Apigenin Increases SHIP-1 Expression, Promotes Tumoricidal Macrophages and Anti-Tumor Immune Responses in Murine Pancreatic Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
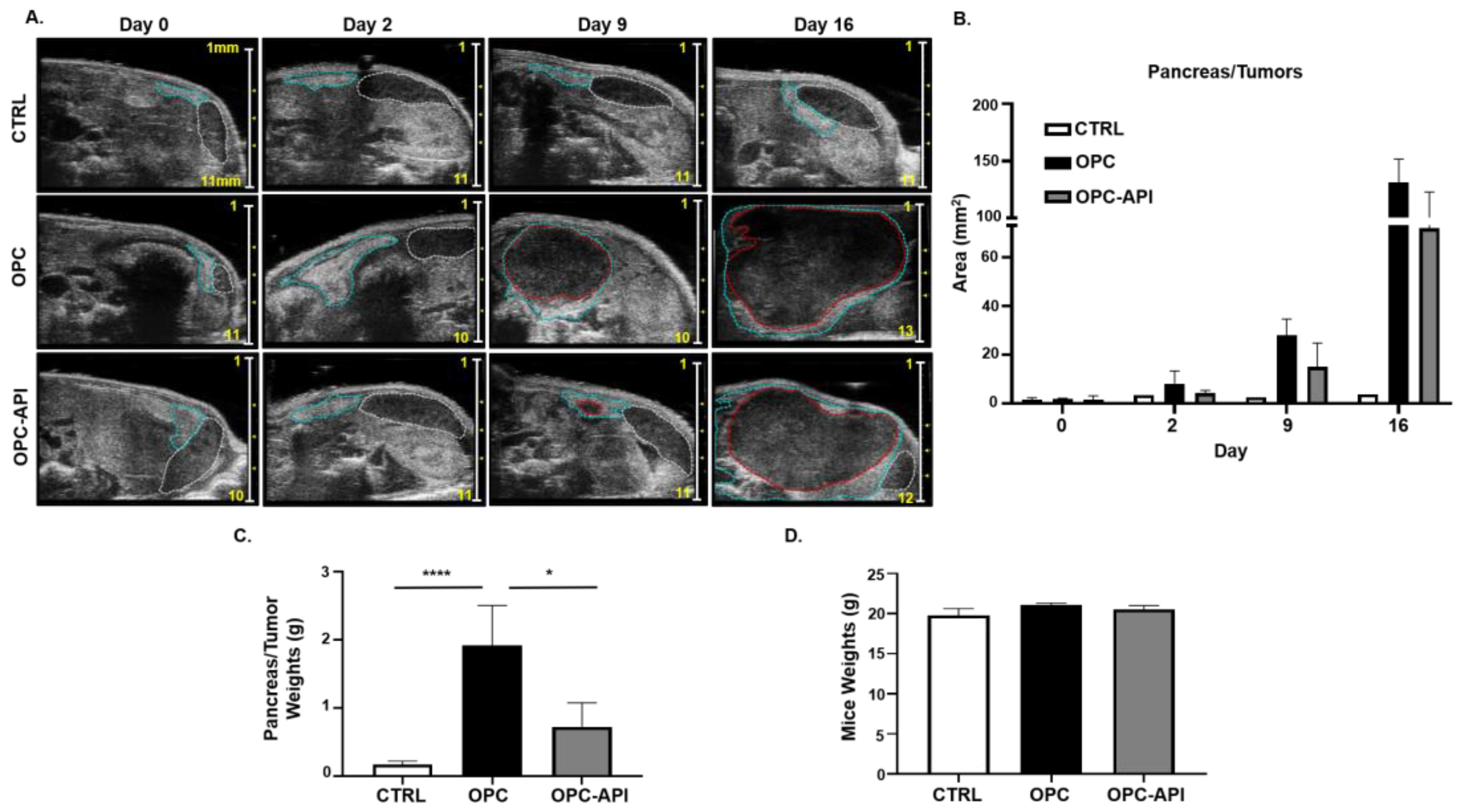
2.1. API Reduces Tumor Burden in an Orthotopic Mouse Model of PC (OPC)
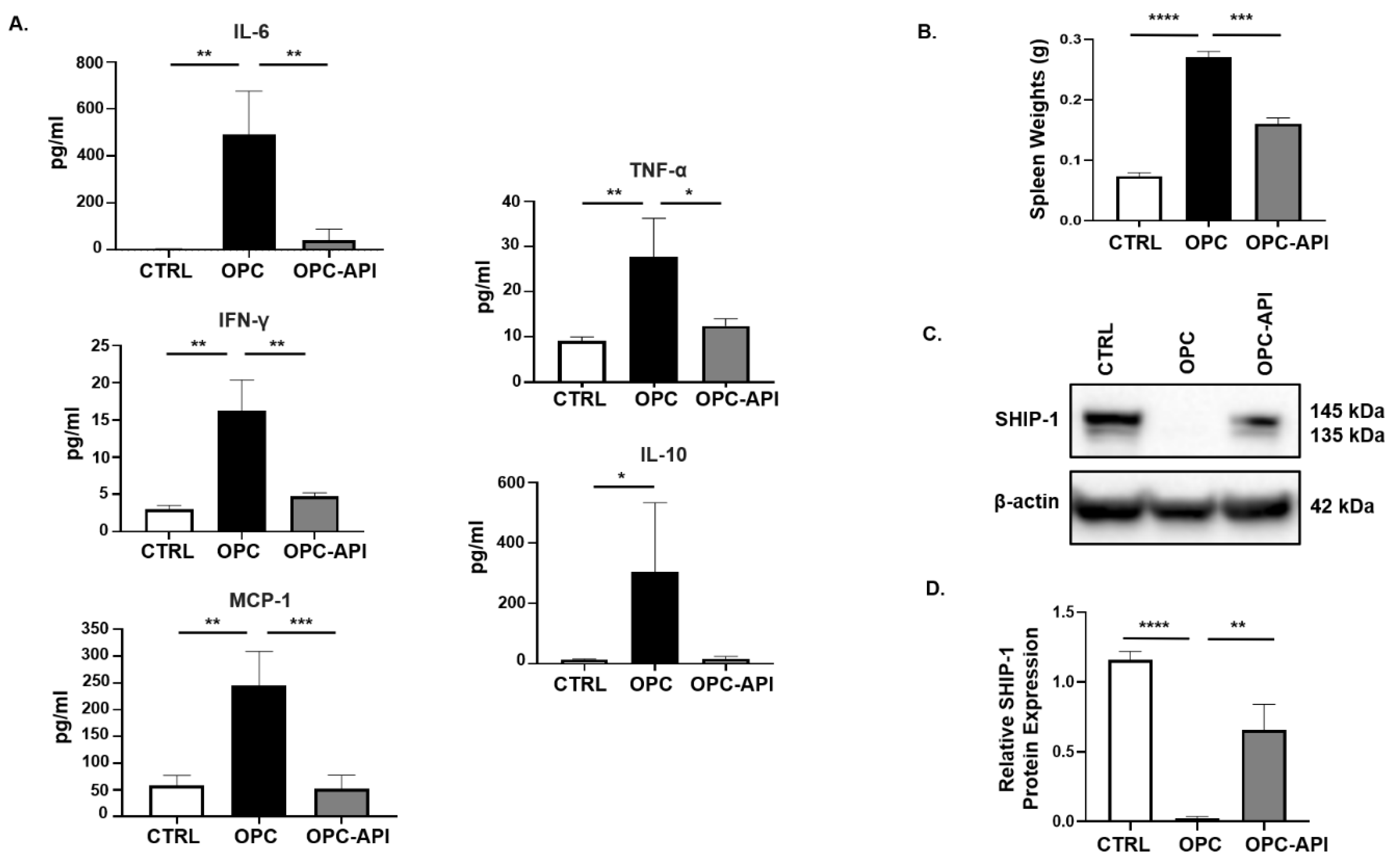
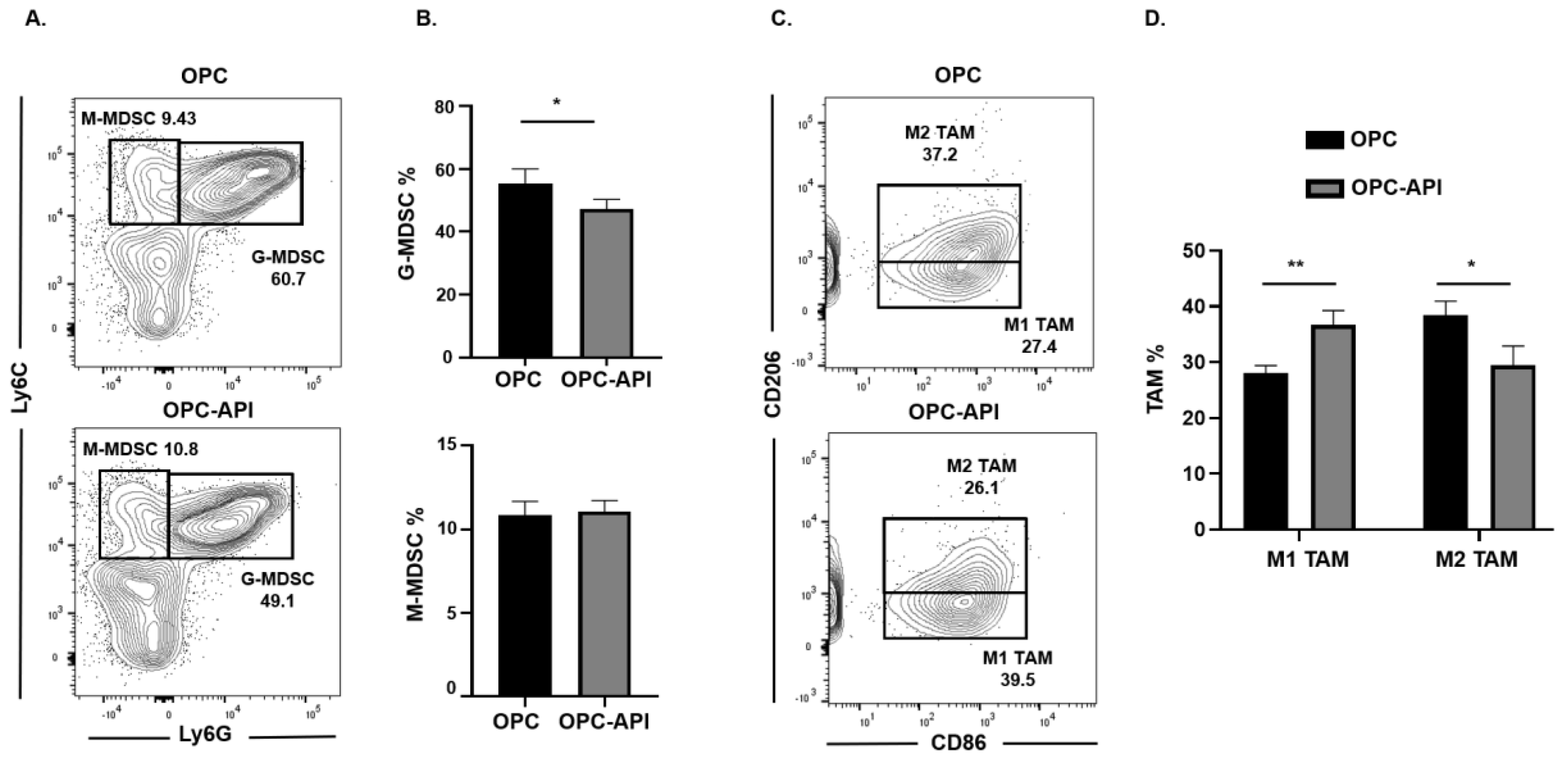
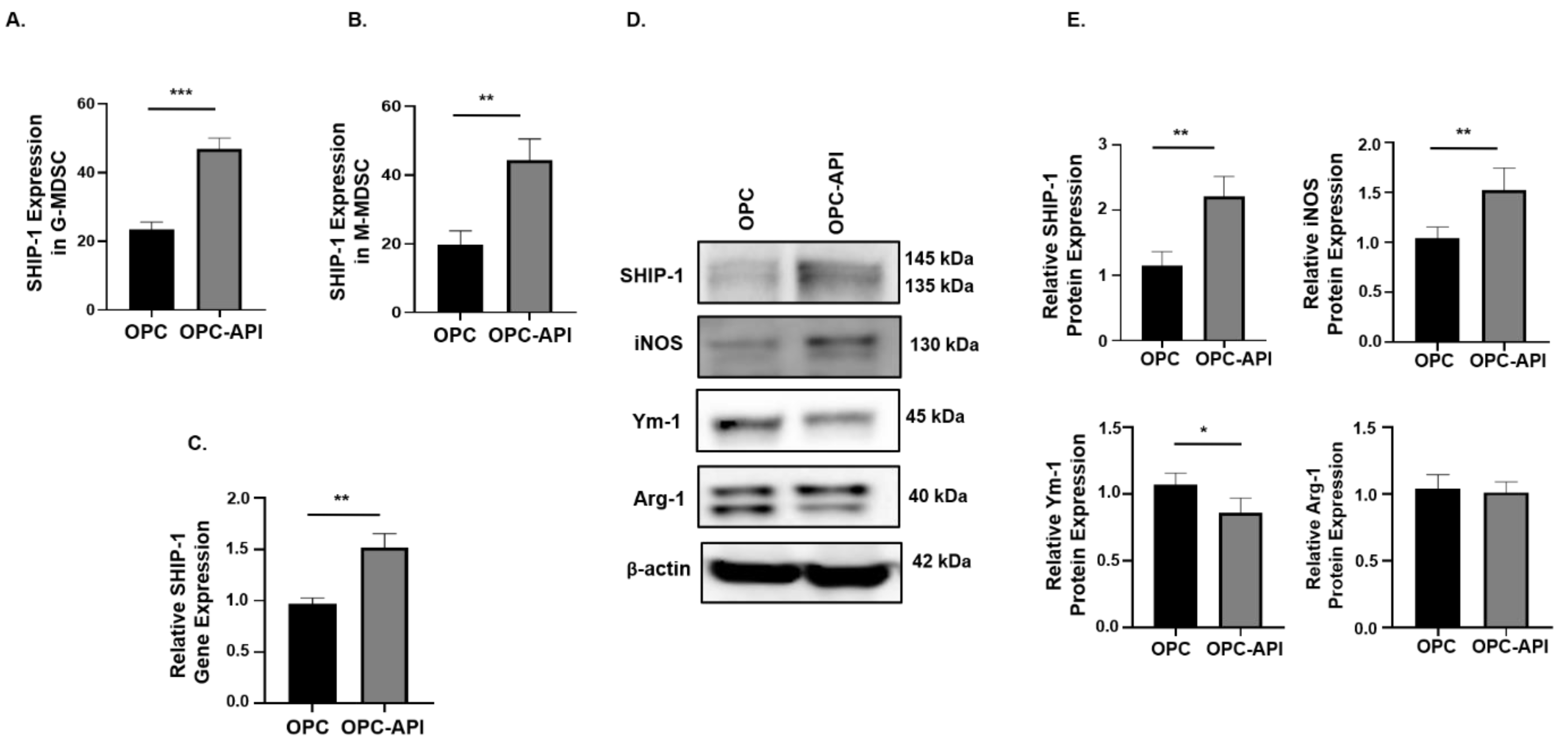
2.2. API Suppresses Inflammation, Induces SHIP-1 Expression, and Reduces MDSC Expansion in OPC Mice
2.3. API Increases M1 TAM in the TME of OPC Mice
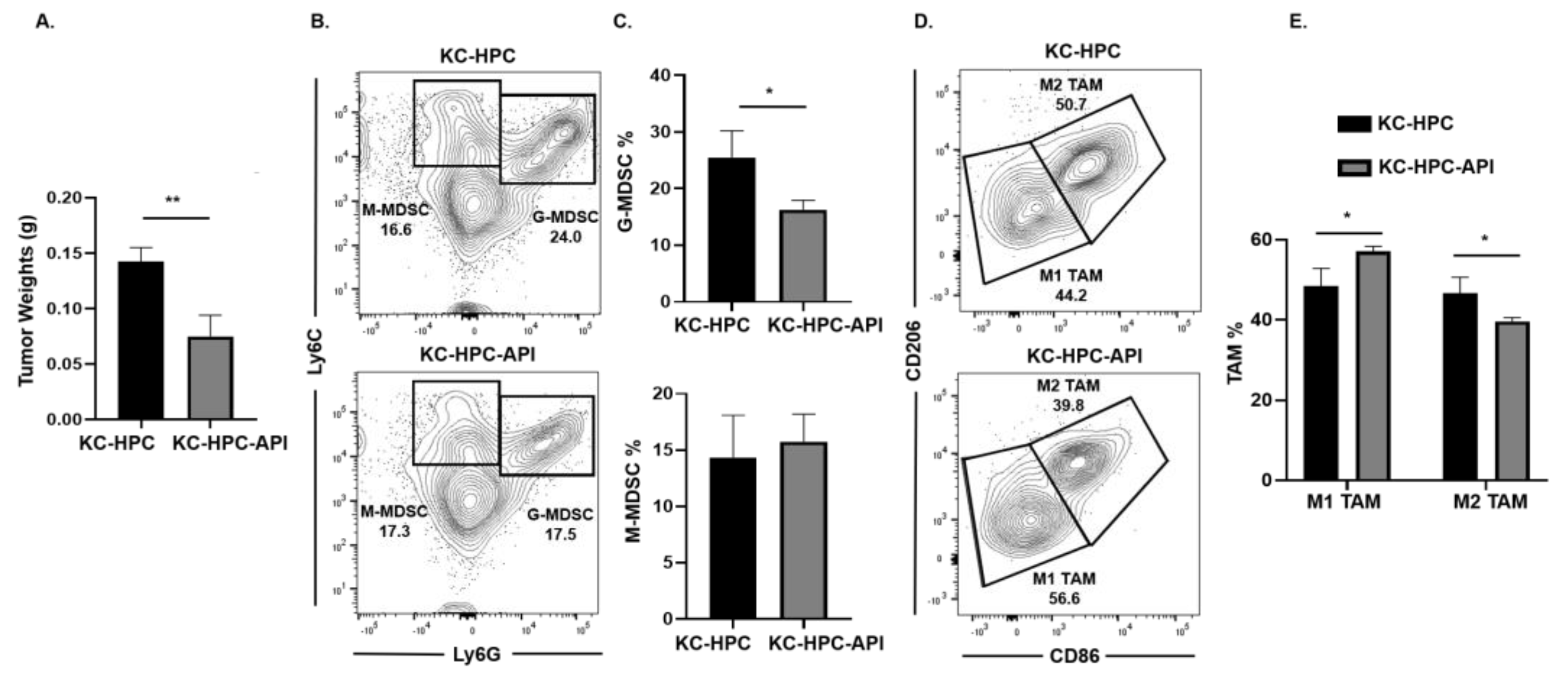
2.4. API Increases M1 TAM in Heterotopic KC-PC Mice
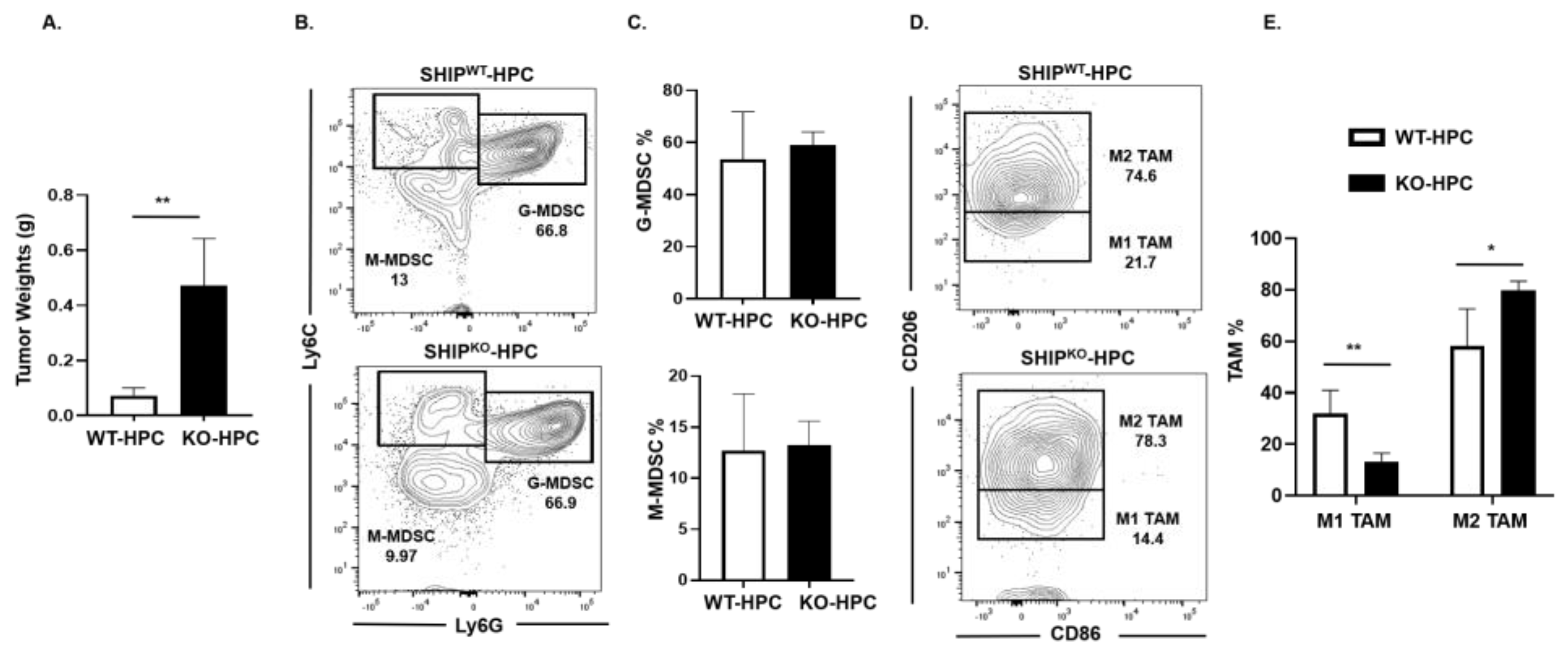
2.5. M1 TAM are Reduced in Heterotopic PC SHIPKO Mice
2.6. API Induces SHIP-1 Expression and Skews TAM to an M1 Phenotype in the TME of OPC Mice
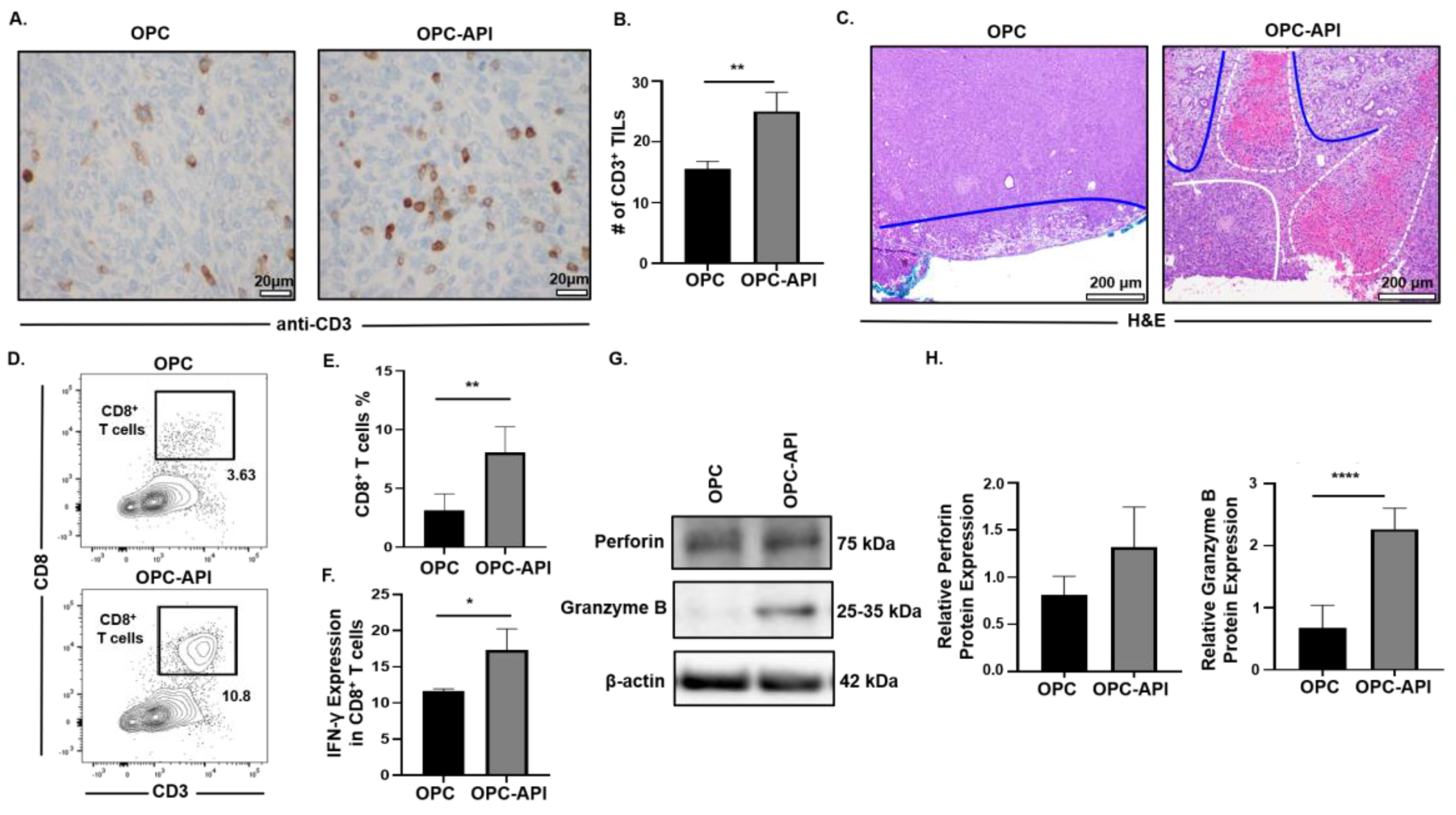
2.7. API Improves Anti-Tumor Immune Responses in the TME of OPC Mice
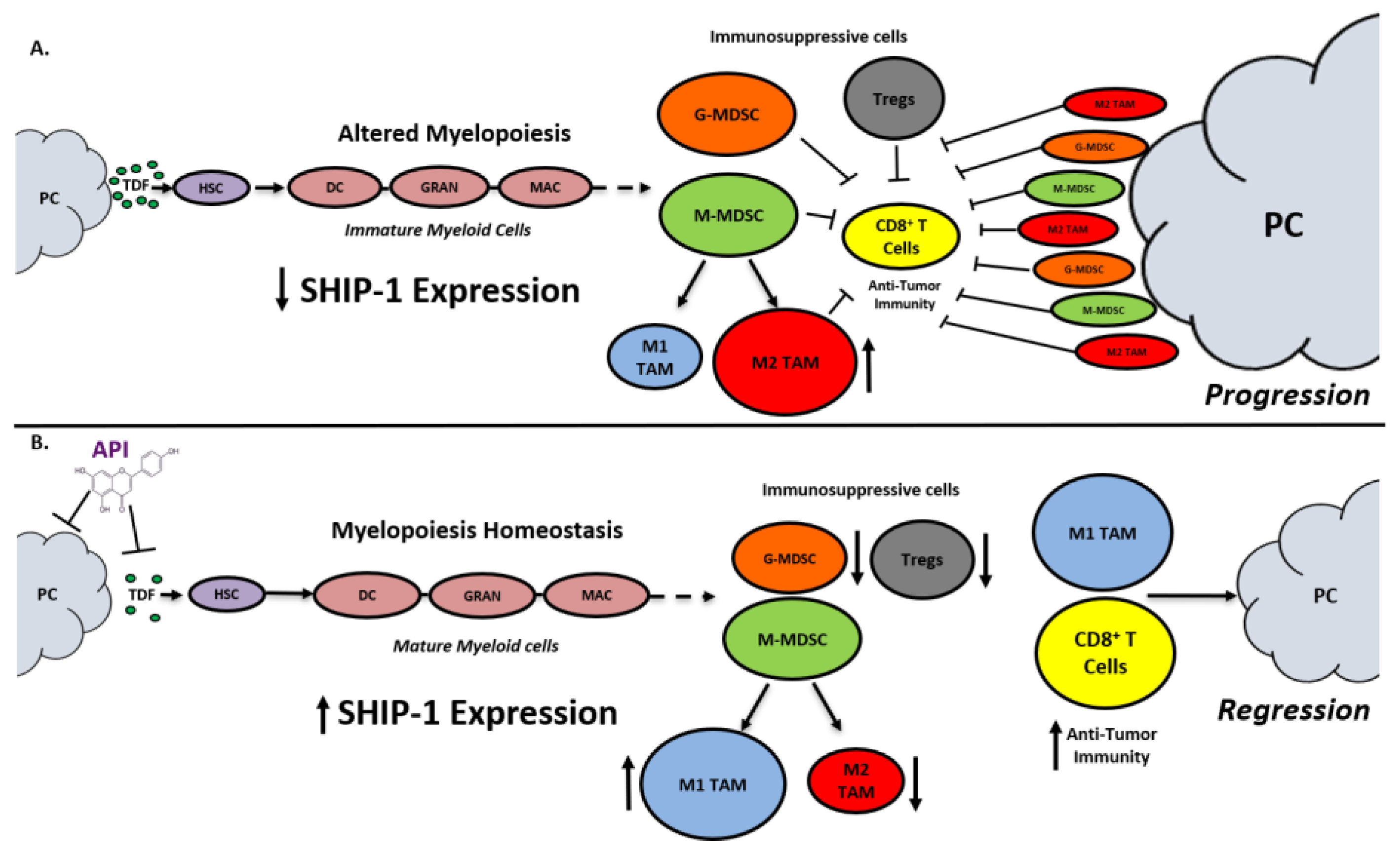
3. Discussion
4. Materials and Methods
4.1. Pancreatic Cancer Cell Lines
4.2. Bioflavonoid
4.3. Pancreatic Cancer Murine Models
4.4. Ultrasound Imaging
4.5. Flow Cytometry
4.6. T Cell Activation Assay
4.7. Western Blot
4.8. Quantitative PCR (qPCR)
4.9. Cytometric Bead Array
4.10. Immunohistochemistry Staining
4.11. Indirect Immunofluorescence
4.12. Hematoxylin and Eosin Staining
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| API | Apigenin |
| Bcl-2 | B-cell lymphoma 2 |
| CK2 | Casein kinase II |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMSO | Dimethyl sulfoxide |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| HPC | Heterotopic pancreatic cancer |
