Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies

 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
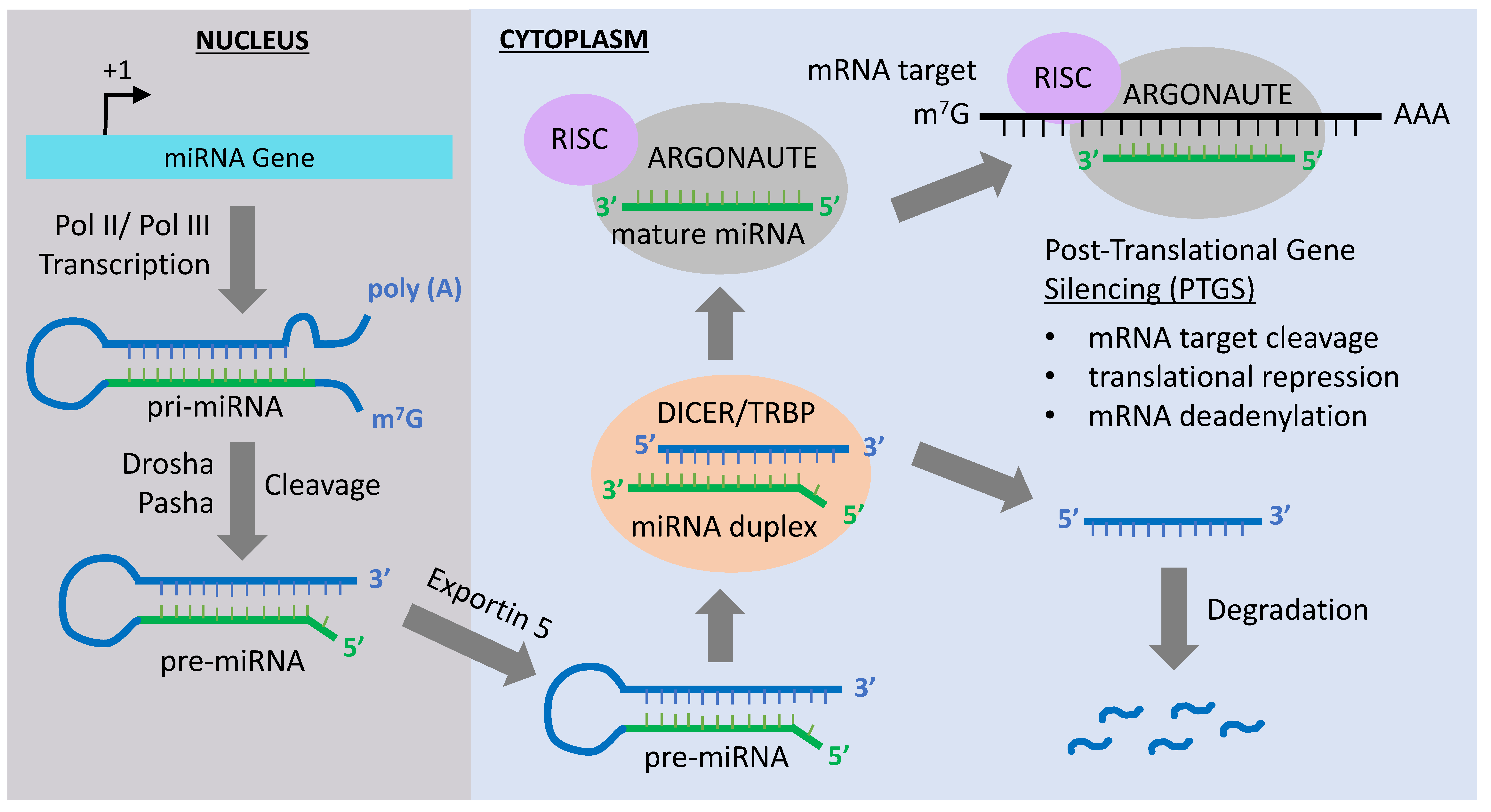
2. MiRNA-Mediated Epigenetic Mechanisms
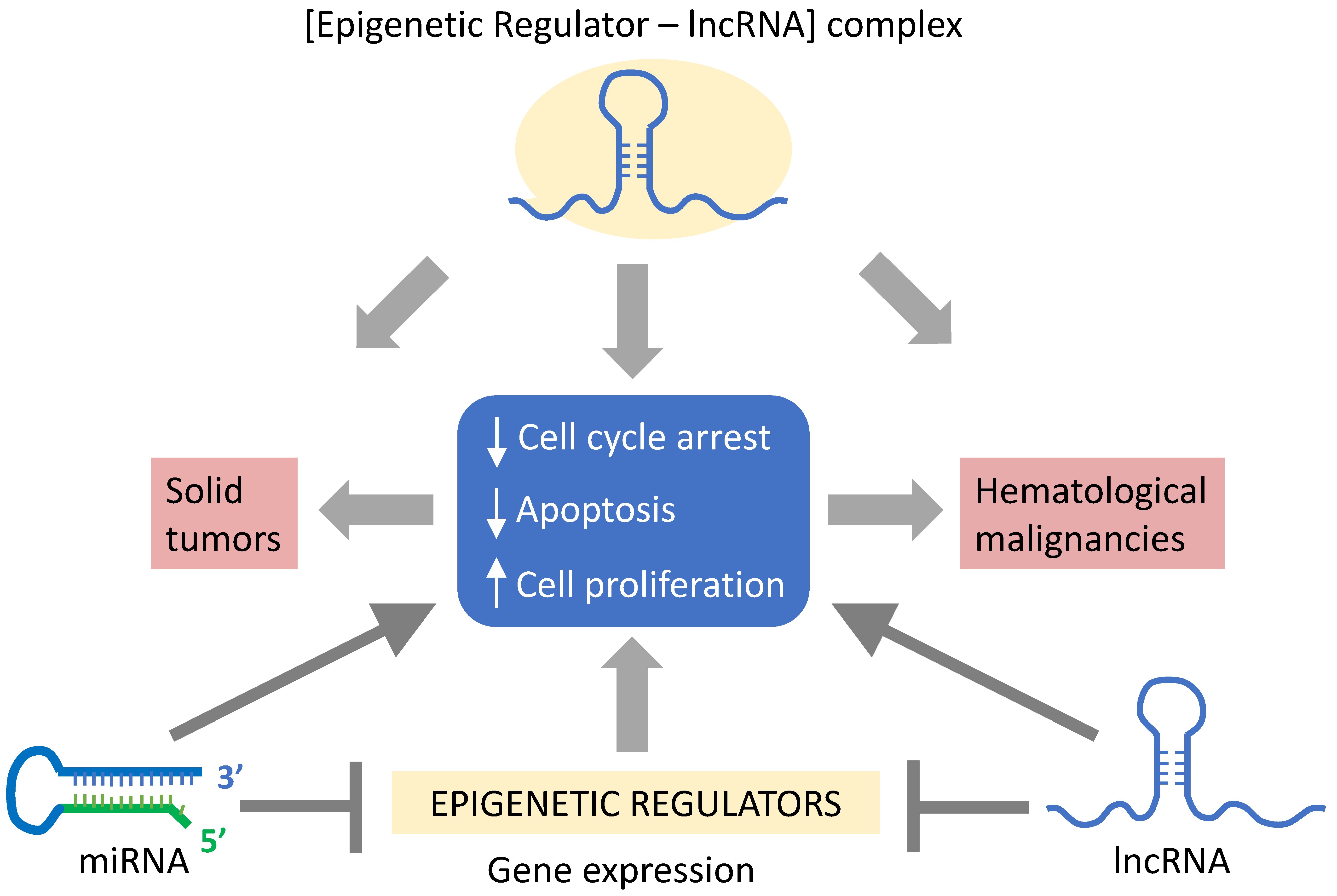
3. LncRNA-Mediated Epigenetic Mechanisms
4. MiRNAs in Solid Tumors
5. MiRNAs in Hematologic Malignancies
6. LncRNAs in Solid Tumors
7. LncRNAs in Hematologic Malignancies
8. Use of ncRNAs in Clinical Therapy
9. Conclusions
Funding
Conflicts of Interest
References
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Tiffon, C. The impact of nutrition and environmental epigenetics on human health and disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Chuang, J.C.; Jones, P.A. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 24–29. [Google Scholar] [CrossRef]
- Kim, Y.J.; Maizel, A.; Chen, X. Traffic into silence: Endomembranes and post-transcriptional RNA silencing. EMBO J. 2014, 33, 968–980. [Google Scholar] [CrossRef]
- Tao, B.B.; Liu, X.Q.; Zhang, W.; Li, S.; Dong, D.; Xiao, M.; Zhong, J. Evidence for the association of chromatin and microRNA regulation in the human genome. Oncotarget 2017, 8, 70958–70966. [Google Scholar] [CrossRef]
- Karijolich, J.; Yu, Y.T. Spliceosomal snRNA modifications and their function. RNA Biol. 2010, 7, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Valadkhan, S. snRNAs as the catalysts of pre-mRNA splicing. Curr. Opin. Chem. Biol. 2005, 9, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, L.B.; Steitz, J.A. Guided tours: From precursor snoRNA to functional snoRNP. Curr. Opin. Cell. Biol. 1999, 11, 378–384. [Google Scholar] [CrossRef]
- Kiss, T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001, 20, 3617–3622. [Google Scholar] [CrossRef]
- Matera, A.G.; Terns, R.M.; Terns, M.P. Non-coding RNAs: Lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. 2007, 8, 209–220. [Google Scholar] [CrossRef]
- Ono, M.; Yamada, K.; Avolio, F.; Scott, M.S.; van Koningsbruggen, S.; Barton, G.J.; Lamond, A.I. Analysis of human small nucleolar RNAs (snoRNA) and the development of snoRNA modulator of gene expression vectors. Mol. Biol. Cell 2010, 21, 1569–1584. [Google Scholar] [CrossRef]
- Maden, B.E.H.; Hughes, J.M. Eukaryotic ribosomal RNA: The recent excitement in the nucleotide modification problem. Chromosoma 1997, 105, 391–400. [Google Scholar] [CrossRef]
- Brimacombe, R.; Stiege, W. Structure and function of ribosomal RNA. Biochem. J. 1985, 229, 1–17. [Google Scholar] [CrossRef]
- O’Donoghue, P.; Ling, J.; Söll, D. Transfer RNA function and evolution. RNA Biol. 2018, 15, 423–426. [Google Scholar] [CrossRef]
- Ishizu, H.; Siomi, H.; Siomi, M.C. Biology of PIWI-interacting RNAs: New insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012, 26, 2361–2373. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.Y.; Lin, H. PIWI proteins and their interactors in piRNA biogenesis, germline development and gene expression. Natl. Sci. Rev. 2014, 1, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, S.; Cheng, B. Epigenetic roles of PIWI-interacting RNAs (piRNAs) in cancer metastasis. Oncol. Rep. 2018, 40, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Cochella, L. A framework for understanding the roles of miRNAs in animal development. Development 2017, 144, 2548–2559. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Nykänen, A.; Haley, B.; Zamore, P.D. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell 2001, 107, 309–321. [Google Scholar] [CrossRef]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduc. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Iki, T.; Yoshikawa, M.; Nishikiori, M.; Jaudal, M.C.; Matsumoto-Yokoyama, E.; Mitsuhara, I.; Meshi, T.; Ishikawa, M. In vitro assembly of plant RNA-induced silencing complexes facilitated by molecular chaperone HSP90. Mol. Cell 2010, 39, 282–291. [Google Scholar] [CrossRef]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6047–6052. [Google Scholar] [CrossRef]
- Miyagishi, M.; Taira, K. U6 promoter–driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat. Biotechnol. 2002, 20, 497–500. [Google Scholar] [CrossRef]
- Abbas-Terki, T.; Blanco-Bose, W.; Déglon, N.; Pralong, W.; Aebischer, P. Lentiviral-mediated RNA interference. Hum. Gene Ther. 2002, 13, 2197–2201. [Google Scholar] [CrossRef]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.; Taxman, D.J. Short hairpin RNA (shRNA): Design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [PubMed]
- Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated actions of microRNAs with other epigenetic factors regulate skeletal muscle development and adaptation. Int. J. Mol. Sci. 2017, 18, 840. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef]
- Duursma, A.M.; Kedde, M.; Schrier, M.; Le Sage, C.; Agami, R. miR-148 targets human DNMT3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. Epigenetic alteration and microRNA dysregulation in cancer. Front. Genet. 2013, 4, 258. [Google Scholar] [CrossRef]
- Wang, X.X.; Zhang, H.; Li, Y. Preliminary study on the role of miR-148a and DNMT1 in the pathogenesis of acute myeloid leukemia. Mol. Med. Rep. 2019, 4, 2943–2952. [Google Scholar] [CrossRef]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef]
- Yong-Ming, H.; Ai-Jun, J.; Xiao-Yue, X.; Jian-Wei, L.; Chen, Y.; Ye, C. miR-449: A potential therapeutic agent for cancer. Anticancer Drugs 2017, 28, 1067–1078. [Google Scholar] [CrossRef]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pinter, S.F. A Tale of Two Cities: How Xist and its partners localize to and silence the bicompartmental X. Semin. Cell Dev. Biol. 2016, 56, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Qu, J.; Li, M.; Zhong, W.; Hu, C. Competing endogenous RNA in cancer: A new pattern of gene expression regulation. Int. J. Clin. Exp. Med. 2015, 8, 17110–17116. [Google Scholar]
- Dhanoa, J.K.; Sethi, R.S.; Verma, R.; Arora, J.S.; Mukhopadhyay, C.S. Long non-coding RNA: Its evolutionary relics and biological implications in mammals: A review. J. Anim. Sci. Technol. 2018, 60, 25. [Google Scholar] [CrossRef]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafreniere, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Mlynarczyk, S.K.; Panning, B. X inactivation: Tsix and Xist as yin and yang. Curr. Biol. 2000, 10, 899–903. [Google Scholar] [CrossRef]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; Young, A.N.; Cerase, A. Function by structure: Spotlights on Xist long non-coding RNA. Front. Mol. Biosci. 2017, 4, 90. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Blanco, M.; Jackson, C.; Aznauryan, E.; Ollikainen, N.; Surka, C.; Chow, A.; Cerase, A.; McDonel, P.; Guttman, M. Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 2016, 354, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Song, X.Q.; Cai, J.P.; Zhang, S. HOTAIR: A cancer-related long non-coding RNA. Neoplasma 2014, 61, 379–391. [Google Scholar] [CrossRef]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Raab, J.R.; Smith, K.N.; Spear, C.C.; Manner, C.J.; Calabrese, J.M.; Magnuson, T. SWI/SNF remains localized to chromatin in the presence of SCHLAP1. Nat. Genet. 2019, 51, 26–29. [Google Scholar] [CrossRef]
