Lamin A/C: Function in Normal and Tumor Cells



Abstract
Simple Summary
Abstract
1. Introduction
2. Lamin Proteins
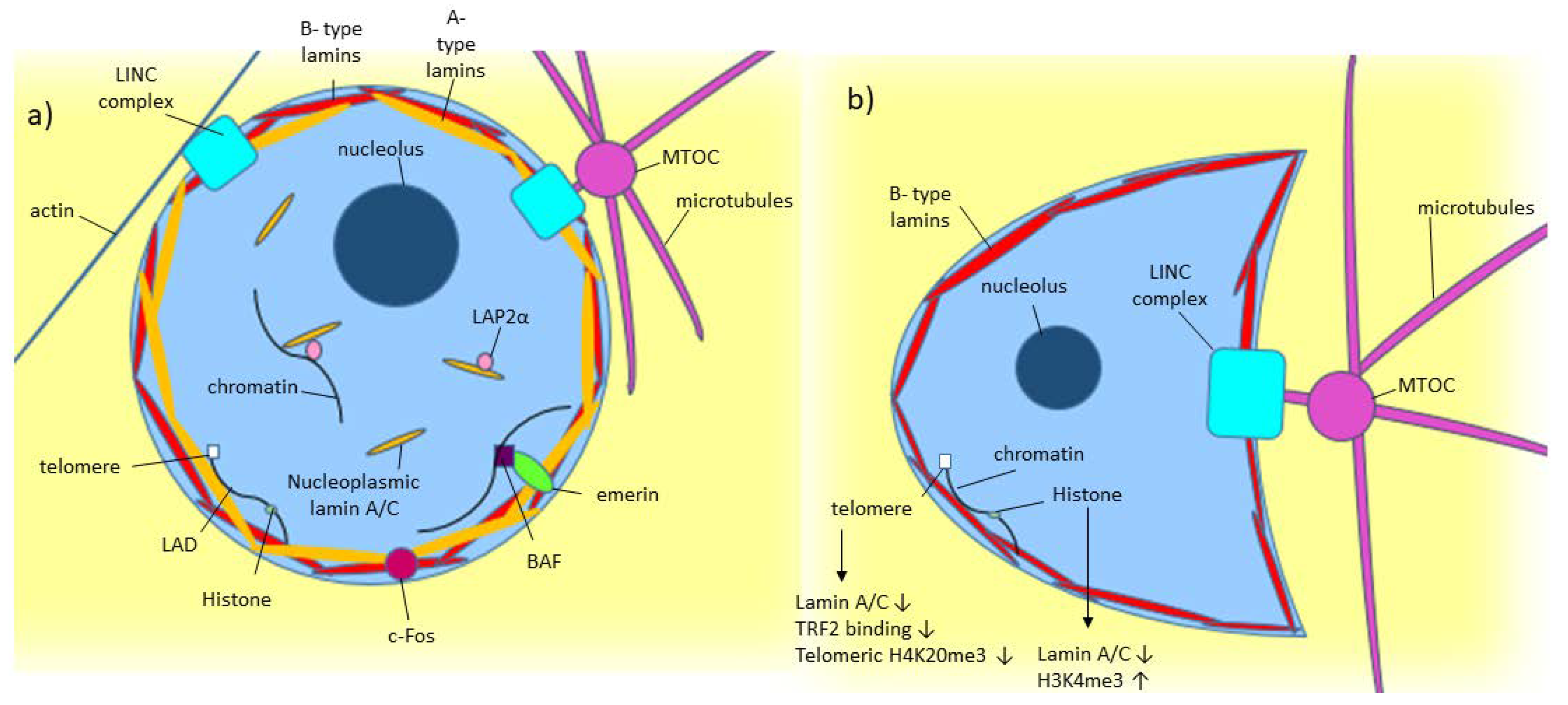
3. Lamin A/C: Gene, Protein, and Nuclear Lamina Structure
4. Lamin A/C: Role in Structural Stability
5. Lamin A/C: Role in Mechanosensing, Mechanosignalling, and Cell Movement
6. Lamin A/C: Role in Genomic Organization and Stability
7. Lamin A/C: Role in Gene Expression and Regulation
8. Lamin A/C in Disease and Cancer
8.1. Laminopathies
8.2. Cancer
9. Deregulated Lamin A/C Expression in Cancer Cells
10. Nuclear Morphology and Cancer
11. Cell Motility/Migration and Cancer
12. Cell Differentiation and Cancer
13. Lamin A/C: Genomic Instability and Senescence in Cancer
14. Lamin A/C Binding Partners and Cancer
15. Prostate Cancer
16. Deregulation of Lamin A/C and Interactive Partners in Prostate Cancer
17. Lamin A/C: Role in Genomic Organization, Expression, and Instability in Prostate Cancer
18. Conclusions
Funding
Conflicts of Interest
References
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior-life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef]
- Wang, X.; Zabell, A.; Koh, W.; Tang, W.H. Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 21. [Google Scholar] [CrossRef]
- Saarinen, I.; Mirtti, T.; Seikkula, H.; Boström, P.J.; Taimen, P. Differential Predictive Roles of A- and B-Type Nuclear Lamins in Prostate Cancer Progression. PLoS ONE 2015, 10, e0140671. [Google Scholar] [CrossRef]
- Meaburn, K.J.; Misteli, T. Assessment of the Utility of Gene Positioning Biomarkers in the Stratification of Prostate Cancers. Front. Genet. 2019, 10, 1029. [Google Scholar] [CrossRef]
- Preisner, H.; Habicht, J.; Garg, S.G.; Gould, S.B. Intermediate filament protein evolution and protists. Cytoskeleton 2018, 75, 231–243. [Google Scholar] [CrossRef]
- Krüger, A.; Batsios, P.; Baumann, O.; Luckert, E.; Schwarz, H.; Stick, R.; Meyer, I.; Gräf, R. Characterization of NE81, the first lamin-like nucleoskeleton protein in a unicellular organism. Mol. Biol. Cell 2012, 23, 360–370. [Google Scholar] [CrossRef]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [PubMed]
- Stewart, C.L.; Kozlov, S.; Fong, L.G.; Young, S.G. Mouse models of the laminopathies. Exp. Cell Res. 2007, 313, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Lee, J.M.; Yang, S.H.; Young, S.G.; Fong, L.G. Nuclear lamins in the brain-new insights into function and regulation. Mol. Neurobiol. 2013, 47, 290–301. [Google Scholar] [CrossRef]
- Kim, Y.; Zheng, Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun. 2013, 440, 8–13. [Google Scholar] [CrossRef]
- Zwerger, M.; Medalia, O. From lamins to lamina: A structural perspective. Histochem. Cell Biol. 2013, 140, 3–12. [Google Scholar] [CrossRef]
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- Goldmann, W.H. Intermediate filaments and cellular mechanics. Cell Biol. Int. 2018, 42, 132–138. [Google Scholar] [CrossRef]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Medalia, O. Lamins: The structure and protein complexes. Curr. Opin. Cell Biol. 2015, 32, 7–12. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, Y.; Keyimu, R.; Hao, J.; Zhao, Z.; Ye, R. The role of lamin A/C in mesenchymal stem cell differentiation. J. Physiol. Biochem. 2019, 75, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Al-Saaidi, R.; Bross, P. Do lamin A and lamin C have unique roles? Chromosoma 2015, 124, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pugh, G.E.; Coates, P.J.; Lane, E.B.; Raymond, Y.; Quinlan, R.A. Distinct nuclear assembly pathways for lamins A and C lead to their increase during quiescence in Swiss 3T3 cells. J. Cell Sci. 1997, 110, 2483–2493. [Google Scholar]