| Inf-MoDC | Inflammatory monocyte-derived dendritic cell |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MDSC | Myeloid-derived suppressor cells |
| OPC | Orthotopic pancreatic cancer |
| PI3K | Phosphatidylinositol 3-kinase |
| RIPA | Radioimmunoprecipitation assay |
| SHIP-1 | Src homology 2 (SH2) domain-containing inositol 5′-phosphatase-1 |
| TAM | Tumor-associated macrophages |
| TDF | Tumor-derived factors |
| TIL | Tumor infiltrating lymphocytes |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
References
- Mizrahi:, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Lafaro, K.J.; Melstrom, L.G. The paradoxical web of pancreatic cancer tumor microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and pancreatic cancer: Unique challenges and potential opportunities. Ther. Adv. Med Oncol. 2018, 10, 1758835918816281. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Valilou, S.F.; Keshavarz-Fathi, M.; Silvestris, N.; Argentiero, A.; Rezaei, N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018, 39, 46–61. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, D.R.; Cripe, J.C.; Hawkins, W.; Goedegebuure, S. Radioprotective properties of myeloid-derived suppressor cells. Transl. Cancer Res. 2016, 5, S923–S925. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Ho, V.W.; Kuroda, E.; Ruschmann, J.; Antignano, F.; Lam, V.; Krystal, G. Role of SHIP in cancer. Exp. Hematol. 2011, 39, 2–13. [Google Scholar] [CrossRef]