- Cristofaro, M.F.D.; Betz, B.L.; Rorie, C.J.; Reisman, D.N.; Wang, W.; Weissman, B.E. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell Physiol. 2001, 186, 136–145. [Google Scholar] [CrossRef]
- Neve, B.; Jonckheere, N.; Vincent, A.; Van Seuningen, I. Epigenetic regulation by lncRNAs: An overview focused on UCA1 in colorectal cancer. Cancers 2018, 10, 440. [Google Scholar] [CrossRef]
- Chiba, H.; Muramatsu, M.; Nomoto, A.; Kato, H. Two human homologues of Saccharomyces cerevisiae SWI2/SNF2 and Drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res. 1994, 22, 1815–1820. [Google Scholar] [CrossRef]
- Chi, J.S.; Li, J.Z.; Jia, J.J.; Zhang, T.; Liu, X.M.; Yi, L. Long non-coding RNA ANRIL in gene regulation and its duality in atherosclerosis. Curr. Med. Sci. 2017, 37, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Meseure, D.; Vacher, S.; Alsibai, K.D.; Nicolas, A.; Chemlali, W.; Caly, M.; Lidereau, R.; Pasmant, E.; Callens, C.; Bieche, I. Expression of ANRIL–polycomb complexes–CDKN2A/B/ARF genes in breast tumors: Identification of a two-gene (EZH2/CBX7) signature with independent prognostic value. Mol. Cancer Res. 2016, 14, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Li, L.; He, Y.; Zheng, D.; Yu, P.; Yu, L.; Tang, L.; Wang, Y.; Wang, Z. EZH2 RIP-seq identifies tissue-specific long non-coding RNAs. Curr. Gene Ther. 2018, 18, 275–285. [Google Scholar] [CrossRef]
- Su, M.; Xiao, Y.; Tang, J.; Wu, J.; Ma, J.; Tian, B.; Zhou, Y.; Wang, H.; Yang, D.; Liao, Q.J.; et al. Role of lncRNA and EZH2 interaction/regulatory network in lung cancer. J. Cancer 2018, 9, 4156–4165. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, M.; Liu, J.; Xu, B.; Yang, J.; Wang, N.; Yan, S.; Wang, F.; He, X.; Ji, G.; et al. Long non-coding RNA PVT1 promotes cell proliferation and migration by silencing ANGPTL4 expression in cholangiocarcinoma. Mol. Ther. Nucleic Acids 2018, 13, 503–513. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Cardenas, C.L.L.; Kessinger, C.W.; Cheng, Y.; MacDonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Ashish, S.Y.; Jaffer, F.A.; et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Puvvula, P.K.; Desetty, R.D.; Pineau, P.; Marchio, A.; Moon, A.; Dejean, A.; Bischof, O. Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat. Commun. 2014, 5, 5323. [Google Scholar] [CrossRef]
- Xia, L.; Zhang, D.; Du, R.; Pan, Y.; Zhao, L.; Sun, S.; Hong, L.; Liu, J.; Fan, D. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int. J. Cancer 2008, 123, 372–379. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Kugel, S.; Sebastián, C.; Fitamant, J.; Ross, K.N.; Saha, S.K.; Jain, E.; Gladden, A.; Arora, K.S.; Kato, Y.; Rivera, M.N.; et al. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 2016, 165, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Xiong, J.; Xu, X.; Lu, W.; Liu, L.; Xiao, D.; Wang, D. Functional elucidation of MiR-34 in osteosarcoma cells and primary tumor samples. Biochem. Biophys. Res. Commun. 2009, 388, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Hasakova, K.; Reis, R.; Vician, M.; Zeman, M.; Herichova, I. Expression of miR-34a-5p is up-regulated in human colorectal cancer and correlates with survival and clock gene PER2 expression. PLoS ONE 2019, 14, e0224396. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Boil. 2011, 8, 706–713. [Google Scholar] [CrossRef]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tan, Z.; Hu, H.; Liu, H.; Wu, T.; Zheng, C.; Wang, X.; Luo, Z.; Wang, J.; Liu, S.; et al. microRNA-21 promotes breast cancer proliferation and metastasis by targeting LZTFL1. BMC Cancer 2019, 19, 738. [Google Scholar] [CrossRef] [PubMed]
- Jesionek-Kupnicka, D.; Braun, M.; Trąbska-Kluch, B.; Czech, J.; Szybka, M.; Szymańska, B.; Kulczycka-Wojdala, D.; Bieńkowski, M.; Kordek, R.; Zawlik, I. MiR-21, miR-34a, miR-125b, miR-181d and miR-648 levels inversely correlate with MGMT and TP53 expression in primary glioblastoma patients. Arch. Med. Sci. 2019, 15, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R.; Yang, C.H.; Pfeffer, L.M. The role of miR-21 in cancer. Drug Dev. Res. 2015, 76, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Harris, M.H.; Zhou, B.; Lodish, H.F. MicroRNA miR-125b causes leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 21558–21563. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; So, A.Y.L.; Mehta, A.; Minisandram, A.; Sinha, N.; Jonsson, V.D.; Rao, D.S.; O’Connell, R.M.; Baltimore, D. Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc. Natl. Acad. Sci. USA 2012, 109, 4233–4238. [Google Scholar] [CrossRef]
- Wu, N.; Lin, X.; Zhao, X.; Zheng, L.; Xiao, L.; Liu, J.; Ge, L.; Cao, S. MiR-125b acts as an oncogene in glioblastoma cells and inhibits cell apoptosis through p53 and p38MAPK-independent pathways. Br. J. Cancer 2013, 109, 2853–2863. [Google Scholar] [CrossRef]
- Romero, P.V.; Cialfi, S.; Palermo, R.; De Blasio, C.; Checquolo, S.; Bellavia, D.; Chiaretti, S.; Foà, R.; Amadori, A.; Gulino, A.; et al. The deregulated expression of miR-125b in acute myeloid leukemia is dependent on the transcription factor C/EBPα. Leukemia 2015, 29, 2442–2445. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Guo, B.; Chen, Z.; Wang, N.; Iacovino, M.; Cheng, J.; Roden, C.; Pan, W.; Khan, S.; Chen, S.; et al. miR-125b promotes MLL-AF9–driven murine acute myeloid leukemia involving a VEGFA-mediated non–cell-intrinsic mechanism. Blood 2017, 129, 1491–1502. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Hoadley, K.; Kushwaha, D.; Ramakrishnan, V.; Li, S.; Kang, C.; You, Y.; Jiang, C.; Song, S.W.; et al. miR-181d: A predictive glioblastoma biomarker that downregulates MGMT expression. Neuro-oncology 2012, 14, 712–719. [Google Scholar] [CrossRef]