- Torvaldson, E.; Kochin, V.; Eriksson, J.E. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus 2015, 6, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Goldman, R.D. Nuclear lamins in cell regulation and disease. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 525–531. [Google Scholar] [CrossRef]
- Kittisopikul, M.; Virtanen, L.; Taimen, P.; Goldman, R.D. Quantitative Analysis of Nuclear Lamins Imaged by Super-Resolution Light Microscopy. Cells 2019, 8, 361. [Google Scholar] [CrossRef]
- González-Granado, J.M.; Navarro-Puche, A.; Molina-Sanchez, P.; Blanco-Berrocal, M.; Viana, R.; Font de Mora, J.; Andrés, V. Sorting nexin 6 enhances lamin a synthesis and incorporation into the nuclear envelope. PLoS ONE 2014, 9, e115571. [Google Scholar] [CrossRef]
- Karoutas, A.; Szymanski, W.; Rausch, T.; Guhathakurta, S.; Rog-Zielinska, E.A.; Peyronnet, R.; Seyfferth, J.; Chen, H.R.; de Leeuw, R.; Herquel, B.; et al. The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat. Cell Biol. 2019, 21, 1248–1260. [Google Scholar] [CrossRef]
- Tariq, Z.; Zhang, H.; Chia-Liu, A.; Shen, Y.; Gete, Y.; Xiong, Z.M.; Tocheny, C.; Campanello, L.; Wu, D.; Losert, W.; et al. Lamin A and microtubules collaborate to maintain nuclear morphology. Nucleus 2017, 8, 433–446. [Google Scholar] [CrossRef]
- Makarov, A.A.; Zou, J.; Houston, D.R.; Spanos, C.; Solovyova, A.S.; Cardenal-Peralta, C.; Rappsilber, J.; Schirmer, E.C. Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nat. Commun. 2019, 10, 3056. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.; Kuijpers, H.J.; Ostlund, C.; Worman, H.J.; Endert, J.; Ramaekers, F.C. Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp. Cell Res. 2005, 304, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, F.; Degano, M. Disease-associated mutations in the coil 2B domain of human lamin A/C affect structural properties that mediate dimerization and intermediate filament formation. J. Struct. Biol. 2013, 181, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef]
- Kim, J.K.; Louhghalam, A.; Lee, G.; Schafer, B.W.; Wirtz, D.; Kim, D.H. Nuclear lamin A/C harnesses the perinuclear apical actin cables to protect nuclear morphology. Nat. Commun. 2017, 8, 2123. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.; Torretta, S.; Procino, G.; Gerbino, A.; Forleo, C.; Favale, S.; Svelto, M. Role of nuclear Lamin A/C in cardiomyocyte functions. Biol. Cell 2014, 106, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; González-Granado, J.M. Nuclear envelope lamin-A as a coordinator of T cell activation. Nucleus 2014, 5, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Ondrej, V.; Lukásová, E.; Krejcí, J.; Matula, P.; Kozubek, S. Lamin A/C and polymeric actin in genome organization. Mol. Cells 2008, 26, 356–361. [Google Scholar] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef]
- Samson, C.; Petitalot, A.; Celli, F.; Herrada, I.; Ropars, V.; Le Du, M.H.; Nhiri, N.; Jacquet, E.; Arteni, A.A.; Buendia, B.; et al. Structural analysis of the ternary complex between lamin A/C, BAF and emerin identifies an interface disrupted in autosomal recessive progeroid diseases. Nucleic Acids Res. 2018, 46, 10460–10473. [Google Scholar] [CrossRef]