- Antignano, F.; Ibaraki, M.; Kim, C.; Ruschmann, J.; Zhang, A.; Helgason, C.D.; Krystal, G. SHIP is required for dendritic cell maturation. J. Immunol. 2010, 184, 2805–2813. [Google Scholar] [CrossRef]
- Kalesnikoff, J.; Sly, L.M.; Hughes, M.R.; Buchse, T.; Rauh, M.J.; Cao, L.P.; Lam, V.; Mui, A.; Huber, M.; Krystal, G. The role of SHIP in cytokine-induced signaling. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 87–103. [Google Scholar] [CrossRef]
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Sly, L.M.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP represses the generation of alternatively activated macrophages. Immunity 2005, 23, 361–374. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Nelson, N.; Vohra, N.; Jerald, M.; Pendleton, L.; Szekeres, K.; Ghansah, T. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS ONE 2011, 6, e27729. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Gloire, G.; Piette, J. Enzymatic and non-enzymatic activities of SHIP-1 in signal transduction and cancer. Biochem. Pharmacol. 2011, 82, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.; Pohnert, S.; Shelton, J.; Franklin, R.; Bertrand, F.; McCubrey, J. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Futami, M.; Carroll, M.; Feng, Y.; Wang, Z.; Fernandez, M.; Whichard, Z.; Chen, Y.; Kornblau, S.; Shpall, E.J.; et al. Loss of SHIP-1 protein expression in high-risk myelodysplastic syndromes is associated with miR-210 and miR-155. Oncogene 2012, 31, 4085–4094. [Google Scholar] [CrossRef]
- Krystal, G.; Hamilton, M.J.; Bennewith, K.L. SHIP prevents metastasis. Aging (Albany NY) 2016, 8, 837–838. [Google Scholar] [CrossRef][Green Version]