- Yang, L.; Ma, Y.; Xin, Y.; Han, R.; Li, R.; Hao, X. Role of the microRNA 181 family in glioma development. Mol. Med. Rep. 2018, 17, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Kreth, S.; Limbeck, E.; Hinske, L.C.; Schütz, S.V.; Thon, N.; Hoefig, K.; Egensperger, R.; Kreth, F.W. In human glioblastomas transcript elongation by alternative polyadenylation and miRNA targeting is a potent mechanism of MGMT silencing. Acta Neuropathol. 2013, 125, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Andreeff, M.; Kantarjian, H.M.; Garcia-Manero, G.; Calin, G.A. MicroRNAs and noncoding RNAs in hematological malignancies: Molecular, clinical and therapeutic implications. Leukemia 2008, 22, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Morenos, L.; Phipson, B.; Willis, S.N.; Brumatti, G.; Eggers, S.; Lalaoui, N.; Brown, L.M.; Kosasih, H.J.; Bartolo, R.C.; et al. Functionally distinct roles for different miR-155 expression levels through contrasting effects on gene expression, in acute myeloid leukaemia. Leukemia 2017, 31, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Witten, L.; Slack, F.J. miR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef]
- Kluiver, J.; Poppema, S.; de Jong, D.; Blokzijl, T.; Harms, G.; Jacobs, S.; Kroesen, B.J.; van den Berg, A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J. Pathol. 2005, 207, 243–249. [Google Scholar] [CrossRef]
- Narayan, N.; Bracken, C.P.; Ekert, P.G. MicroRNA-155 expression and function in AML: An evolving paradigm. Exp. Hematol. 2018, 62, 1–6. [Google Scholar] [CrossRef]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Cui, J.; Li, Y.Y.; Culicchia, F. Up-regulation of micro-RNA-221 (miRNA-221; chr Xp11. 3) and caspase-3 accompanies down-regulation of the survivin-1 homolog BIRC1 (NAIP) in glioblastoma multiforme (GBM). J. Neurooncol. 2009, 91, 27–32. [Google Scholar] [CrossRef]
- Wei, W.; Yang, Y.; Cai, J.; Cui, K.; Li, R.X.; Wang, H.; Shang, X.; Wei, D. MiR-30a-5p suppresses tumor metastasis of human colorectal cancer by targeting ITGB3. Cell Physiol. Biochem. 2016, 39, 1165–1176. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, G.; Wu, J.H.; Jiang, C.P. Diverse roles of miR-29 in cancer. Oncol. Rep. 2014, 31, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.Z.; O’Connor, S.M.; van Holst Pellekaan, N.G.; Young, G.P.; James, R.J. Reduced Accumulation of Specific MicroRNAs in Colorectal Neoplasia11Note: Susan M. O’Connor and Nicholas G. van Holst Pellekaan contributed equally to this work. Mol. Cancer Res. 2003, 1, 882–891. [Google Scholar] [PubMed]
- Sheng, N.; Tan, G.; You, W.; Chen, H.; Gong, J.; Chen, D.; Zhang, H.; Wang, Z. MiR-145 inhibits human colorectal cancer cell migration and invasion via PAK4-dependent pathway. Cancer Med. 2017, 6, 1331–1340. [Google Scholar] [CrossRef]
- De Luca, L.; Trino, S.; Laurenzana, I.; Tagliaferri, D.; Falco, G.; Grieco, V.; Bianchino, G.; Nozza, F.; Campia, V.; D’Alessio, F.; et al. Knockdown of miR-128a induces Lin28a expression and reverts myeloid differentiation blockage in acute myeloid leukemia. Cell Death Dis. 2017, 8, e2849. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, C.; Olive, V.; Lykken, E.; Feng, F.; Sevilla, J.; Wan, Y.; He, L.; Li, Q.J. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 2011, 118, 5487–5497. [Google Scholar] [CrossRef]
- Moussay, E.; Wang, K.; Cho, J.H.; van Moer, K.; Pierson, S.; Paggetti, J.; Nazarov, P.V.; Palissot, V.; Hood, L.E.; Berchem, G.; et al. MicroRNA as biomarkers and regulators in B-cell chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2011, 108, 6573–6578. [Google Scholar] [CrossRef]
- Willimott, S.; Wagner, S.D. Stromal cells and CD40 ligand (CD154) alter the miRNome and induce miRNA clusters including, miR-125b/miR-99a/let-7c and miR-17-92 in chronic lymphocytic leukaemia. Leukemia 2012, 26, 1113–1116. [Google Scholar] [CrossRef]
- He, M.; Wang, Q.Y.; Yin, Q.Q.; Tang, J.; Lu, Y.; Zhou, C.X.; Duan, C.W.; Hong, D.L.; Tanaka, T.; Chen, G.Q.; et al. HIF-1 α downregulates miR-17/20a directly targeting p21 and STAT3: A role in myeloid leukemic cell differentiation. Cell Death Differ. 2013, 20, 408–418. [Google Scholar] [CrossRef]
- Luo, J.W.; Wang, X.; Yang, Y.; Mao, Q. Role of micro-RNA (miRNA) in pathogenesis of glioblastoma. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1630–1639. [Google Scholar]
- Zhang, Z.; Tang, H.; Wang, Z.; Zhang, B.; Liu, W.; Lu, H.; Xiao, L.; Liu, X.; Wang, R.; Li, X.; et al. MiR-185 targets the DNA methyltransferases 1 and regulates global DNA methylation in human glioma. Mol. Cancer 2011, 10, 124. [Google Scholar] [CrossRef]
- Mekala, J.R.; Naushad, S.M.; Ponnusamy, L.; Arivazhagan, G.; Sakthiprasad, V.; Pal-Bhadra, M. Epigenetic regulation of miR-200 as the potential strategy for the therapy against triple-negative breast cancer. Gene 2018, 641, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, H.; Li, Z.; Li, Y.; Wang, X.; Gurbuxani, S.; Chen, P.; He, C.; You, D.; Zhang, S.; et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell 2012, 22, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Teshima, K.; Ikeda, S.; Kitadate, A.; Watanabe, A.; Nara, M.; Yamashita, J.; Ohshima, K.; Sawada, K.; Tagawa, H. MicroRNA-150 inhibits tumor invasion and metastasis by targeting the chemokine receptor CCR6, in advanced cutaneous T-cell lymphoma. Blood 2014, 123, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Abe, F.; Kitadate, A.; Ikeda, S.; Yamashita, J.; Nakanishi, H.; Takahashi, N.; Asaka, C.; Teshima, K.; Miyagaki, T.; Sugaya, M.; et al. Histone deacetylase inhibitors inhibit metastasis by restoring a tumor suppressive microRNA-150 in advanced cutaneous T-cell lymphoma. Oncotarget 2017, 8, 7572–7585. [Google Scholar] [CrossRef]
- Qian, B.; Katsaros, D.; Lu, L.; Preti, M.; Durando, A.; Arisio, R.; Mu, L.; Yu, H. High miR-21 expression in breast cancer associated with poor disease-free survival in early stages disease and high TGF-beta1. Breast Cancer Res. Treat. 2009, 117, 131–140. [Google Scholar] [CrossRef]
- Han, R.; Zhao, J.; Lu, L. MicroRNA-34a expression affects breast cancer invasion in vitro and patient survival via downregulation of E2F1 and E2F3 expression. Oncol. Rep. 2020, 43, 2062–2072. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Han, Y.; Meng, F.; Venter, J.; Wu, N.; Wan, Y.; Standeford, H.; Francis, H.; Meininger, C.; Greene, J., Jr.; Trzeciakowski, J.P.; et al. miR-34a-dependent overexpression of Per1 decreases cholangiocarcinoma growth. J. Hepatol. 2016, 64, 1295–1304. [Google Scholar] [CrossRef]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [PubMed]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef]
- Natarajan, A.T.; Vermeulen, S.; Darroudi, F.; Valentine, M.B.; Brent, T.P.; Mitra, S.; Tano, K. Chromosomal localization of human O6-methylguanine-DNA methyltransferase (MGMT) gene by in situ hybridization. Mutagenesis 1992, 7, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef]