- Ranade, D.; Pradhan, R.; Jayakrishnan, M.; Hegde, S.; Sengupta, K. Lamin A/C and Emerin depletion impacts chromatin organization and dynamics in the interphase nucleus. BMC Mol. Cell Biol. 2019, 20, 11. [Google Scholar] [CrossRef]
- Bronshtein, I.; Kepten, E.; Kanter, I.; Berezin, S.; Lindner, M.; Redwood, A.B.; Mai, S.; Gonzalo, S.; Foisner, R.; Shav-Tal, Y.; et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015, 6, 8044. [Google Scholar] [CrossRef]
- Pradhan, R.; Nallappa, M.J.; Sengupta, K. Lamin A/C modulates spatial organization and function of the Hsp70 gene locus via nuclear myosin I. J. Cell Sci. 2020, 133, jcs236265. [Google Scholar] [CrossRef]
- Wood, A.M.; Rendtlew Danielsen, J.M.; Lucas, C.A.; Rice, E.L.; Scalzo, D.; Shimi, T.; Goldman, R.D.; Smith, E.D.; Le Beau, M.M.; Kosak, S.T. TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends. Nat. Commun. 2014, 5, 5467. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytom. A 2005, 67, 144–150. [Google Scholar] [CrossRef]
- Ikegami, K.; Secchia, S.; Almakki, O.; Lieb, J.D.; Moskowitz, I.P. Phosphorylated Lamin A/C in the Nuclear Interior Binds Active Enhancers Associated with Abnormal Transcription in Progeria. Dev. Cell 2020, 52, 699–713.e11. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M.A.; Bergmann, J.H.; Lazar, Z.; Spector, D.L. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 2013, 4, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S.; et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Yoon, M.H.; Park, B.J. Laminopathies; Mutations on single gene and various human genetic diseases. BMB Rep. 2018, 51, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, A.; Procino, G.; Svelto, M.; Carmosino, M. Role of Lamin A/C Gene Mutations in the Signaling Defects Leading to Cardiomyopathies. Front. Physiol. 2018, 9, 1356. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef]
- Vigouroux, C.; Guénantin, A.C.; Vatier, C.; Capel, E.; Le Dour, C.; Afonso, P.; Bidault, G.; Béréziat, V.; Lascols, O.; Capeau, J.; et al. Lipodystrophic syndromes due to LMNA mutations: Recent developments on biomolecular aspects, pathophysiological hypotheses and therapeutic perspectives. Nucleus 2018, 9, 235–248. [Google Scholar] [CrossRef]
- Kim, Y.; Bayona, P.W.; Kim, M.; Chang, J.; Hong, S.; Park, Y.; Budiman, A.; Kim, Y.J.; Choi, C.Y.; Kim, W.S.; et al. Macrophage Lamin A/C Regulates Inflammation and the Development of Obesity-Induced Insulin Resistance. Front. Immunol. 2018, 9, 696. [Google Scholar] [CrossRef]
- Méndez-López, I.; Blanco-Luquin, I.; Sánchez-Ruiz de Gordoa, J.; Urdánoz-Casado, A.; Roldán, M.; Acha, B.; Echavarri, C.; Zelaya, V.; Jericó, I.; Mendioroz, M. Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 878. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Sakthivel, K.M.; Sehgal, P. A Novel Role of Lamins from Genetic Disease to Cancer Biomarkers. Oncol. Rev. 2016, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Lochs, S.; Kefalopoulou, S.; Kind, J. Lamina Associated Domains and Gene Regulation in Development and Cancer. Cells 2019, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.R.; Robson, J.L.; Simon, W.J.; Twigg, J.; Cruikshank, D.; Wilson, R.G.; Hutchison, C.J. The role of Lamin A in cytoskeleton organization in colorectal cancer cells: A proteomic investigation. Nucleus 2011, 2, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.F.; Krivosheyev, V.; de Souza, A.; Chin, K.; Gaetz, H.P.; Chaudhary, N.; Worman, H.J.; Holt, P.R. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 1999, 45, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.R.; Przyborski, S.A.; Wilson, R.G.; Hutchison, C.J. Lamins as cancer biomarkers. Biochem. Soc. Trans. 2010, 38, 297–300. [Google Scholar] [CrossRef]
- Contu, F.; Rangel-Pozzo, A.; Trokajlo, P.; Wark, L.; Klewes, L.; Johnson, N.A.; Petrogiannis-Haliotis, T.; Gartner, J.G.; Garini, Y.; Vanni, R.; et al. Distinct 3D Structural Patterns of Lamin A/C Expression in Hodgkin and Reed-Sternberg Cells. Cancers 2018, 10, 286. [Google Scholar] [CrossRef]
- Wang, A.S.; Kozlov, S.V.; Stewart, C.L.; Horn, H.F. Tissue specific loss of A-type lamins in the gastrointestinal epithelium can enhance polyp size. Differentiation 2015, 89, 11–21. [Google Scholar] [CrossRef]
- Gong, G.; Chen, P.; Li, L.; Tan, H.; Zhou, J.; Zhou, Y.; Yang, X.; Wu, X. Loss of lamin A but not lamin C expression in epithelial ovarian cancer cells is associated with metastasis and poor prognosis. Pathol. Res. Pract. 2015, 211, 175–182. [Google Scholar] [CrossRef]
- Aljada, A.; Doria, J.; Saleh, A.M.; Al-Matar, S.H.; AlGabbani, S.; Shamsa, H.B.; Al-Bawab, A.; Ahmed, A.A. Altered Lamin A/C splice variant expression as a possible diagnostic marker in breast cancer. Cell. Oncol. 2016, 39, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kaspi, E.; Frankel, D.; Guinde, J.; Perrin, S.; Laroumagne, S.; Robaglia-Schlupp, A.; Ostacolo, K.; Harhouri, K.; Tazi-Mezalek, R.; Micallef, J.; et al. Low lamin A expression in lung adenocarcinoma cells from pleural effusions is a pejorative factor associated with high number of metastatic sites and poor Performance status. PLoS ONE 2017, 12, e0183136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Huang, W.; Cai, J.; Ba, J.; Wang, Y.; Ke, Q.; Huang, Y.; Liu, X.; Qiu, Y.; et al. Nuclear Nestin deficiency drives tumor senescence via lamin A/C-dependent nuclear deformation. Nat. Commun. 2018, 9, 3613. [Google Scholar] [CrossRef]
- Moiseeva, O.; Lessard, F.; Acevedo-Aquino, M.; Vernier, M.; Tsantrizos, Y.S.; Ferbeyre, G. Mutant lamin A links prophase to a p53 independent senescence program. Cell Cycle 2015, 14, 2408–2421. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mitchell, M.J.; Denais, C.; Chan, M.F.; Wang, Z.; Lammerding, J.; King, M.R. Lamin A/C deficiency reduces circulating tumor cell resistance to fluid shear stress. Am. J. Physiol. Cell Physiol. 2015, 309, C736–C746. [Google Scholar] [CrossRef]
- Willis, N.D.; Cox, T.R.; Rahman-Casañs, S.F.; Smits, K.; Przyborski, S.A.; van den Brandt, P.; van Engeland, M.; Weijenberg, M.; Wilson, R.G.; de Bruïne, A.; et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS ONE 2008, 3, e2988. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, J.; He, L.; Gong, G.; Wu, X. Effect of lamin-A expression on migration and nuclear stability of ovarian cancer cells. Gynecol. Oncol. 2019, 152, 166–176. [Google Scholar] [CrossRef]
- Reis-Sobreiro, M.; Chen, J.F.; Novitskaya, T.; You, S.; Morley, S.; Steadman, K.; Gill, N.K.; Eskaros, A.; Rotinen, M.; Chu, C.Y.; et al. Emerin Deregulation Links Nuclear Shape Instability to Metastatic Potential. Cancer Res. 2018, 78, 6086–6097. [Google Scholar] [CrossRef]