- Ghansah, T.; Paraiso, K.H.; Highfill, S.; Desponts, C.; May, S.; McIntosh, J.K.; Wang, J.W.; Ninos, J.; Brayer, J.; Cheng, F.; et al. Expansion of myeloid suppressor cells in SHIP-deficient mice represses allogeneic T cell responses. J. Immunol. 2004, 173, 7324–7330. [Google Scholar] [CrossRef]
- Ghansah, T. A novel strategy for modulation of MDSC to enhance cancer immunotherapy. Oncoimmunology 2012, 1, 984–985. [Google Scholar] [CrossRef][Green Version]
- Fendrich, V. Chemoprevention of pancreatic cancer—one step closer. Langenbeck’s Arch. Surg. 2012, 397, 495–505. [Google Scholar] [CrossRef]
- Orfali, G.; Duarte, A.C.; Bonadio, V.; Martinez, N.P.; de Araujo, M.E.; Priviero, F.B.; Carvalho, P.O.; Priolli, D.G. Review of anticancer mechanisms of isoquercitin. World J. Clin. Oncol. 2016, 7, 189–199. [Google Scholar] [CrossRef]
- Jiang, B.-H.; Qiu, J.-G.; Wang, L.; Liu, W.-J.; Wang, J.-F.; Zhao, E.-J.; Zhou, F.-M.; Ji, X.-B.; Wang, L.-H.; Xia, Z.-K. Apigenin inhibits IL-6 transcription and suppresses esophageal carcinogenesis. Front. Pharmacol. 2019, 10, 1002. [Google Scholar]
- Chen, X.J.; Wu, M.Y.; Li, D.H.; You, J. Apigenin inhibits glioma cell growth through promoting microRNA-16 and suppression of BCL-2 and nuclear factor-κB/MMP-9. Mol. Med. Rep. 2016, 14, 2352–2358. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Gonzalez de Mejia, E. Interactions between dietary flavonoids apigenin or luteolin and chemotherapeutic drugs to potentiate anti-proliferative effect on human pancreatic cancer cells, in vitro. Food Chem. Toxicol. 