- Jacinto, F.V.; Esteller, M. MGMT hypermethylation: A prognostic foe, a predictive friend. DNA Repair 2007, 6, 1155–1160. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Kushwaha, D.; Ramakrishnan, V.; Ng, K.; Steed, T.; Nguyen, T.; Futalan, D.; Akers, J.C.; Sarkaria, J.; Jiang, T.; Chowdhury, D.; et al. A genome-wide miRNA screen revealed miR-603 as a MGMT-regulating miRNA in glioblastomas. Oncotarget 2014, 5, 4026–4039. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2010, 17, 2141–2151. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; Greco, S.; Rameshwar, P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015, 6, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.G.; Tang, L. Conservation of the heterochronic regulator Lin-28, its developmental expression and microRNA complementary sites. Dev. Biol. 2003, 258, 432–442. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Daley, G.Q. Lin28: Primal regulator of growth and metabolism in stem cells. Cell Stem Cell 2013, 12, 395–406. [Google Scholar] [CrossRef]
- Jiang, S.; Baltimore, D. RNA-binding protein Lin28 in cancer and immunity. Cancer Lett. 2016, 375, 108–113. [Google Scholar] [CrossRef]
- Huang, X.A.; Lin, H. The microRNA regulation of stem cells. Wiley Interdisciplinary Reviews: Dev. Biol. 2012, 1, 83–95. [Google Scholar] [CrossRef]
- Cui, M.; Yao, X.; Lin, Y.; Zhang, D.; Cui, R.; Zhang, X. Interactive functions of microRNAs in the miR-23a-27a-24-2 cluster and the potential for targeted therapy in cancer. J. Cell Physiol. 2020, 235, 6–16. [Google Scholar] [CrossRef]
- Georgantas, R.W.; Hildreth, R.; Morisot, S.; Alder, J.; Liu, C.G.; Heimfeld, S.; Calin, G.A.; Croce, C.M.; Civin, C.I. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: A circuit diagram of differentiation control. Proc. Natl. Acad. Sci. USA 2007, 104, 2750–2755. [Google Scholar] [CrossRef]
- Nguyen, T.; Rich, A.; Dahl, R. MiR-24 promotes the survival of hematopoietic cells. PLoS ONE 2013, 8, e55406. [Google Scholar] [CrossRef]
- Yin, J.Y.; Tang, Q.; Qian, W.; Qian, J.; Lin, J.; Wen, X.M.; Zhou, J.D.; Zhang, Y.Y.; Zhu, X.W.; Deng, Z.Q. Increased expression of miR-24 is associated with acute myeloid leukemia with t (8; 21). Int. J. Clin. Exp. Pathol. 2014, 7, 8032–8038. [Google Scholar]
- Yuan, Y.; Kluiver, J.; Koerts, J.; de Jong, D.; Rutgers, B.; Razak, F.R.A.; Terpstra, M.; Plaat, B.E.; Nolte, I.M.; Diepstra, A.; et al. miR-24-3p is overexpressed in Hodgkin lymphoma and protects Hodgkin and Reed-Sternberg cells from apoptosis. Am. J. Pathol. 2017, 187, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; So, A.Y.L.; Sookram, R.; Wong, S.; Wang, J.K.; Ouyang, Y.; He, P.; Su, Y.; Casellas, R.; Baltimore, D. Epigenetic silencing of miR-125b is required for normal B-cell development. Blood 2018, 131, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. MiR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Croce, C.M. Micro RNA s play a central role in molecular dysfunctions linking inflammation with cancer. Immunol. Rev. 2013, 253, 167–184. [Google Scholar] [CrossRef]
- Nerlov, C. C/EBPα mutations in acute myeloid leukaemias. Nat. Rev. Cancer 2004, 4, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Porse, B.T.; Bryder, D.; Theilgaard-Mönch, K.; Hasemann, M.S.; Anderson, K.; Damgaard, I.; Jacobsen, S.E.W.; Nerlov, C. Loss of C/EBPα cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J. Exp. Med. 2005, 202, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.W.J.; Yuita, H.; Rao, A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 2019, 134, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef]
- He, N.; Chan, C.K.; Sobhian, B.; Chou, S.; Xue, Y.; Liu, M.; Alber, T.; Benkirane, M.; Zhou, Q. Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. USA 2011, 108, 636–645. [Google Scholar] [CrossRef]
- Prange, K.H.; Mandoli, A.; Kuznetsova, T.; Wang, S.Y.; Sotoca, A.M.; Marneth, A.E.; van der Reijden, B.A.; Stunnenberg, H.G.; Martens, J.H. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene 2017, 36, 3346–3356. [Google Scholar] [CrossRef]
- Mishra, A.; Garzon, R. The (miR) e of CTCL. Blood 2014, 123, 1438. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The miR-17-92 family of microRNA clusters in development and disease. Cancer J. (Sudbury Mass.) 2012, 18, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Dal Bo, M.; Bomben, R.; Hernández, L.; Gattei, V. The MYC/miR-17-92 axis in lymphoproliferative disorders: A common pathway with therapeutic potential. Oncotarget 2015, 6, 19381–19392. [Google Scholar] [CrossRef] [PubMed]
- Pospisil, V.; Vargova, K.; Kokavec, J.; Rybarova, J.; Savvulidi, F.; Jonasova, A.; Necas, E.; Zavadil, J.; Laslo, P.; Stopka, T. Epigenetic silencing of the oncogenic miR-17-92 cluster during PU. 1-directed macrophage differentiation. EMBO J. 2011, 30, 4450–4464. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.H.; Nielsen, A.L.; Helgstrand, C.; Lees, M.; Cloos, P.; Kastrup, J.S.; Helin, K.; Olsen, L.; Gajhede, M. Studies of H3K4me3 demethylation by KDM5B/Jarid1B/PLU1 reveals strong substrate recognition in vitro and identifies 2, 4-pyridine-dicarboxylic acid as an in vitro and in cell inhibitor. FEBS J. 2012, 279, 1905–1914. [Google Scholar] [CrossRef]
- Harmeyer, K.M.; Facompre, N.D.; Herlyn, M.; Basu, D. JARID1 histone demethylases: Emerging targets in cancer. Trends Cancer 2017, 3, 713–725. [Google Scholar] [CrossRef]
- Fang, L.L.; Wang, X.H.; Sun, B.F.; Zhang, X.D.; Zhu, X.H.; Yu, Z.J.; Luo, H. Expression, regulation and mechanism of action of the miR-17-92 cluster in tumor cells. Int. J. Mol. Med. 2017, 40, 1624–1630. [Google Scholar] [CrossRef]
- Xiang, J.; Wu, J. Feud or friend? The role of the miR-17-92 cluster in tumorigenesis. Curr. Genom. 2010, 11, 129–135. [Google Scholar] [CrossRef]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17~92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long noncoding RNA and cancer: A new paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef]