- Nardella, M.; Guglielmi, L.; Musa, C.; Iannetti, I.; Maresca, G.; Amendola, D.; Porru, M.; Carico, E.; Sessa, G.; Camerlingo, R.; et al. Down-regulation of the Lamin A/C in neuroblastoma triggers the expansion of tumor initiating cells. Oncotarget 2015, 6, 32821–32840. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, Y. Suspension state increases reattachment of breast cancer cells by up-regulating lamin A/C. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2272–2282. [Google Scholar] [CrossRef]
- Kong, L.; Schäfer, G.; Bu, H.; Zhang, Y.; Zhang, Y.; Klocker, H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 2012, 33, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhao, H.; Yang, R.; Wang, L.; Ma, H.; Xu, X.; Zhou, P.; Kong, L. Lamin A/C might be involved in the EMT signalling pathway. Gene 2018, 663, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Capo-chichi, C.D.; Cai, K.Q.; Smedberg, J.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.X. Loss of A-type lamin expression compromises nuclear envelope integrity in breast cancer. Chin. J. Cancer 2011, 30, 415–425. [Google Scholar] [CrossRef]
- Matsumoto, A.; Hieda, M.; Yokoyama, Y.; Nishioka, Y.; Yoshidome, K.; Tsujimoto, M.; Matsuura, N. Global loss of a nuclear lamina component, lamin A/C, and LINC complex components SUN1, SUN2, and nesprin-2 in breast cancer. Cancer Med. 2015, 4, 1547–1557. [Google Scholar] [CrossRef]
- Wang, J.; Kondo, T.; Nakazawa, T.; Oishi, N.; Mochizuki, K.; Katoh, R. Constitutional abnormality of nuclear membrane proteins in small cell lung carcinoma. Virchows Arch. 2019, 475, 407–414. [Google Scholar] [CrossRef]
- Wazir, U.; Ahmed, M.H.; Bridger, J.M.; Harvey, A.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. The clinicopathological significance of lamin A/C, lamin B1 and lamin B receptor mRNA expression in human breast cancer. Cell. Mol. Biol. Lett. 2013, 18, 595–611. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, L.; Weng, D.; Xu, D.; Geng, J.; Zhao, F. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 8. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2017, 8, 7935–7945. [Google Scholar] [CrossRef] [PubMed]
- Capo-chichi, C.D.; Aguida, B.; Chabi, N.W.; Cai, Q.K.; Offrin, G.; Agossou, V.K.; Sanni, A.; Xu, X.X. Lamin A/C deficiency is an independent risk factor for cervical cancer. Cell. Oncol. 2016, 39, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Belt, E.J.; Fijneman, R.J.; van den Berg, E.G.; Bril, H.; Delis-van Diemen, P.M.; Tijssen, M.; van Essen, H.F.; de Lange-de Klerk, E.S.; Beliën, J.A.; Stockmann, H.B.; et al. Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence. Eur. J. Cancer 2011, 47, 1837–1845. [Google Scholar] [CrossRef]
- Skvortsov, S.; Schäfer, G.; Stasyk, T.; Fuchsberger, C.; Bonn, G.K.; Bartsch, G.; Klocker, H.; Huber, L.A. Proteomics profiling of microdissected low- and high-grade prostate tumors identifies Lamin A as a discriminatory biomarker. Proteome Res. 2011, 10, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Machiels, B.M.; Hopman, A.H.; Broers, J.L.; Bot, F.J.; Arends, J.W.; Ramaekers, F.C.; Schouten, H.C. Comparison of A and B-type lamin expression in reactive lymph nodes and nodular sclerosing Hodgkin’s disease. Histopathology 1997, 31, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Alhudiri, I.M.; Nolan, C.C.; Ellis, I.O.; Elzagheid, A.; Rakha, E.A.; Green, A.R.; Chapman, C.J. Expression of Lamin A/C in early-stage breast cancer and its prognostic value. Breast Cancer Res. Treat. 