2013, 60, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin: A promising molecule for cancer prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; de Mejia, E.G. Flavonoid apigenin modified gene expression associated with inflammation and cancer and induced apoptosis in human pancreatic cancer cells through inhibition of GSK-3beta/NF-kappaB signaling cascade. Mol. Nutr. Food Res. 2013, 57, 2112–2127. [Google Scholar] [CrossRef]
- King, J.C.; Lu, Q.Y.; Li, G.; Moro, A.; Takahashi, H.; Chen, M.; Go, V.L.; Reber, H.A.; Eibl, G.; Hines, O.J. Evidence for activation of mutated p53 by apigenin in human pancreatic cancer. Biochim. Biophys. Acta 2012, 1823, 593–604. [Google Scholar] [CrossRef]
- Madunić, J.; Madunić, I.V.; Gajski, G.; Popić, J.; Garaj-Vrhovac, V. Apigenin: A dietary flavonoid with diverse anticancer properties. Cancer Lett. 2018, 413, 11–22. [Google Scholar] [CrossRef]
- Nelson, N.; Szekeres, K.; Iclozan, C.; Rivera, I.O.; McGill, A.; Johnson, G.; Nwogu, O.; Ghansah, T. Apigenin: Selective CK2 inhibitor increases Ikaros expression and improves T cell homeostasis and function in murine pancreatic cancer. PLoS ONE 2017, 12, e0170197. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic. Biol. Med. 2008, 44, 1833–1845. [Google Scholar] [CrossRef]
- Mukherjee, O.; Weingarten, L.; Padberg, I.; Pracht, C.; Sinha, R.; Hochdörfer, T.; Kuppig, S.; Backofen, R.; Reth, M.; Huber, M. The SH2-domain of SHIP1 interacts with the SHIP1 C-terminus: Impact on SHIP1/Ig-α interaction. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 206–214. [Google Scholar] [CrossRef]