- Martínez-Barriocanal, Á.; Arango, D.; Dopeso, H. PVT1 long non-coding RNA in gastrointestinal cancer. Front. Oncol. 2020, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, R.; Wu, Y.; Liu, Y.; Su, W.; Xiong, W.; Zeng, Z. PVT1 promotes cancer progression via MicroRNAs. Front. Oncol. 2019, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Gadad, S.S.; Kim, D.S.; Kraus, W.L. Discovery, annotation, and functional analysis of long noncoding RNAs controlling cell-cycle gene expression and proliferation in breast cancer cells. Mol. Cell 2015, 59, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Braga, E.A.; Fridman, M.V.; Moscovtsev, A.A.; Filippova, E.A.; Dmitriev, A.A.; Kushlinskii, N.E. LncRNAs in ovarian cancer progression, metastasis, and main pathways: crRNA and alternative mechanisms. Int. J. Mol. Sci. 2020, 21, 8855. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huo, X.; Yang, X.R.; He, J.; Cheng, L.; Wang, N.; Deng, X.; Jin, H.; Wang, N.; Wang, C.; et al. STAT3-mediated upregulation of lncRNA HOXD-AS1 as a ceRNA facilitates liver cancer metastasis by regulating SOX4. Mol. Cancer 2017, 16, 136. [Google Scholar] [CrossRef]
- Chi, C.; Mao, M.; Shen, Z.; Chen, Y.; Chen, J.; Hou, W. HOXD-AS1 exerts oncogenic functions and promotes chemoresistance in cisplatin-resistant cervical cancer cells. Hum. Gene. Ther. 2018, 29, 1438–1448. [Google Scholar] [CrossRef]
- Yang, M.H.; Zhao, L.; Wang, L.; Ou-Yang, W.; Hu, S.S.; Li, W.L.; Ai, M.L.; Wang, Y.Q.; Han, Y.; Li, T.T.; et al. Nuclear lncRNA HOXD-AS1 suppresses colorectal carcinoma growth and metastasis via inhibiting HOXD3-induced integrin β3 transcriptional activating and MAPK/AKT signalling. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, M.; Lu, K.; Liu, J.; Zhang, M.; Wu, W.; De, W.; Wang, Z.; Wang, R. The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregualtion of p21WAF1/CIP1 expression. PLoS ONE 2013, 8, e77293. [Google Scholar] [CrossRef]
- Liu, X.H.; Sun, M.; Nie, F.Q.; Ge, Y.B.; Zhang, E.B.; Yin, D.D.; Kong, R.; Xia, R.; Lu, K.H.; Li, J.H.; et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol. Cancer 2014, 13, 92. [Google Scholar] [CrossRef]
- Xing, C.Y.; Hu, X.Q.; Xie, F.Y.; Yu, Z.J.; Li, H.Y.; Wu, J.B.; Tang, L.Y.; Gao, S.M. Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett. 2015, 589, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Mandal, S.S. LncRNA HOTAIR: A master regulator of chromatin dynamics and cancer. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, X.; Liang, H.; Jin, Z. Long non-coding RNA HOTAIR and STAT3 synergistically regulate the cervical cancer cell migration and invasion. Chem. Biol. Interact. 2018, 286, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Hajjari, M.; Salavaty, A. HOTAIR: An oncogenic long non-coding RNA in different cancers. Cancer Biol. Med. 2015, 12, 1–9. [Google Scholar]
- Li, J.; Chen, Y.; Chen, Z.; He, A.; Xie, H.; Zhang, Q.; Cai, Z.; Liu, Y.; Huang, W. SPRY4-IT1: A novel oncogenic long non-coding RNA in human cancers. Tumor. Biol. 2017, 39, 1010428317711406. [Google Scholar] [CrossRef]
- Shi, Y.; Li, J.; Liu, Y.; Ding, J.; Fan, Y.; Tian, Y.; Wang, L.; Lian, Y.; Wang, K.; Shu, Y. The long noncoding RNA SPRY4-IT1 increases the proliferation of human breast cancer cells by upregulating ZNF703 expression. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef]
- Pei, J.; Wang, B. Notch-1 promotes breast cancer cells proliferation by regulating LncRNA GAS5. Int. J. Clin. Exp. Med. 2015, 8, 14464–14471. [Google Scholar]
- Xu, C.; Zhang, Y.; Wang, Q.; Xu, Z.; Jiang, J.; Gao, Y.; Gao, M.; Kang, J.; Wu, M.; Xiong, J.; et al. Long non-coding RNA GAS5 controls human embryonic stem cell self-renewal by maintaining NODAL signalling. Nat. Commun. 2016, 7, 1–18. [Google Scholar] [CrossRef]
- Ji, J.; Dai, X.; Yeung, S.C.J.; He, X. The role of long non-coding RNA GAS5 in cancers. Cancer Manag. Res. 2019, 11, 2729. [Google Scholar] [CrossRef]
- Sang, Y.; Tang, J.; Li, S.; Li, L.; Tang, X.; Cheng, C.; Luo, Y.; Qian, X.; Deng, L.M.; Liu, L.; et al. LncRNA PANDAR regulates the G1/S transition of breast cancer cells by suppressing p16 INK4A expression. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Zou, Y.; Zhong, Y.; Wu, J.; Xiao, H.; Zhang, X.; Liao, X.; Li, J.; Mao, X.; Liu, Y.; Zhang, F. Long non-coding PANDAR as a novel biomarker in human cancer: A systemic review. Cell Prolif. 2018, 51, e12422. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Hatzimichael, E.; Dasoula, A.; Dranitsaris, G.; Tsiara, S.; Syrrou, M.; Georgiou, I.; Bourantas, K.L. CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leuk. Res. 2010, 34, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Modali, S.D.; Parekh, V.I.; Kebebew, E.; Agarwal, S.K. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol. Endocrinol. 2015, 29, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Zhang, S. Long non-coding RNA MEG-3 suppresses gastric carcinoma cell growth, invasion and migration via EMT regulation. Mol. Med. Rep. 2019, 20, 2685–2693. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Li, L.; Liu, Y.; Geng, P.; Li, G.; Song, H. LncRNA SNHG1 enhances cell proliferation, migration, and invasion in cervical cancer. Biochem. Cell Biol. 2018, 96, 38–43. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, G.; Ma, Y.; Qu, H. lncRNA CCAT1 contributes to the growth and invasion of gastric cancer via targeting miR-219-1. J. Cell Biochem. 2019, 120, 19457–19468. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Yang, F. The role of long non-coding RNA H19 in breast cancer. Oncol. Lett. 2020, 19, 7–16. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Esmaeili, M.; Taheri, M. H19 lncRNA: Roles in tumorigenesis. Biomed. Pharm. 2020, 123, 109774. [Google Scholar] [CrossRef]
- Hughes, J.M.; Legnini, I.; Salvatori, B.; Masciarelli, S.; Marchioni, M.; Fazi, F.; Morlando, M.; Bozzoni, I.; Fatica, A. C/EBPα-p30 protein induces expression of the oncogenic long non-coding RNA UCA1 in acute myeloid leukemia. Oncotarget 2015, 6, 18534–18544. [Google Scholar] [CrossRef]
- Sun, R.; Qin, C.; Jiang, B.; Fang, S.; Pan, X.; Peng, L.; Liu, Z.; Li, W.; Li, Y.; Li, G. Down-regulation of MALAT1 inhibits cervical cancer cell invasion and metastasis by inhibition of epithelial–mesenchymal transition. Mol. Biosyst. 2016, 12, 952–962. [Google Scholar] [CrossRef]
- Yang, L.; Bai, H.S.; Deng, Y.; Fan, L. High MALAT1 expression predicts a poor prognosis of cervical cancer and promotes cancer cell growth and invasion. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3187–3193. [Google Scholar] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Liu, S.; Lu, S.; Yu, X.; Lai, J.; Wu, Y.; Chen, S.; Wang, L.; Yu, Z.; Luo, G.; et al. The c-Myc-regulated lncRNA NEAT1 and paraspeckles modulate imatinib-induced apoptosis in CML cells. Mol. Cancer 2018, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, S.; Piao, H.Y.; Wang, Y.; Wu, Y.; Meng, X.Y.; Yang, D.; Zheng, Z.C.; Zhao, Y. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J. Physiol. Biochem. 2019, 75, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, M.; Niu, Q.; Zhang, F.; Yang, Y.; Jiang, X. Knockdown of long non-coding RNA ANRIL inhibits tumorigenesis in human gastric cancer cells via microRNA-99a-mediated down-regulation of BMI1. Braz. J. Med. Biol. Res. 2018, 51, e6839. [Google Scholar] [CrossRef]
