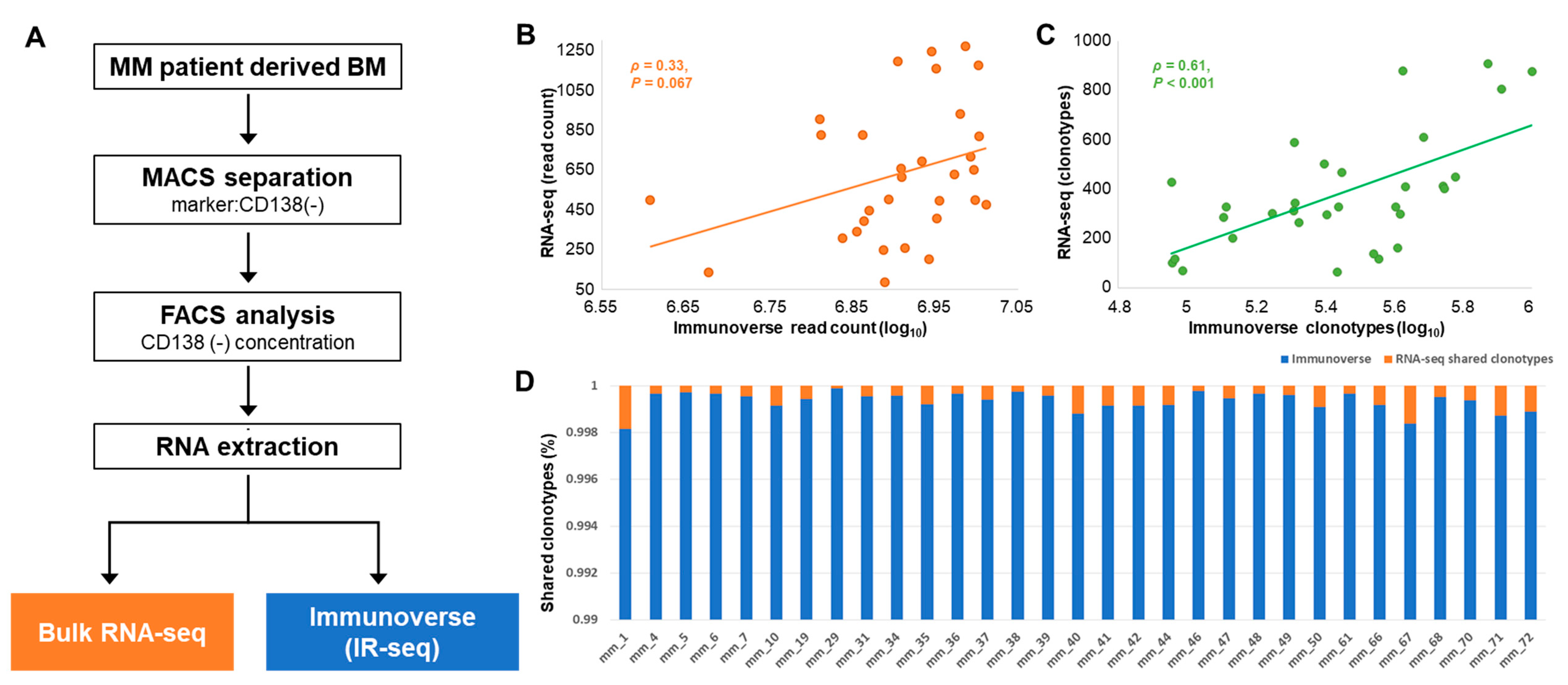
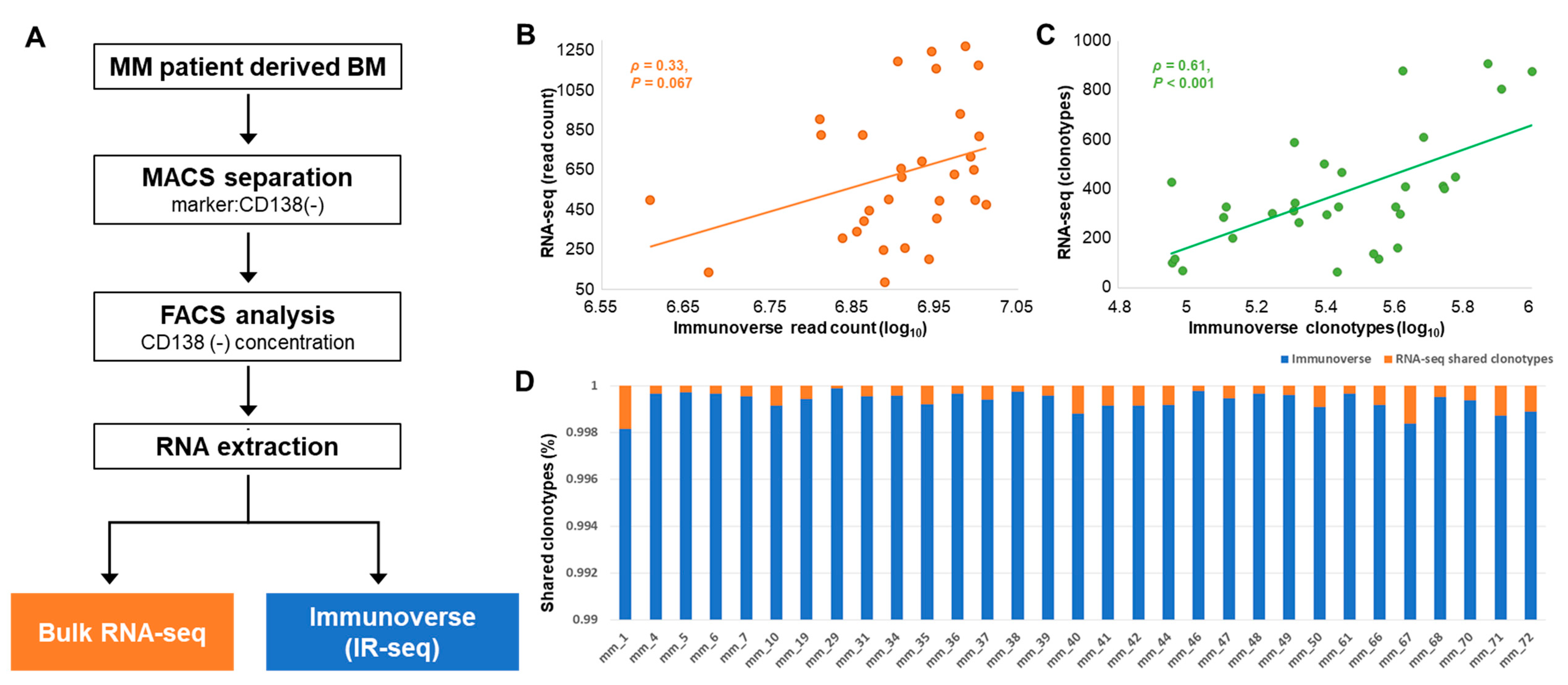
2.1. Study Population and Basic Characteristics of TCR Sequences
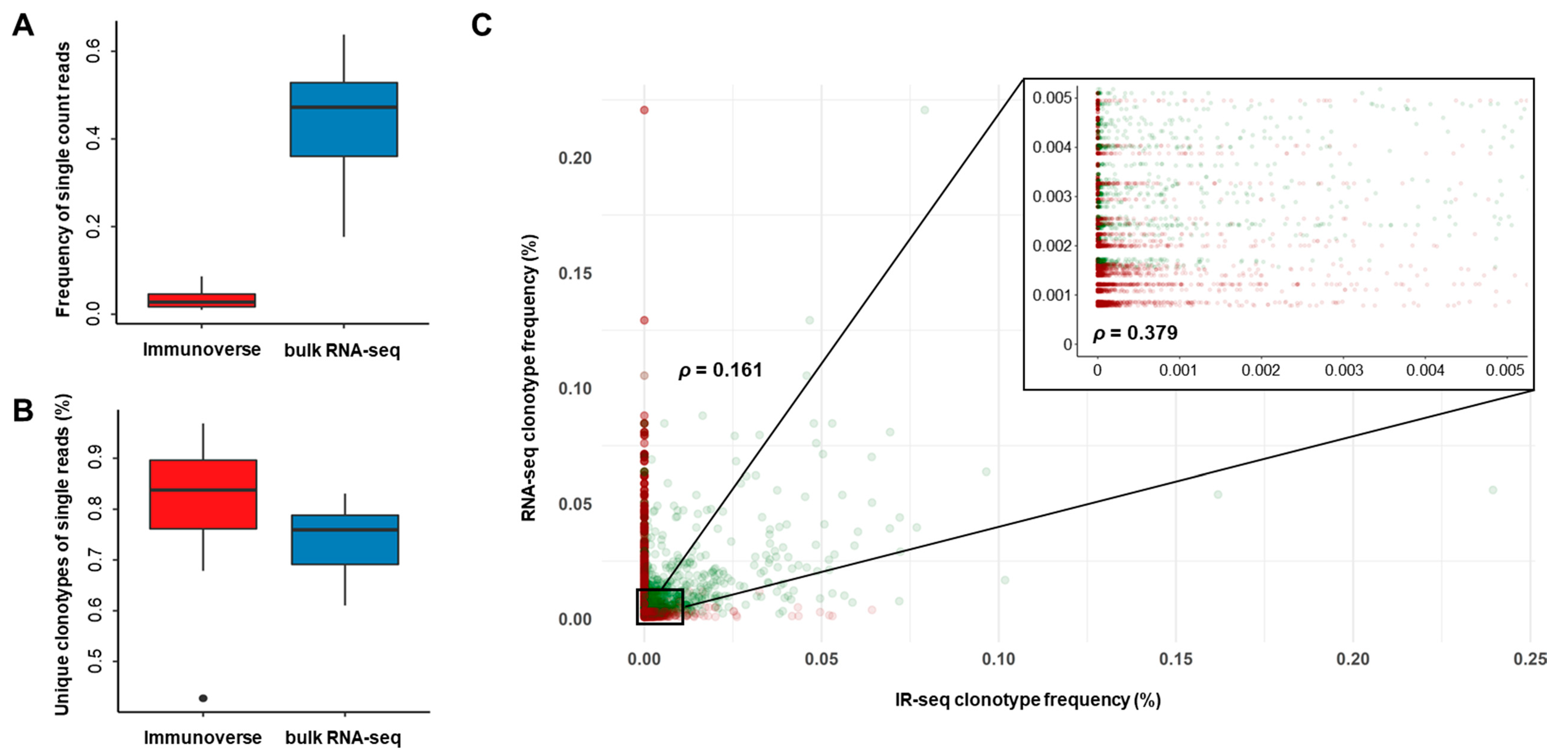
2.2. TCR Repertoire Diversity in MM Samples from IR-seq and RNA-seq
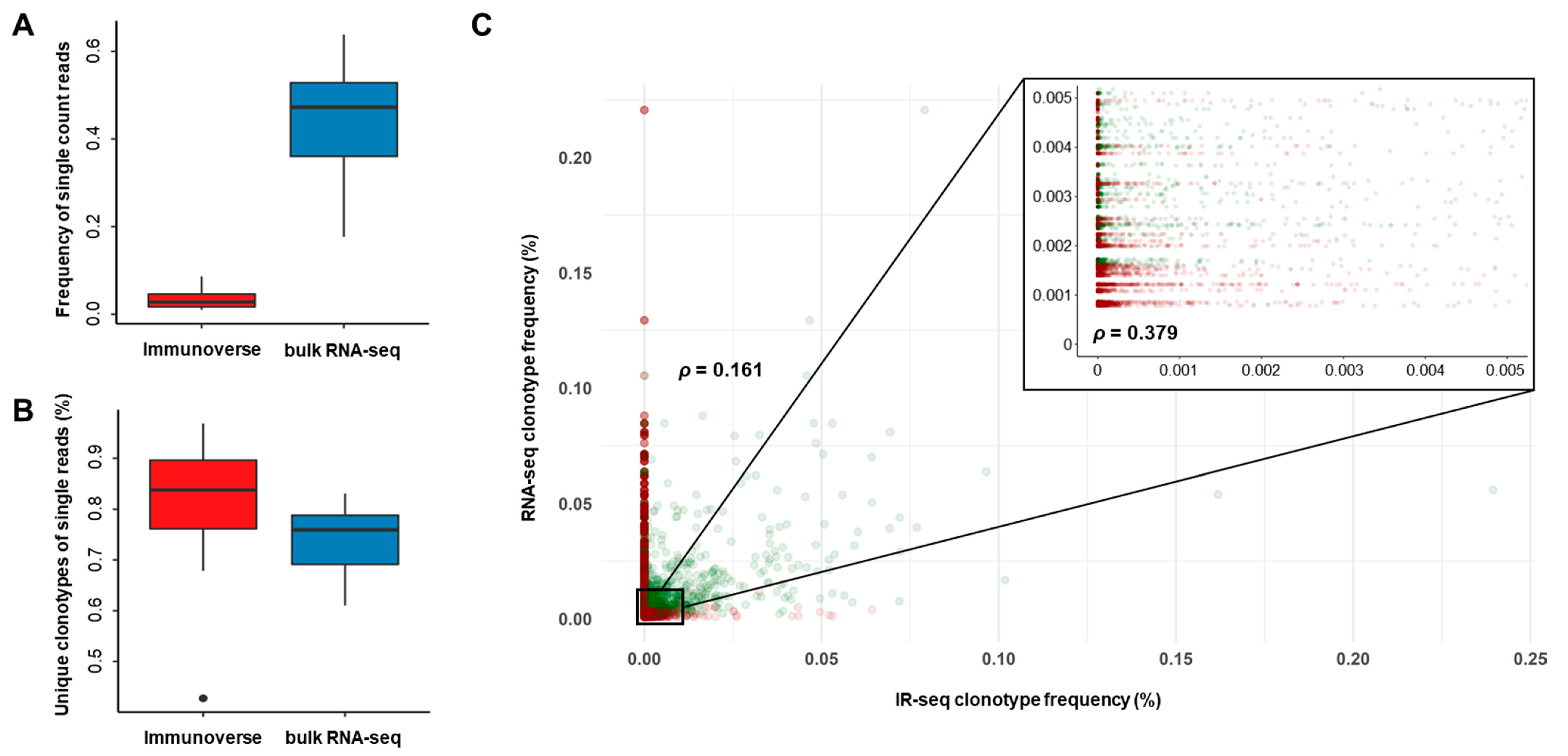
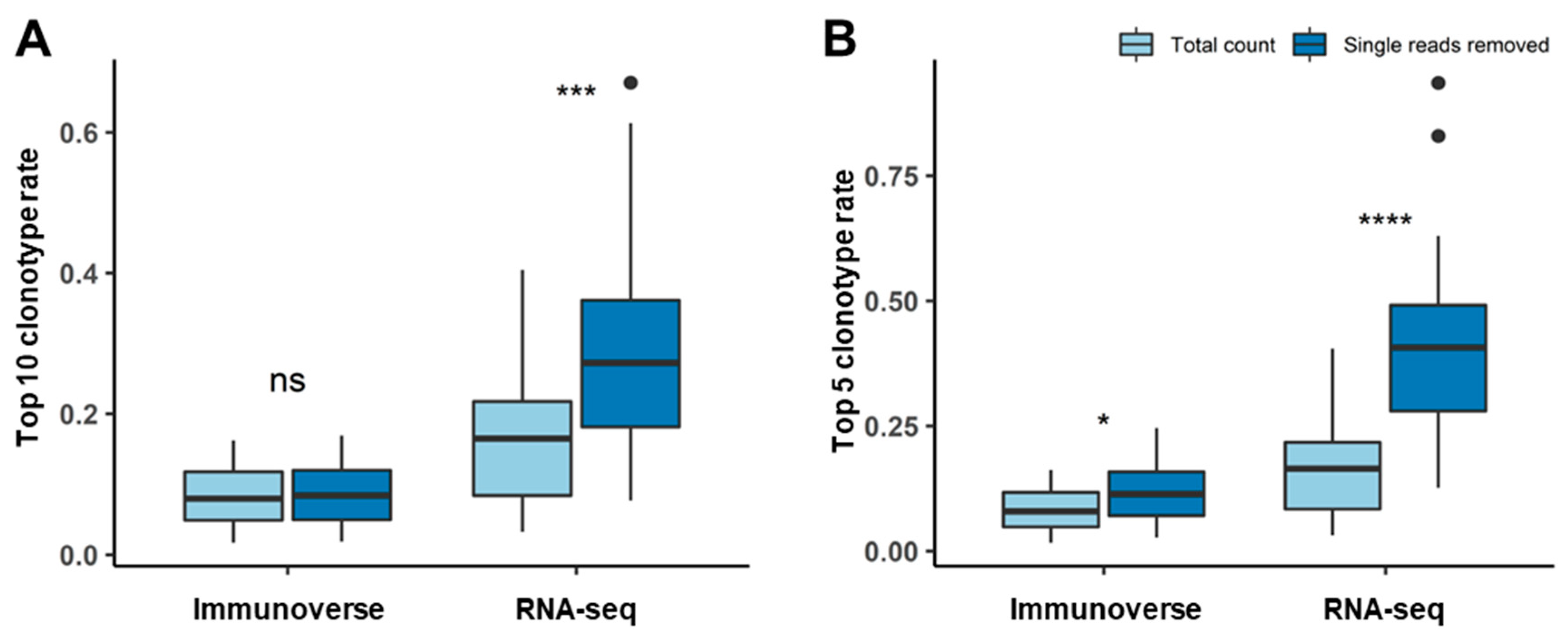
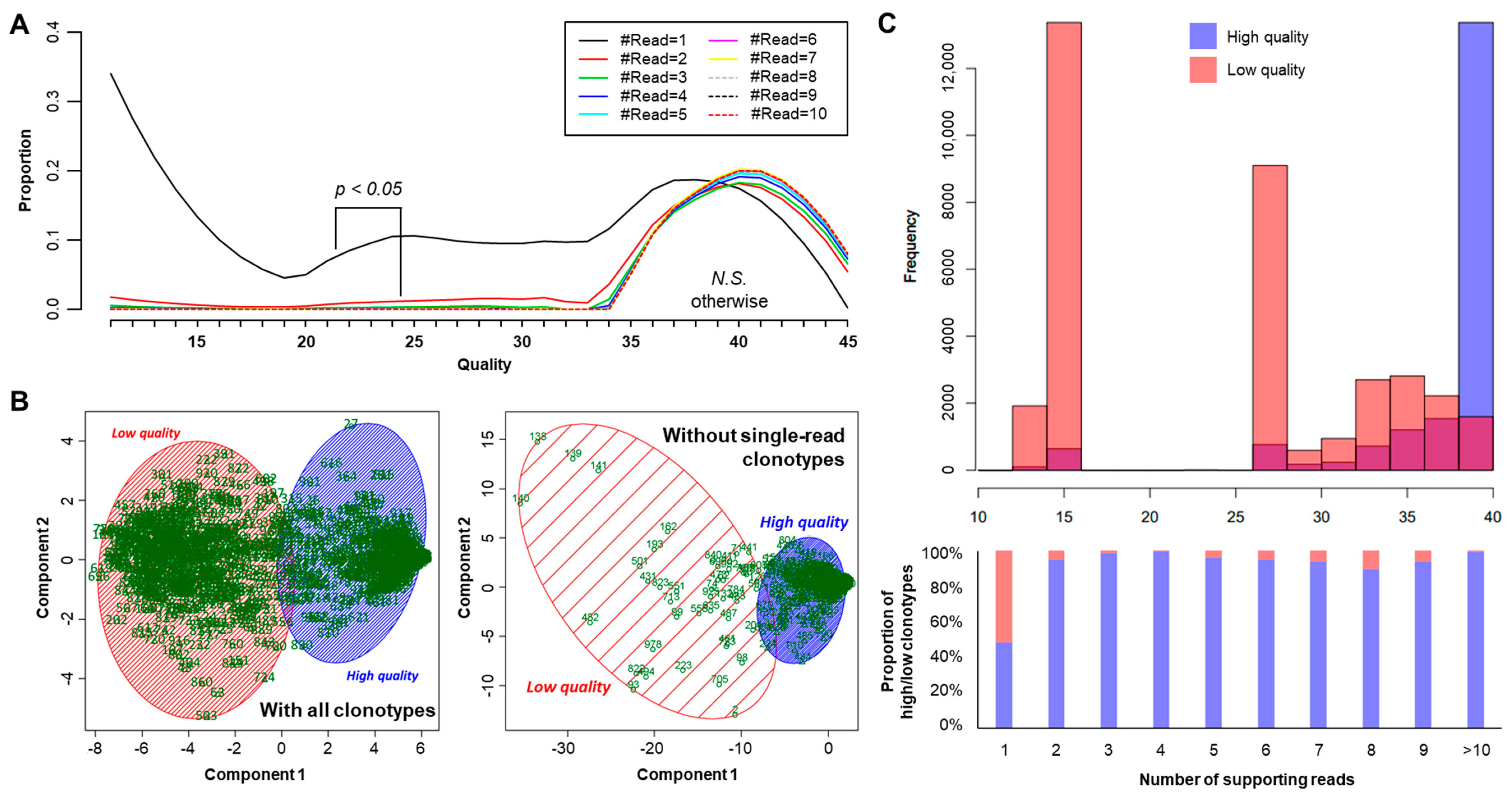
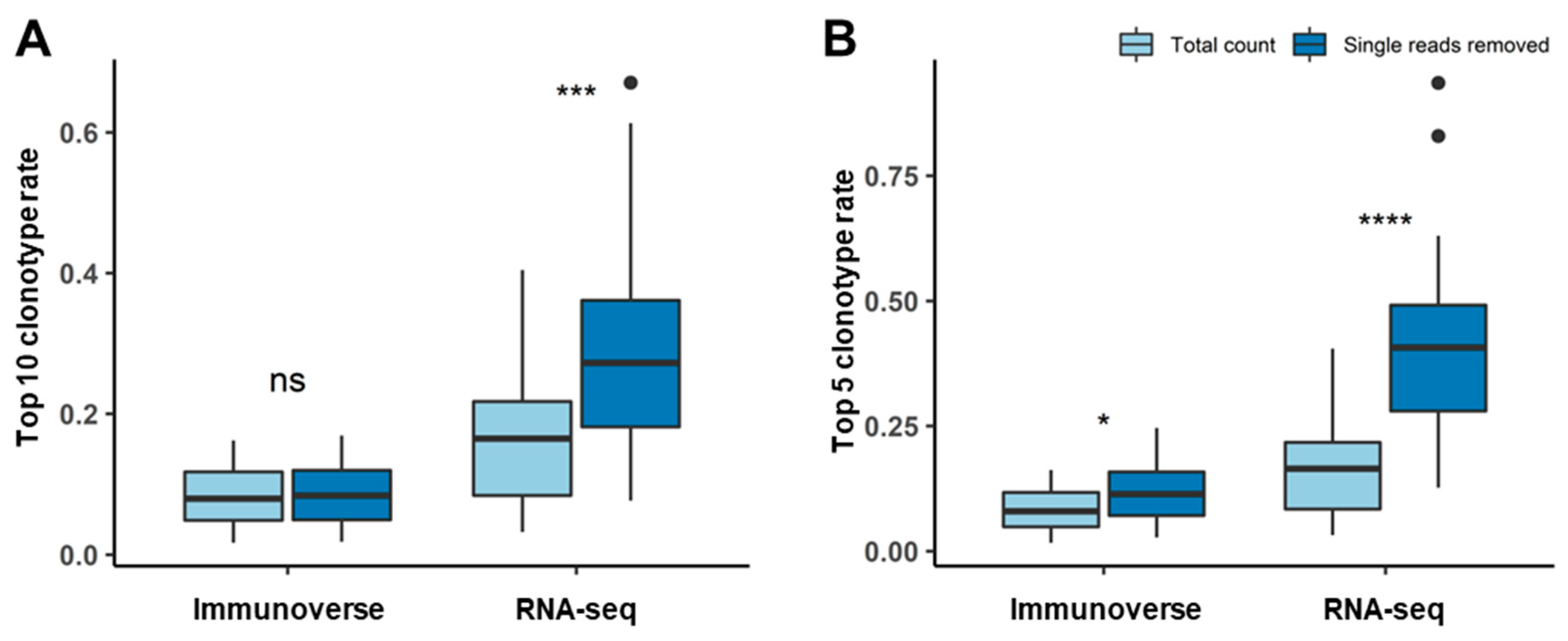
2.3. Proper Read Filtering and Clonotype Abundances at the Single-Patient Level
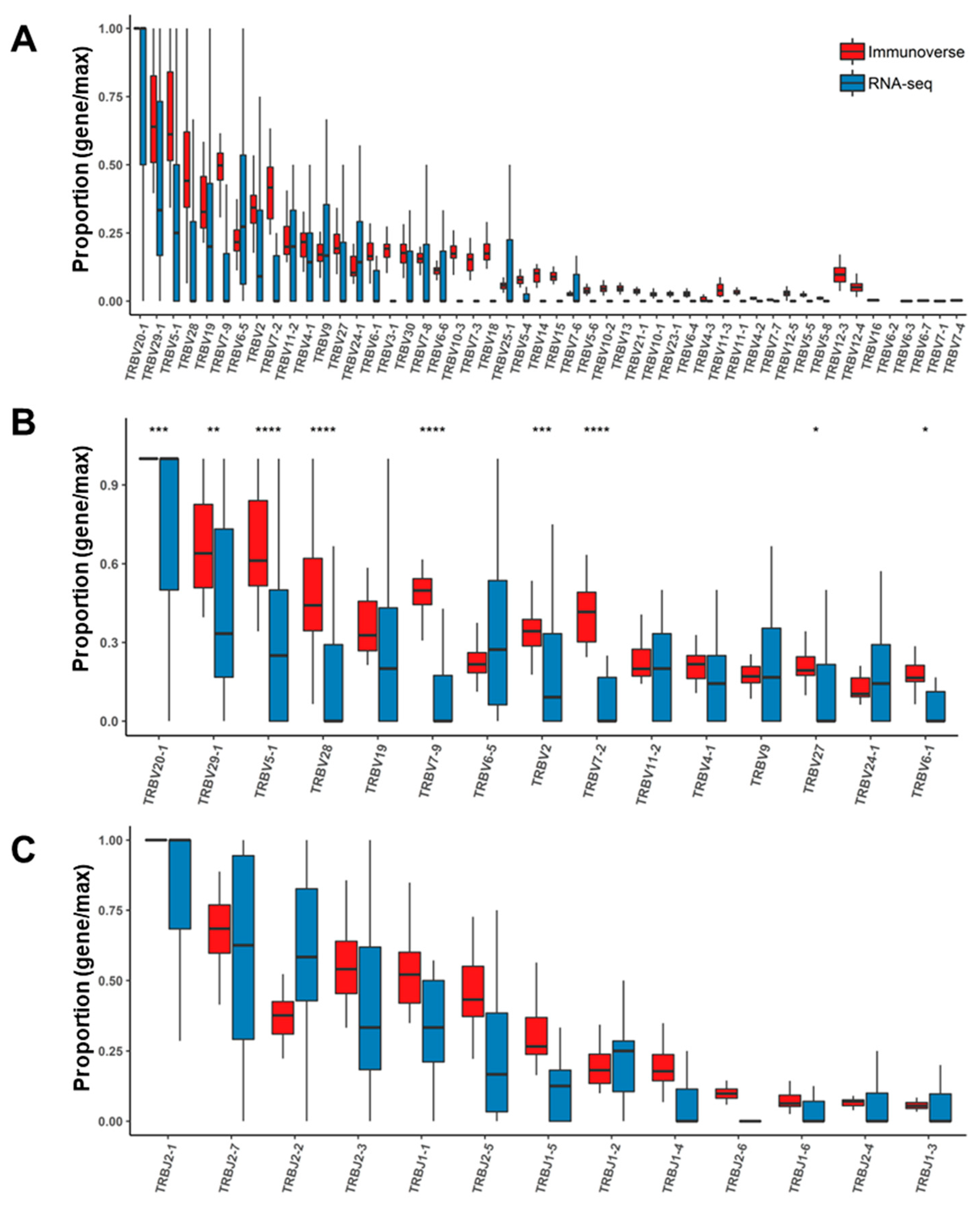
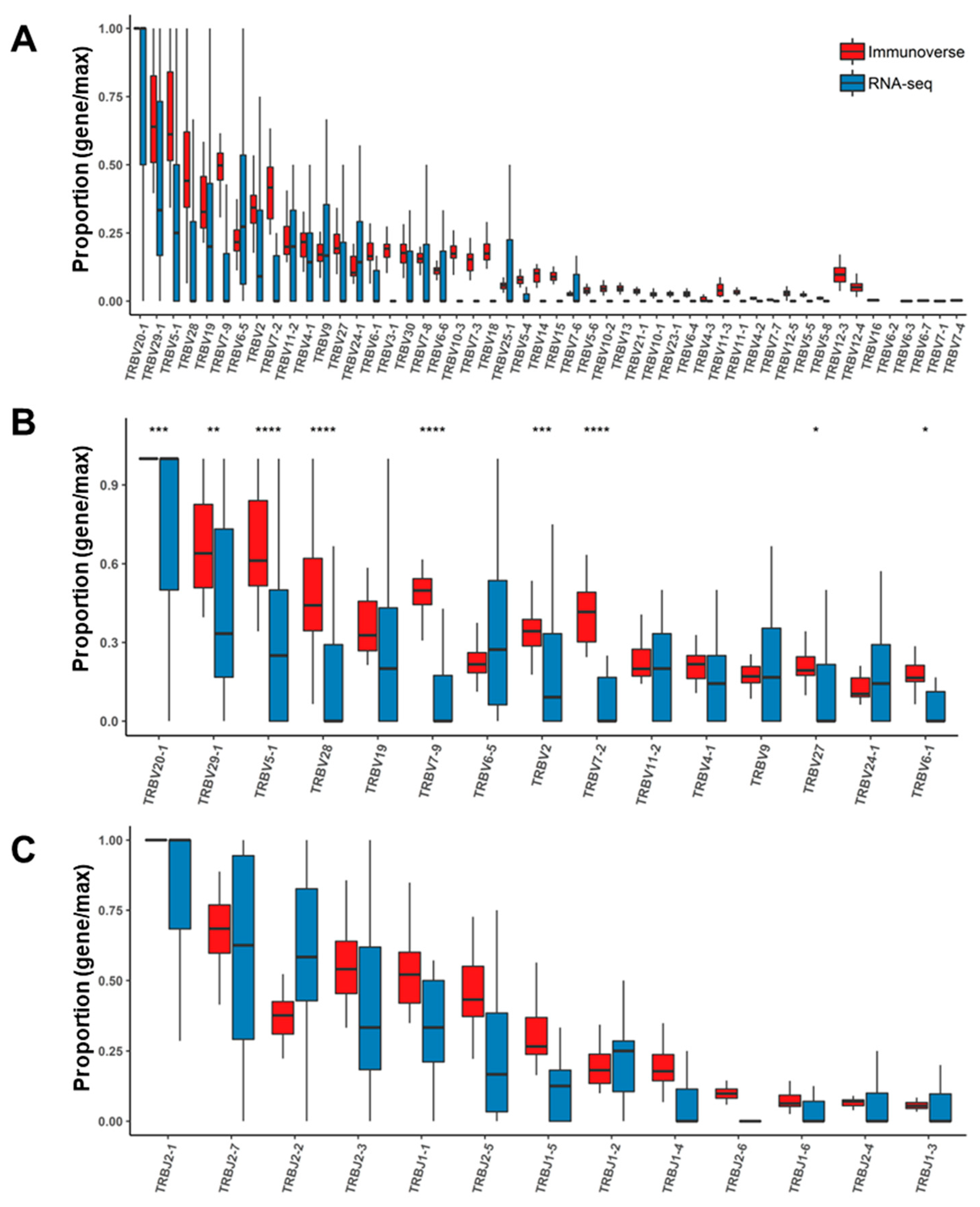
2.4. Assessment of TCR Genes from IR-seq and RNA-seq after Read Filtering
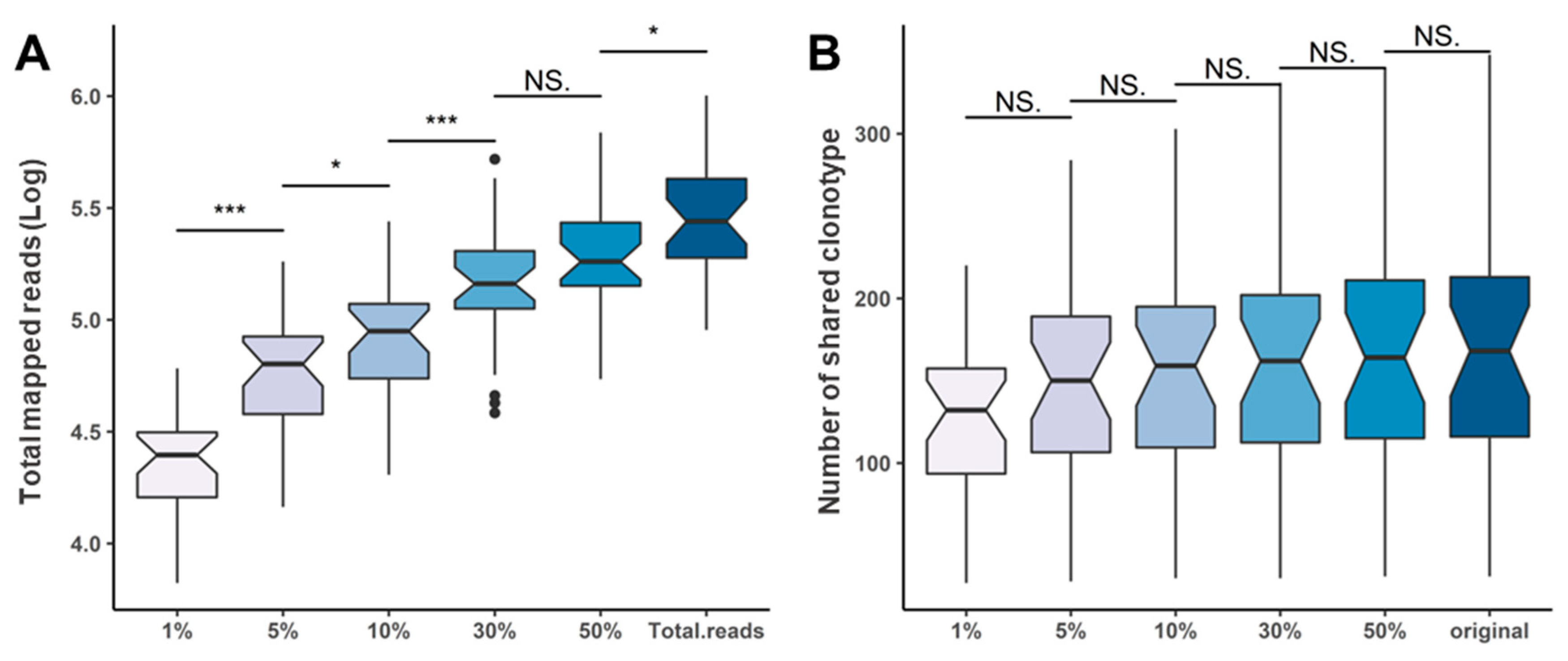
2.5. Repertoire Inference Using Random Sampling
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Processing
4.2. RNA Sequencing
4.3. Immunoverse Sequencing
4.4. Data Analysis and TCR Repertoire Extraction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Ethics Statements
References
- Dudley, D.J. The immune system in health and disease. Baillière’s Clin. Obstet. Gynaecol. 1992, 6, 393–416. [Google Scholar] [CrossRef]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How tcrs bind mhcs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naive to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Vanhanen, R.; Heikkilä, N.; Aggarwal, K.; Hamm, D.; Tarkkila, H.; Pätilä, T.; Jokiranta, T.S.; Saramäki, J.; Arstila, T.P. T cell receptor diversity in the human thymus. Mol. Immunol. 