2019, 174, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Redwood, A.B.; Perkins, S.M.; Vanderwaal, R.P.; Feng, Z.; Biehl, K.J.; Gonzalez-Suarez, I.; Morgado-Palacin, L.; Shi, W.; Sage, J.; Roti-Roti, J.L.; et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle 2011, 10, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Rauschert, I.; Aldunate, F.; Preussner, J.; Arocena-Sutz, M.; Peraza, V.; Looso, M.; Benech, J.C.; Agrelo, R. Promoter hypermethylation as a mechanism for Lamin A/C silencing in a subset of neuroblastoma cells. PLoS ONE 2017, 12, e0175953. [Google Scholar] [CrossRef] [PubMed]
- Capo-Chichi, C.D.; Cai, K.Q.; Xu, X.X. Overexpression and cytoplasmic localization of caspase-6 is associated with lamin A degradation in set of ovarian cancers. Biomark. Res. 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; George, S.H.; Kobetz, E.; Xu, X.X. New biological research and understanding of Papanicolaou’s test. Diagn. Cytopathol. 2018, 46, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Infante, E.; Chavrier, P. Nucleus-Invadopodia Duo During Cancer Invasion. Trends Cell Biol. 2019, 29, 93–96. [Google Scholar] [CrossRef]
- Broers, J.L.; Ramaekers, F.C. The role of the nuclear lamina in cancer and apoptosis. Adv. Exp. Med. Biol. 2014, 773, 27–48. [Google Scholar] [CrossRef]
- Smith, E.R.; Capo-Chichi, C.D.; Xu, X.X. Defective Nuclear Lamina in Aneuploidy and Carcinogenesis. Front. Oncol. 2018, 8, 529. [Google Scholar] [CrossRef]
- Maynard, S.; Keijzers, G.; Akbari, M.; Ezra, M.B.; Hall, A.; Morevati, M.; Scheibye-Knudsen, M.; Gonzalo, S.; Bartek, J.; Bohr, V.A. Lamin A/C promotes DNA base excision repair. Nucleic Acids Res. 2019, 47, 11709–11728. [Google Scholar] [CrossRef]
- Fujiwara, C.; Muramatsu, Y.; Nishii, M.; Tokunaka, K.; Tahara, H.; Ueno, M.; Yamori, T.; Sugimoto, Y.; Seimiya, H. Cell-based chemical fingerprinting identifies telomeres and lamin A as modifiers of DNA damage response in cancer cells. Sci. Rep. 2018, 8, 14827. [Google Scholar] [CrossRef]
- Kiran, K.G.; Palaniswamy, M.; Angayarkanni, J. Human telomerase inhibitors from microbial source. World J. Microbiol. Biotechnol. 2015, 31, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Xu, B. Telomerase Inhibitors from Natural Products and Their Anticancer Potential. Int. J. Mol. Sci. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Man, R.J.; Chen, L.W.; Zhu, H.L. Telomerase inhibitors: A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.H.; Wang, P.H.; Niu, S.W.; Zhen, Y.Y.; Huang, M.S.; Hsiao, M.; Yang, C.J. Inhibition of FAK Signaling Elicits Lamin A/C-Associated Nuclear Deformity and Cellular Senescence. Front. Oncol. 2019, 9, 22. [Google Scholar] [CrossRef]
- Matralis, A.N.; Xanthopoulos, D.; Huot, G.; Lopes-Paciencia, S.; Cole, C.; de Vries, H.; Ferbeyre, G.; Tsantrizos, Y.S. Molecular tools that block maturation of the nuclear lamin A and decelerate cancer cell migration. Bioorg. Med. Chem. 2018, 26, 5547–5554. [Google Scholar] [CrossRef]
- Brachner, A.; Foisner, R. Lamina-associated polypeptide (LAP)2α and other LEM proteins in cancer biology. Adv. Exp. Med. Biol. 2014, 773, 143–163. [Google Scholar] [CrossRef]