- Song, Z.; Wu, W.; Chen, M.; Cheng, W.; Yu, J.; Fang, J.; Xu, L.; Yasunaga, J.I.; Matsuoka, M.; Zhao, T. Long noncoding RNA ANRIL supports proliferation of adult T-cell leukemia cells through cooperation with EZH2. J. Virol. 2018, 92, e00909-18. [Google Scholar] [CrossRef]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef]
- Sun, J.; Li, W.; Sun, Y.; Yu, D.; Wen, X.; Wang, H.; Cui, J.; Wang, G.; Hoffman, A.R.; Hu, J.F. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014, 42, 9588–9601. [Google Scholar] [CrossRef]
- Huppi, K.; Volfovsky, N.; Runfola, T.; Jones, T.L.; Mackiewicz, M.; Martin, S.E.; Mushinski, J.F.; Stephens, R.; Caplen, N.J. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol. Cancer Res. 2008, 6, 212–221. [Google Scholar] [CrossRef]
- Zheng, J.; Yu, F.; Dong, P.; Wu, L.; Zhang, Y.; Hu, Y.; Zheng, L. Long non-coding RNA PVT1 activates hepatic stellate cells through competitively binding microRNA-152. Oncotarget 2016, 7, 62886–62897. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Xu, L.J.; Zhao, L.F.; Jia, C.W.; Xu, M.Y. Long noncoding RNA PVT1 enhances the viability and invasion of papillary thyroid carcinoma cells by functioning as ceRNA of microRNA-30a through mediating expression of insulin like growth factor 1 receptor. Biomed. Pharm. 2018, 104, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Yan, X.; Li, Z.; Xu, X.; Mao, Q.; Ma, W.; Hong, Z.; Chen, X.; Yuan, Y. Long non-coding RNA PVT1 serves as a competing endogenous RNA for miR-186-5p to promote the tumorigenesis and metastasis of hepatocellular carcinoma. Tumor Biol. 2017, 39, 1010428317705338. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Song, Z.; Chen, H.; Chen, Z.; Yang, P.; Li, W.; Yang, Z.; Zhang, T.; Wang, F.; Wei, J.; et al. Long noncoding RNA PVT1-214 promotes proliferation and invasion of colorectal cancer by stabilizing Lin28 and interacting with miR-128. Oncogene 2019, 38, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Colombo, T.; Farina, L.; Macino, G.; Paci, P. PVT1: A rising star among oncogenic long noncoding RNAs. Biomed. Res. Int. 2015, 2015, 304208. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Cao, W.; de Oliveira Ribeiro, R.; Liu, D.; Saintigny, P.; Xia, R.; Xue, Y.; Lin, R.; Mao, L.; Ren, H. EZH2 promotes malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PLoS ONE 2012, 7, e52984. [Google Scholar] [CrossRef]
- Mozdarani, H.; Ezzatizadeh, V.; Rahbar Parvaneh, R. The emerging role of the long non-coding RNA HOTAIR in breast cancer development and treatment. J. Transl. Med. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Portoso, M.; Ragazzini, R.; Brenčič, Ž.; Moiani, A.; Michaud, A.; Vassilev, I.; Wassef, M.; Servant, N.; Sargueil, B.; Margueron, R. PRC 2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. 2017, 36, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Sui, Y.; Zheng, X. MiR-331-3p inhibits proliferation and promotes apoptosis by targeting HER2 through the PI3K/Akt and ERK1/2 pathways in colorectal cancer. Oncol. Rep. 2016, 35, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Y.; Yu, Q.M.; Du, Y.A.; Yang, L.T.; Dong, R.Z.; Huang, L.; Yu, P.F.; Cheng, X.D. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 2013, 9, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011, 147, 773–788. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef]
- Cimadamore, A.; Gasparrini, S.; Mazzucchelli, R.; Doria, A.; Cheng, L.; Lopez-Beltran, A.; Santoni, M.; Scarpelli, M.; Montironi, R. Long non-coding RNAs in prostate cancer with emphasis on second chromosome locus associated with prostate-1 expression. Front. Oncol. 2017, 7, 305. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, L.; Wang, Y.; Zhao, G.; Xie, R.; Liu, C.; Xiao, X.; Wu, K.; Nie, Y.; Zhang, H.; et al. KLF8 involves in TGF-beta-induced EMT and promotes invasion and migration in gastric cancer cells. J. Cancer Res. Clin. Oncol. 2013, 139, 1033–1042. [Google Scholar] [CrossRef]
- Alvarez-Dominguez, J.R.; Lodish, H.F. Emerging mechanisms of long noncoding RNA function during normal and malignant hematopoiesis. Blood 2017, 130, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, L.; Mathur, R.; Braun, F.K.; Wise, J.F.; Berkova, Z.; Neelapu, S.; Kwak, L.W.; Samaniego, F. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia 2014, 28, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Shao, Z. HOTAIR is upregulated in acute myeloid leukemia and that indicates a poor prognosis. Int. J. Clin. Exp. Pathol. 2015, 8, 7223–7228. [Google Scholar] [PubMed]
- Yao, H.; Duan, M.; Lin, L.; Wu, C.; Fu, X.; Wang, H.; Guo, L.; Chen, W.; Huang, L.; Liu, D.; et al. TET2 and MEG3 promoter methylation is associated with acute myeloid leukemia in a Hainan population. Oncotarget 2017, 8, 18337–18347. [Google Scholar] [CrossRef]
- Chen, L.; Wang, W.; Cao, L.; Li, Z.; Wang, X. Long non-coding RNA CCAT1 acts as a competing endogenous RNA to regulate cell growth and differentiation in acute myeloid leukemia. Mol. Cells 2016, 39, 330–336. [Google Scholar]
- Watanabe, T. Adult T-cell leukemia: Molecular basis for clonal expansion and transformation of HTLV-1–infected T cells. Blood 2017, 129, 1071–1081. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for cancer therapy: Hopes and challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicities. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotecnol. 2018, 46, 274–283. [Google Scholar] [CrossRef]
- Young, S.W.S.; Stenzel, M.; Jia-Lin, Y. Nanoparticle-siRNA: A potential cancer therapy? Crit. Rev. Oncol. Hematol. 2016, 98, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Huang, M.; Guo, W.W.; Huang, Q.; Zhang, L.Z.; Jiang, G. Nano-based delivery of RNAi in cancer therapy. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, L.; Yang, M. Lipid-based Vehicles for siRNA Delivery in Biomedical Field. Curr. Pharm. Biotechnol. 2020, 21, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.Y.; Guo, P.; Wen, W.C.; Wong, H.L. Lipid-based nanocarriers for RNA delivery. Curr. Pharm. Des. 2015, 21, 3140–3147. [Google Scholar] [CrossRef]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Sühs, K.W.; Stangel, M.; et al. Lipid nanoparticle-mediated siRNA delivery for safe targeting of human CML in vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.E.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 2010, 5, e8697. [Google Scholar] [CrossRef]