2016, 76, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Gazzola, A.; Mannu, C.; Rossi, M.; Laginestra, M.A.; Sapienza, M.R.; Fuligni, F.; Etebari, M.; Melle, F.; Sabattini, E.; Agostinelli, C.; et al. The evolution of clonality testing in the diagnosis and monitoring of hematological malignancies. Ther. Adv. Hematol. 2014, 5, 35–47. [Google Scholar] [CrossRef]
- Ban Dongen, J.J.M.; Wolvers-Tettero, I.L.M. Analysis of immunoglobulin and T cell receptor genes. Part I: Basic and technical aspects. Clin. Chim. Acta 1991, 198, 1–92. [Google Scholar] [CrossRef]
- Mahe, E.; Pugh, T.; Kamel-Reid, S. T cell clonality assessment: Past, present and future. J. Clin. Pathol. 2018, 71, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Greiff, V.; Miho, E.; Menzel, U.; Reddy, S.T. Bioinformatic and Statistical Analysis of Adaptive Immune Repertoires. Trends Immunol. 2015, 36, 738–749. [Google Scholar] [CrossRef]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of methodologies for T-cell receptor repertoire analysis. Bmc Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Zhang, Q.; Liu, C.-J.; Guo, A.-Y. An ultrasensitive T-cell receptor detection method for TCR-Seq and RNA-Seq data. bioRxiv 2019, 740340. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Miyama, T.; Kawase, T.; Honjo, Y.; Kitaura, K.; Sato, H.; Shin-I, T.; Suzuki, R. Next-Generation Immune Repertoire Sequencing as a Clue to Elucidate the Landscape of Immune Modulation by Host–Gut Microbiome Interactions. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benichou, J.; Ben-Hamo, R.; Louzoun, Y.; Efroni, S. Rep-Seq: Uncovering the immunological repertoire through next-generation sequencing. Immunology 2012, 135, 183–191. [Google Scholar] [CrossRef]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour–immune cell interactions. Nat. Rev. Genet. 2016, 17, 441. [Google Scholar] [CrossRef] [PubMed]
- Farmanbar, A.; Kneller, R.; Firouzi, S. RNA sequencing identifies clonal structure of T-cell repertoires in patients with adult T-cell leukemia/lymphoma. Npj Genom. Med. 2019, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.; Raeburn, L.A.; Holt, R.A. Profiling tissue-resident T cell repertoires by RNA sequencing. Genome Med. 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, D.A.; Poslavsky, S.; Davydov, A.N.; Frenkel, F.E.; Fanchi, L.; Zolotareva, O.I.; Hemmers, S.; Putintseva, E.V.; Obraztsova, A.S.; Shugay, M.; et al. Antigen receptor repertoire profiling from RNA-seq data. Nat. Biotechnol. 2017, 35, 908–911. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, D.; Li, W.; Huang, Y.; Ye, X.; Waite, J.; Barry, T.; Edelmann, K.H.; Levenkova, N.; Guo, C.; et al. Evaluation of the capacities of mouse TCR profiling from short read RNA-seq data. PLoS ONE 2018, 13, e0207020. [Google Scholar] [CrossRef]