- Grozescu, T.; Popa, F. Prostate cancer between prognosis and adequate/proper therapy. J. Med. Life 2017, 10, 5–12. [Google Scholar]
- Drachenberg, D.; Awe, J.A.; Rangel Pozzo, A.; Saranchuk, J.; Mai, S. Advancing Risk Assessment of Intermediate Risk Prostate Cancer Patients. Cancers 2019, 11, 855. [Google Scholar] [CrossRef]
- Debes, J.D.; Sebo, T.J.; Heemers, H.V.; Kipp, B.R.; Haugen, D.L.; Lohse, C.M.; Tindall, D.J. p300 modulates nuclear morphology in prostate cancer. Cancer Res. 2005, 65, 708–712. [Google Scholar] [PubMed]
- Khan, Z.S.; Santos, J.M.; Hussain, F. Aggressive prostate cancer cell nuclei have reduced stiffness. Biomicrofluidics 2018, 12, 014102. [Google Scholar] [CrossRef]
- Helfand, B.T.; Wang, Y.; Pfleghaar, K.; Shimi, T.; Taimen, P.; Shumaker, D.K. Chromosomal regions associated with prostate cancer risk localize to lamin B-deficient microdomains and exhibit reduced gene transcription. J. Pathol. 2012, 226, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, Y.; Jiang, H.; Nie, D. Promotion of tumor development in prostate cancer by progerin. Cancer Cell Int. 2010, 10, 47. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell and Cancer Type | Lamin A/C Expression | Effect on Cancer | Reference |
|---|---|---|---|
| Lung cancer cell lines A549, and H1299 | Decrease in lamin A/C | Increased tumor senescence | [73] |
| Human non-small cell lung carcinoma H 1299, human osteosarcoma U-2 OS, human cervix carcinoma HeLa | Lamin A/C mutated to S22A-progerin | Increased cellular senescence | [74] |
| Breast adenocarcinoma cell lines MDA-MB-231 and MDA-MB-468 | Lamin A/C knockout | Decreased cell’s resistance to fluid shear stress | [75] |
| Human pre-metastatic colon adenocarcinoma cell line SW480 | Transfected with GFP-lamin A | Downregulated E-cadherin and increased cell motility | [76] |
| Human pre-metastatic colon adenocarcinoma cell line SW480 | Lamin A/C upregulated | Increased cell motility | [65] |
| Human ovarian cancer cell line H08910 | Lamin A/C overexpressed, Lamin A/C 30–40% inhibited, lamin A/C 70–80% inhibited | Lamin A/C overexpressed: Decreased migration through 3 μm pore. 30–40% inhibited: Increased migration through 3 μm pore. 70–80% inhibited: Decreased migration through 3 μm pore | [77] |
| Prostate cancer cell line DU145 and breast cancer cell line BT-549 | Lamin A/C depleted | Emerin mislocalized and nuclear membrane had blebbing | [78] |
| Neuroblastoma cell line SH-SY5Y | Lamin A/C downregulated | Tumor initiating cells developed | [79] |
| Breast cancer cell line MDA-MB-231 in suspension culture | Lamin A/C downregulated | Decreased adhesion and reattachment of cells | [80] |
| Prostate cancer cell line PC3, DU145, and LNCaP | Lamin A/C upregulated | Increased PI3k subunit, p110, and p85 expression and increased growth and invasive capabilities | [81] |
| Prostate cancer cell line PC-3M-1E8 and PC-3M-2B4 | Lamin A/C knockdown | Cell growth was inhibited and colony formation was decreased. E-cadherin downregulated. Vimentin, snail, and slug upregulated | [82] |
| Cancer Type | Lamin A/C Expression | Reference |
| 56 invasive ductal carcinoma samples | Majority of samples heterogeneous for lamin A/C expression, 38% had no lamin A/C expressed | [83] |
| 656 colorectal adenocarcinoma tumor samples | 70% positive for lamin A/C, 30% negative | [76] |