- Torres, M.P.; Rachagani, S.; Souchek, J.J.; Mallya, K.; Johansson, S.L.; Batra, S.K. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: Applications in diagnosis and therapy. PLoS ONE 2013, 8, e80580. [Google Scholar] [CrossRef]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Sasaki, S.-i.; Baba, T. Two-Faced Roles of Tumor-Associated Neutrophils in Cancer Development and Progression. Int. J. Mol. Sci. 2020, 21, 3457. [Google Scholar] [CrossRef] [PubMed]
- Collazo, M.M.; Wood, D.; Paraiso, K.H.; Lund, E.; Engelman, R.W.; Le, C.-T.; Stauch, D.; Kotsch, K.; Kerr, W.G. SHIP limits immunoregulatory capacity in the T-cell compartment. Blood 2009, 113, 2934–2944. [Google Scholar] [CrossRef]
- Varone, A.; Spano, D.; Corda, D. Shp1 in solid cancers and their therapy. Front. Oncol. 2020, 10, 935. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, S.; Huang, Y.; Song, L.; Yin, Y.; Kong, W.; Chen, X.; Ouyang, X. Expression and prognosis value of SHP2 in patients with pancreatic ductal adenocarcinoma. Tumor Biol. 2016, 37, 7853–7859. [Google Scholar] [CrossRef]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1-and RAS-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Ruckert, M.T.; de Andrade, P.V.; Santos, V.S.; Silveira, V.S. Protein tyrosine phosphatases: Promising targets in pancreatic ductal adenocarcinoma. Cell. Mol. Life Sci. 2019, 76, 2571–2592. [Google Scholar] [CrossRef]