- Eberlein, J.; Harrison, T.; Sims, J.; McKittrick, I.; Wemmer, M.; Griffin, L.M.; Culver, B.P.; Johnson, L.; Kudlow, B.A. Characterization of B- and T-cell immune repertoires using Anchored Multiplex PCR and Next-Generation Sequencing. J. Immunol. 2017, 198, 123–157. [Google Scholar]
- Ma, K.-Y.; He, C.; Wendel, B.S.; Williams, C.M.; Xiao, J.; Yang, H.; Jiang, N. Immune Repertoire Sequencing Using Molecular Identifiers Enables Accurate Clonality Discovery and Clone Size Quantification. Front. Immunol. 2018, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.J.; Doherty, P.C.; McCluskey, J.; Rossjohn, J. Structural determinants of T-cell receptor bias in immunity. Nat. Rev. Immunol. 2006, 6, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Bashford-Rogers, R.J.; Palser, A.L.; Idris, S.F.; Carter, L.; Epstein, M.; Callard, R.E.; Douek, D.C.; Vassiliou, G.S.; Follows, G.A.; Hubank, M.; et al. Capturing needles in haystacks: A comparison of B-cell receptor sequencing methods. Bmc Immunol. 2014, 15, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niklas, N.; Pröll, J.; Weinberger, J.; Zopf, A.; Wiesinger, K.; Krismer, K.; Bettelheim, P.; Gabriel, C. Qualifying high-throughput immune repertoire sequencing. Cell. Immunol. 2014, 288, 31–38. [Google Scholar] [CrossRef]