| 73 breast cancer tumor samples | 84.9% had low lamin A/C expression (low defined as less than 50% of cancer cells stained positive for lamin A/C) | [84] |
| 33 small cell lung carcinomas (SCLC), 72 non-small cell lung carcinomas (34 adenocarcinoma, 30 squamous cell carcinoma, 8 large cell carcinoma) | 91% of SCLC had negative or low lamin A/C expression, 3% of non-SCLC had negative or low lamin A/C expression | [85] |
| 115 breast cancer tissue samples | Lower lamin A/C than found in non-cancerous breast tissue. | [86] |
| 126 gastric carcinoma samples | 70 positive for lamin A/C 56 negative for lamin A/C (lamin A/C associated with poorer prognosis and lower differentiation) | [87] |
| 87 endometrial cancer samples | Lamin A was reduced in all high-grade endometrial cancer samples | [88] |
| 61 epithelial ovarian cancer samples | Lower lamin A expression than normal and benign controls | [70] |
| 17 primary colorectal carcinomas, 18 adenomatous polyps | Lamin A/C reduced or absent in all samples | [66] |
| 128 breast adenocarcinomas | Lamin C expression increased, Lamin A expression decreased | [71] |
| 76 cervical uterine smears (CUS) | 39% normal expression, 28% weak, 33% none. Oncogenic HPV infection rate highest in group with no lamin A/C staining. | [89] |
| 219 stage II and 151 stage III colon cancer samples | 17.8% low lamin A/C expression, Reoccurrence of cancer 45.5% in low lamin A/C expression group compared to 29.6% in high lamin A/C expression group | [90] |
| 94 prostate tumor tissue microarrays | Lamin A/C had low expression in tumor regions with a Gleason pattern (GP) less than 3 and higher expression in regions with a GP of 4 or 5 | [81] |
| 4 prostate adenocarcinoma cohorts | Lamin A/C mRNA reduced when the Gleason score is 8, but the level increases above Gleason score 8 and in metastatic regions | [82] |
| Tissue microarray from 501 prostate cancer patients | Low lamin A/C associated with increased lymph node metastasis and disease-specific death | [7] |
| Tissue cores from 94 prostate tumor samples | Lamin A expression higher in higher Gleason score tumors | [91] |
| Biopsies from 9 patients with Hodgkin’s disease | Most Reed-Sternberg and Hodgkin cells expressed lamin A/C while surrounding B and T lymphocytes did not. | [92] |
| Cancer Type | Lamin A/C Expression | Effect on Cancer | Reference |
|---|---|---|---|
| Invasive breast carcinoma | Reduced lamin A/C expression | More aggressive phenotype than tumors with high lamin A/C expression | [93] |
| Stage II and Stage III colon cancer tumors | Low lamin A/C expression | Increased disease recurrence | [90] |
| Colorectal adenocarcinoma tumors | Sample either did or did not express lamin A/C | Mortality risk twice as high as tumors not expressing lamin A/C | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubik, N.; Mai, S. Lamin A/C: Function in Normal and Tumor Cells. Cancers 2020, 12, 3688. https://doi.org/10.3390/cancers12123688
Dubik N, Mai S. Lamin A/C: Function in Normal and Tumor Cells. Cancers. 2020; 12(12):3688. https://doi.org/10.3390/cancers12123688
Chicago/Turabian StyleDubik, Niina, and Sabine Mai. 2020. "Lamin A/C: Function in Normal and Tumor Cells" Cancers 12, no. 12: 3688. https://doi.org/10.3390/cancers12123688
APA StyleDubik, N., & Mai, S. (2020). Lamin A/C: Function in Normal and Tumor Cells. Cancers, 12(12), 3688. https://doi.org/10.3390/cancers12123688