- Saurer, L.; Reber, P.; Schaffner, T.; Büchler, M.W.; Buri, C.; Kappeler, A.; Walz, A.; Friess, H.; Mueller, C. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. Gastroenterology 2000, 118, 356–367. [Google Scholar] [CrossRef]
- Singh, P.; Mishra, S.K.; Noel, S.; Sharma, S.; Rath, S.K. Acute exposure of apigenin induces hepatotoxicity in Swiss mice. PLoS ONE 2012, 7, e31964. [Google Scholar] [CrossRef]
- Caltagirone, S.; Rossi, C.; Poggi, A.; Ranelletti, F.O.; Natali, P.G.; Brunetti, M.; Aiello, F.B.; Piantelli, M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int. J. Cancer 2000, 87, 595–600. [Google Scholar] [CrossRef]
- Ai, X.-Y.; Qin, Y.; Liu, H.-J.; Cui, Z.-H.; Li, M.; Yang, J.-H.; Zhong, W.-L.; Liu, Y.-R.; Chen, S.; Sun, T. Apigenin inhibits colonic inflammation and tumorigenesis by suppressing STAT3-NF-κB signaling. Oncotarget 2017, 8, 100216. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Bhaskaran, N.; Babcook, M.A.; Fu, P.; MacLennan, G.T.; Gupta, S. Apigenin inhibits prostate cancer progression in TRAMP mice via targeting PI3K/Akt/FoxO pathway. Carcinogenesis 2014, 35, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, J.-L.; Liu, R.; Li, X.-X.; Li, J.-F.; Zhang, L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules 2013, 18, 9949–9965. [Google Scholar] [CrossRef] [PubMed]
- Corbett, T.H.; Roberts, B.J.; Leopold, W.R.; Peckham, J.C.; Wilkoff, L.J.; Griswold, D.P., Jr.; Schabel, F.M., Jr. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984, 44, 717–726. [Google Scholar]
- Helgason, C.D.; Damen, J.E.; Rosten, P.; Grewal, R.; Sorensen, P.; Chappel, S.M.; Borowski, A.; Jirik, F.; Krystal, G.; Humphries, R.K. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998, 12, 1610–1620. [Google Scholar] [CrossRef]