- Levin, M.; Levander, F.; Palmason, R.; Greiff, L.; Ohlin, M. Antibody-encoding repertoires of bone marrow and peripheral blood—A focus on IgE. J. Allergy Clin. Immunol. 2017, 139, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Georgiou, G.; Ippolito, G.C.; Beausang, J.; Busse, C.E.; Wardemann, H.; Quake, S.R. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 2014, 32, 158–168. [Google Scholar] [CrossRef]
- Li, S.; Lefranc, M.-P.; Miles, J.J.; Alamyar, E.; Giudicelli, V.; Duroux, P.; Freeman, J.D.; Corbin, V.D.A.; Scheerlinck, J.-P.; Frohman, M.A.; et al. IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat. Commun. 2013, 4, 2333. [Google Scholar] [CrossRef]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for comprehensive adaptive immunity profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Nazarov, V.I.; Pogorelyy, M.V.; Komech, E.A.; Zvyagin, I.V.; Bolotin, D.A.; Shugay, M.; Chudakov, D.M.; Lebedev, Y.B.; Mamedov, I.Z. tcR: An R package for T cell receptor repertoire advanced data analysis. BMC Bioinform. 2015, 16, 175. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | |
|---|---|
| N = 31 | N (%) |
| Sample N | 31 |
| Gender | |
| Male | 17 (54.8) |
| Female | 14 (45.2) |
| Median Age (Years) | |
| ≤65 | 13 (41.9) |
| >65 | 18 (58.1) |
| Average CD138 (-) rate (range) | 0.75 (0.1–22.4) |