- Kaur, J.; Daoud, A.; Eblen, S.T. Targeting Chromatin Remodeling for Cancer Therapy. Curr. Mol. Pharmacol. 2019, 12, 215–229. [Google Scholar] [CrossRef]
- Gonzalez-Fierro, A.; Dueñas-González, A. Emerging DNA methylation inhibitors for cancer therapy: Challenges and prospects. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 27–35. [Google Scholar] [CrossRef]
- Giri, A.K.; Aittokallio, T. DNMT inhibitors increase methylation in the cancer genome. Front. Pharmacol. 2019, 10, 385. [Google Scholar] [CrossRef]
- Yamagishi, M.; Uchimaru, K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 2017, 29, 375–381. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Wang, W.T.; Han, C.; Sun, Y.M.; Chen, T.Q.; Chen, Y.Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Qian, X.; Wu, L.; Li, B.; Wang, Y.; Kong, X.; Xiong, L. microRNA: The impact of cancer stemness and therapeutic resistance. Cells 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, A.I.; Trinidad, J.R.; Sandiford, O.; Etchegaray, J.P.; Rameshwar, P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020, 39, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Yang, Y.A.; Zhang, A.; Fong, K.W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances ER signalling and confers tamoxifen resistance in breast cancer. Oncogene 2016, 35, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Balatti, V.; Nigita, G.; Veneziano, D.; Drusco, A.; Stein, G.S.; Messier, T.L.; Farina, N.H.; Lian, J.B.; Tomasello, L.; Liu, C.G.; et al. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 8071–8076. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Balatti, V.; Palamarchuk, A.; Rizzotto, L.; Veneziano, D.; Nigita, G.; Rassenti, L.Z.; Pass, H.I.; Kipps, T.J.; Liu, C.G.; et al. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5071–5076. [Google Scholar] [CrossRef]
- Veneziano, D.; Tomasello, L.; Balatti, V.; Palamarchuk, A.; Rassenti, L.Z.; Kipps, T.J.; Pekarsky, Y.; Croce, C.M. Dysregulation of different classes of tRNA fragments in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2019, 116, 24252–24258. [Google Scholar] [CrossRef]
- Park, J.; Ahn, S.H.; Shin, M.G.; Kim, H.K.; Chang, S. tRNA-derived small RNAs: Novel epigenetic regulators. Cancers 2020, 12, 2773. [Google Scholar] [CrossRef]
- Balatti, V.; Pekarsky, Y.; Croce, C.M. Role of the tRNA-derived small RNAs in cancer: New potential biomarkers and target for therapy. Adv. Cancer Res. 2017, 135, 173–187. [Google Scholar]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Rusyn, I. Environmental toxicants, epigenetics, and cancer. In Epigenetic Alterations in Oncogenesis; Springer: New York, NY, USA, 2013; pp. 215–232. [Google Scholar]
- Romani, M.; Pistillo, M.P.; Banelli, B. Environmental epigenetics: Crossroad between public health, lifestyle, and cancer prevention. Biomed. Res. Int. 2015, 2015, 587983. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA damage, mutagenesis and cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef]
- Lu, J.; Tan, T.; Zhu, L.; Dong, H.; Xian, R. Hypomethylation Causes MIR21 Overexpression in Tumors. Mol. Ther. Oncolytics 2020, 18, 47–57. [Google Scholar] [CrossRef]
- Ortiz, I.M.D.P.; Barros-Filho, M.C.; Dos Reis, M.B.; Beltrami, C.M.; Marchi, F.A.; Kuasne, H.; do Canto, L.M.; de Mello, J.B.H.; Abildgaard, C.; Pinto, C.A.L. Loss of DNA methylation is related to increased expression of miR-21 and miR-146b in papillary thyroid carcinoma. Clin. Epigenetics 2018, 10, 144. [Google Scholar] [CrossRef]
- Bhatia, V.; Yadav, A.; Tiwari, R.; Nigam, S.; Goel, S.; Carskadon, S.; Gupta, N.; Goel, A.; Palanisamy, N.; Ateeq, B. Epigenetic silencing of miRNA-338-5p and miRNA-421 drives SPINK1-positive prostate cancer. Clin. Cancer Res. 2019, 25, 2755–2768. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Tachiwana, H.; Yamamoto, T.; Saitoh, N. Gene regulation by non-coding RNAs in the 3D genome architecture. Curr. Opin. Genet. Dev. 2020, 61, 69–74. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Targets | Function | References |
|---|---|---|---|
| miR-29 a, b, c | DNMT3A and DNMT3B | Tumor suppression by repression of de novo DNA methylation. Protects tumor-suppressor genes from been silenced by DNA methylation. | Fabbri et al., 2007 [44] Suzuki et al., 2013 [47] |
| miR-148 | DNMT3B DNMT1 | Negative feedback loop between DNMT1 and miR-148 in AML. Inhibition of cell proliferation and increase of apoptosis. | Duursma et al., 2008 [45] Wang et al., 2019 [48] |
| miR-449a | HDAC1 | Inhibition of tumor growth, invasion and metastasis. Promotes apoptosis and differentiation. | Noonan et al., 2009 [49] Yong-Ming et al., 2017 [50] |
| miR-152 miR-185 miR-342 | DNMT1 | DNA hypomethylation. Promotes the expression of tumor-suppressor genes. | Suzuki et al., 2013 [47] |
| miR-26a miR-98 miR-124 miR-214 let-7 miR-101 miR-137 | EZH2 | Prevents the progression of prostate cancer and metastasis. | Suzuki et al., 2013 [47] |
| lncRNA | Origin/Location | Interactions with Epigenetic Regulators | Function | References |
|---|---|---|---|---|
| HOTAIR (HOX transcript antisense RNA) | Transcribed from antisense strand of homeobox C gene in chromosome 12 | PRC2 LSD1/CoREST | Gene silencing by methylation of H3K27me3 and demethylation of H3K4me2 | Cai et al., 2014 [65] |
| SCHLAP1 (second chromosome locus associated with prostate-1) | From chromosome 2 | SWI/SNF | Partially antagonizes location and function of SWI/SNF | Raab et al., 2019 [67] |
| NEAT1 (nuclear paraspeckle assembly transcript 1) | Transcribed from the multiple endocrine neoplasia locus in chromosome 11 | Subpopulation of SWI/SNF complexes | Nuclear paraspeckle (nuclear bodies) assembly | Neve et al., 2018 [69] |
| XIST (X-inactive specific transcript) | Chromosome X | PRC1 | Silencing one pair of X chromosomes | Pintacuda et al., 2017 [63] |
| ANRIL (antisense non-coding RNA in the INK4 locus) | Transcribed from the CDKN2A/B gene cluster at chromosome 9 in the antisense direction | PRC1 (CBX7), PRC2 (SUZ12) | Transcriptional repression | Chi et al., 2017 [71] |
| GAS5 Growth arrest-specific 5) | From chromosome 1 | PRC2 | Repression of glucocorticoids receptors, IRF4 (interferon regulatory factor 4) | Wang et al., 2018 [73] |
| MEG3 (maternally expressed 3) | Maternally expressed, generates multiple isoforms by alternative splicing, from chromosome 14 | JARID2, EZH2 | Transcriptional repression | Wang et al., 2018 [73] |