| Heavy Chain Isotype | |
| lgG | 21 (67.7) |
| lgA | 6 (19.4) |
| lgM | NA |
| lgD | NA |
| LCD | 1 (3.2) |
| Light Chain Isotype | |
| Kappa | 17 (54.8) |
| Lambda | 11 (35.5) |
| R-ISS | |
| I | 8 (25.8) |
| II | 16 (51.6) |
| III | 7 (22.6) |
| Fish Results | |
| p53 deletion | 4 (12.9) |
| p16 deletion | 3 (9.7) |
| lgH rearrangement | 10 (32.3) |
| 1q trisomy | 9 (29) |
| RB1 deletion | 9 (29) |
| t (14; 16) | 1 (3.2) |
| t (4; 14) | 4 (12.9) |
| mm_# | RNA Sequencing Results | Immunoverse Results | |||||
|---|---|---|---|---|---|---|---|
| STAR Mapped Reads | Total Read Counts | Total Unique Clonotypes | Singleton Reads | Total Read Counts | Total Unique Clonotypes | Singleton Reads | |
| MM_1 | 54,357,923 | 820 | 431 | 299 | 4,050,594 | 90,068 | 63,126 |
| MM_10 | 48,374,912 | 826 | 504 | 403 | 7,293,770 | 250,572 | 199,844 |
| MM_19 | 56,698,751 | 819 | 331 | 250 | 10,073,447 | 275,520 | 252,410 |
| MM_29 | 59,981,954 | 85 | 65 | 54 | 7,761,188 | 272,594 | 272,079 |
| MM_31 | 51,623,766 | 659 | 331 | 241 | 8,120,860 | 403,438 | 373,784 |
| MM_34 | 48,536,509 | 1245 | 809 | 649 | 8,836,571 | 821,579 | 688,017 |
| MM_35 | 49,161,059 | 1195 | 883 | 697 | 8,039,194 | 423,108 | 366,790 |
| MM_36 | 43,189,398 | 652 | 416 | 323 | 9,918,279 | 555,803 | 521,536 |
| MM_37 | 52,095,895 | 827 | 613 | 510 | 6,500,208 | 487,124 | 425,707 |
| MM_38 | 49,316,301 | 258 | 165 | 124 | 8,195,923 | 408,764 | 378,106 |
| MM_39 | 48,288,693 | 716 | 412 | 338 | 9,833,228 | 431,722 | 396,254 |
| MM_4 | 34,559,031 | 615 | 451 | 348 | 8,126,107 | 602,704 | 557,693 |
| MM_40 | 45,488,936 | 499 | 289 | 198 | 9,963,987 | 127,280 | 105,512 |
| MM_41 | 42,258,878 | 202 | 104 | 71 | 8,761,312 | 90,503 | 86,489 |
| MM_42 | 45,563,654 | 340 | 120 | 78 | 7,180,034 | 91,962 | 78,633 |
| MM_44 | 53,663,728 | 693 | 470 | 371 | 8,595,247 | 281,018 | 200,700 |
| MM_46 | 47,093,079 | 408 | 118 | 72 | 8,958,694 | 360,441 | 348,835 |
| MM_47 | 46,913,279 | 477 | 299 | 237 | 10,274,849 | 254,130 | 202,285 |
| MM_48 | 45,465,437 | 933 | 403 | 308 | 9,565,796 | 559,857 | 501,017 |
| MM_49 | 50,302,951 | 628 | 300 | 215 | 9,402,588 | 416,450 | 381,187 |
| MM_5 | 46,521,960 | 248 | 141 | 97 | 7,732,378 | 347,955 | 345,460 |
| MM_50 | 41,180,449 | 392 | 304 | 250 | 7,324,348 | 176,306 | 126,269 |
| MM_6 | 52,868,514 | 1177 | 879 | 712 | 10,053,037 | 1,007,123 | 866,380 |
| MM_61 | 41,841,298 | 136 | 71 | 54 | 4,765,448 | 96,738 | 91,318 |
| MM_66 | 37,559,665 | 503 | 346 | 259 | 7,836,149 | 205,940 | 163,488 |
| MM_67 | 41,723,673 | 904 | 329 | 213 | 6,478,122 | 129,689 | 99,656 |
| MM_68 | 45,666,045 | 495 | 313 | 209 | 9,016,842 | 203,599 | 146,692 |
| MM_7 | 57,602,270 | 1273 | 912 | 718 | 9,700,140 | 749,213 | 664,845 |