| PVT1 (plasmacytoma variant translocation 1) | From chromosome 8 | PRC2 (EZH2) | Oncogene | Yu et al., 2018 [75] |
| MALAT1 (metastasis associated lung adenocarcinoma transcript 1) | Also known as NEAT2 (non-coding nuclear-enriched abundant transcript 2). Infrequently spliced ncRNA, from chromosome 11 | PRC2 (EZH2), HDAC9, BRG1 | Tumorigenesis Vascular disease | Wang et al., 2018 [73] Cardenas et al., 2018 [77] |
| KCNQ1OT1 (KCNQ1 overlapping transcript 1) | Part of an imprinting control region in chromosome 11 | G9a, PRC2 (EZH2) | Gene silencing by H3K9me2 H3K27me3 | Wang et al., 2018 [73] |
| H19 (H19 imprinted maternally expressed transcript) | From imprinted region in chromosome 11 | SAHH, PRC2 (EZH2) | Tumor-suppressor Oncogene | Zhou et al., 2015 [76] |
| UCA1 (urothelial cancer associated 1) | From chromosome 19 | PRC2 (EZH2), SWI/SNF | Tumorigenesis | Neve et al., 2018 [69] |
| PANDAR (promoter of CDKN1A antisense DNA damage activated RNA) | From chromosome 6 | PRC1 PRC2 | Tumorigenesis | Puvvula et al., 2014 [78] |
| miRNA | Cancer/Disease Involvement | References |
|---|---|---|
| miR-15b miR-16 | Upregulated in gastric cancer and downregulated in chronic lymphocytic leukemia (CLL) | Xia et al., 2008 [79] Cimmino et al., 2005 [80] Xia et al., 2008 [79] |
| LET-7 | Downregulated in lung, pancreatic cancer and acute lymphoblastic leukemia (ALL) | Takamizawa et al., 2004 [81] Kugel et al., 2016 [82] |
| miR-34 (a, b and c) | Downregulated in gastric and cervical cancer neuroblastoma. Upregulated in glioblastoma multiforme (GBM) (miR-34b) and colorectal cancer (miR-34a) | Zhang and Liao, 2019 [83] He et al., 2009 [84], Hermeking et al., 2012 [85] Bommer et al., 2007 [86] Tarasov et al., 2007 [87] He et al., 2009 [84] Hasakova et al., 2019 [88] Han et al., 2002 [89] |
| miR-21 | Upregulated in GBM, solid tumors and multiple myeloma | Kumarswamy et al., 2011 [90] Asangani et al., 2008 [91] Wang et al., 2019 [92] Jesionek-Kupnicka et al., 2019 [93] Pfeffer et al., 2015 [94] |
| miR-125 (a and b) | Upregulated in AML and GBM (miR-125b) | Bousquet et al., 2010 [95] Chaudhuri et al., 2012 [96] Wu et al., 2013 [97] Romero et al., 2015 [98] Liu et al., 2017 [99] Jesionek-Kupnicka et al., 2019 [93] |
| miR-181d | Downregulated in GBM | Zhang et al., 2012 [100] Yang et al., 2018 [101] Jesionek-Kupnicka et al., 2019 [93] |
| miR-648 | Downregulated in GBM | Kreth et al., 2013 [102] Jesionek-Kupnicka et al., 2019 [93] |
| miR-155 | Upregulated in AML, colorectal cancer and Hodgkin’s lymphoma | Fabbri et al., 2008 [103] Narayan et al., 2017 [104] Witten and Slack, 2020 [105] Kluiver et al., 2005 [106] Narayan et al., 2018 [107] Eis et al., 2005 [108] |
| miR-221 | Upregulated in GBM | Lukiw et al., 2009 [109] |
| miR-30a-5p | Downregulated in colorectal cancer | Wei et al., 2016 [110] |
| miR-29 family | Upregulated in colorectal and cervical cancer and downregulated in lung cancer and AML | Fabbri et al., 2007 [44] Jiang et al., 2014 [111] |
| miR-145 | Downregulated in colorectal cancer | Michael et al., 2003 [112] Sheng et al., 2017 [113] |
| miR-128a | Upregulated in AML | De Luca et al., 2017 [114] |
| miR-17/92 cluster | Upregulated and downregulated in myeloid leukemias and upregulated in colorectal cancer and CLL | Fabbri et al., 2008 [103] Jiang et al., 2011 [115] Moussay et al., 2011 [116] Willimott and Wagner, 2012 [117] He et al., 2013 [118] |
| miR-7 | Downregulated in GBM | Luo et al., 2015 [119] |
| miR-185 | Downregulated in GBM | Zhang et al., 2011 [120] |
| miR-24a | Upregulated in AML | Fabbri et al., 2008 [103] |
| miR-200 | Downregulated in breast cancer | Mekala et al., 2018 [121] |
| miR-150 | Downregulated in CTCL, AML | Jiang et al., 2012 [122] Ito et al., 2014 [123] Abe et al., 2017 [124] |
| lncRNA | Cancer Involvement | References |
|---|---|---|
| PVT1 | Gastrointestinal, renal, breast cancer, acute promyelocytic leukemia | Martínez-Barriocanal et al., 2020 [171] Wang et al., 2019 [172] Sun et al., 2015 [173] Zeng et al., 2015 [174] |
| HOXD1-AS1 | Bladder, cervical, gastric, ovarian, colorectal, prostate, GBM, melanoma, osteosarcoma, liver and non-small-cell lung cancers | Braga et al., 2020 [175] Wang et al., 2017 [176] Chi et al., 2018 [177] Yang et al., 2019 [178] |
| HOTAIR | Pancreatic, cervical, breast, lung, oral, gastric cancers, AML | Liu et al., 2013 [179] Liu et al., 2014 [180] Xing et al., 2015 [181] Bhan et al., 2015 [182] Zhang et al., 2018 [183] Hajjari and Salavaty, 2015 [184] |
| SPRY4-IT1 | Breast and cervical cancer | Li et al., 2017 [185] Shi et al., 2015 [186] |
| GAS5 | Breast, lung, prostate cancer, blood | Pei et al., 2015 [187] Xu et al., 2016 [188] Ji et al., 2019 [189] |
| PANDAR | Breast, gastric, colorectal, bladder cancer | Sang et al., 2016 [190] Zou et al., 2018 [191] |
| MEG3 | Gastric and pancreatic cancer, AML | Benetatos et al., 2010 [192] Modali et al., 2015 [193] Jiao et al., 2019 [194] Bhan et al., 2017 [170] |
| SNHG1 | Cervical cancer | Liu et al., 2018 [195] |
| CCAT-1 | Colon, gastric cancer, AML | Li et al., 2019 [196] |
| H19 | Breast, gastric cancer | Wang et al., 2020 [197] Ghafouri-Fard et al., 2020 [198] |
| UCA1 | Pancreatic, colorectal cancer, AML | Neve et al., 2018 [69] Hughes et al., 2015 [199] |
| MALAT1 | Lung, cervical, breast cancer, lymphoblastic leukemia | Sun et al., 2016 [200] Yang et al., 2015 [201] Tripathi et al., 2010 [202] |
| SChLAP1 | Prostate cancer | Prensner et al., 2013 [66] |
| NEAT1 | Breast, gastric, colorectal cancer, acute promyelocytic leukemia | Zeng et al., 2014 [203] Zhang et al., 2019 [204] |
| ANRIL | Gastric cancer, breast cancer, adult T-cell leukemia | Meseure et al., 2016 [72] Liu et al., 2018 [205] Song et al., 2018 [206] |
| LUNAR1 | T-ALL, lymphoblastic leukemia | Trimarchi et al., 2014 [207] |
| IRAIN | AML | Sun et al., 2014 [208] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.-P. Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers 2020, 12, 3657. https://doi.org/10.3390/cancers12123657
Kumar S, Gonzalez EA, Rameshwar P, Etchegaray J-P. Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers. 2020; 12(12):3657. https://doi.org/10.3390/cancers12123657
Chicago/Turabian StyleKumar, Subhasree, Edward A. Gonzalez, Pranela Rameshwar, and Jean-Pierre Etchegaray. 2020. "Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies" Cancers 12, no. 12: 3657. https://doi.org/10.3390/cancers12123657
APA StyleKumar, S., Gonzalez, E. A., Rameshwar, P., & Etchegaray, J.-P. (2020). Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers, 12(12), 3657. https://doi.org/10.3390/cancers12123657