| MM_70 | 36,073,087 | 447 | 267 | 199 | 7,430,562 | 210,866 | 154,254 |
| MM_71 | 47,691,240 | 1158 | 591 | 412 | 8,943,282 | 204,495 | 141,742 |
| MM_72 | 38,540,649 | 306 | 203 | 156 | 6,900,306 | 135,644 | 114,276 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Song, S.; Yoon, S.-S.; Koh, Y.; Yun, H. Proper Read Filtering Method to Adequately Analyze Whole-Transcriptome Sequencing and RNA Based Immune Repertoire Sequencing Data for Tumor Milieu Research. Cancers 2020, 12, 3693. https://doi.org/10.3390/cancers12123693
Lee S, Song S, Yoon S-S, Koh Y, Yun H. Proper Read Filtering Method to Adequately Analyze Whole-Transcriptome Sequencing and RNA Based Immune Repertoire Sequencing Data for Tumor Milieu Research. Cancers. 2020; 12(12):3693. https://doi.org/10.3390/cancers12123693
Chicago/Turabian StyleLee, Sungyoung, Seulki Song, Sung-Soo Yoon, Youngil Koh, and Hongseok Yun. 2020. "Proper Read Filtering Method to Adequately Analyze Whole-Transcriptome Sequencing and RNA Based Immune Repertoire Sequencing Data for Tumor Milieu Research" Cancers 12, no. 12: 3693. https://doi.org/10.3390/cancers12123693
APA StyleLee, S., Song, S., Yoon, S.-S., Koh, Y., & Yun, H. (2020). Proper Read Filtering Method to Adequately Analyze Whole-Transcriptome Sequencing and RNA Based Immune Repertoire Sequencing Data for Tumor Milieu Research. Cancers, 12(12), 3693. https://doi.org/10.3390/cancers12123693