- Tsantikos, E.; Lau, M.; Castelino, C.M.; Maxwell, M.J.; Passey, S.L.; Hansen, M.J.; McGregor, N.E.; Sims, N.A.; Steinfort, D.P.; Irving, L.B. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J. Clin. Investig. 2018, 128, 2406–2418. [Google Scholar] [CrossRef]
- Chai, M.G.; Kim-Fuchs, C.; Angst, E.; Sloan, E.K. Bioluminescent orthotopic model of pancreatic cancer progression. JoVE 2013, e50395. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, K.; Wang, C.; Gong, W.; Yoshimura, T.; Wang, J.M.; Liu, M. Isolation of mouse tumor-infiltrating leukocytes by percoll gradient centrifugation. Bio-Protocol 2013, 3, 892e. [Google Scholar] [CrossRef]
- Nelson, N.; Szekeres, K.; Cooper, D.; Ghansah, T. Preparation of myeloid derived suppressor cells (MDSC) from naive and pancreatic tumor-bearing mice using flow cytometry and automated magnetic activated cell sorting (AutoMACS). J. Vis. Exp. 2012, e3875. [Google Scholar] [CrossRef]
- Revell, P.A.; Grossman, W.J.; Thomas, D.A.; Cao, X.; Behl, R.; Ratner, J.A.; Lu, Z.H.; Ley, T.J. Granzyme B and the downstream granzymes C and/or F are important for cytotoxic lymphocyte functions. J. Immunol. 2005, 174, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.J.; House, I.G.; Oliaro, J.; Ramsbottom, K.M.; Hagn, M.; Yagita, H.; Trapani, J.A.; Voskoboinik, I. A method for detecting intracellular perforin in mouse lymphocytes. J. Immunol. 2014, 193, 5744–5750. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalobos-Ayala, K.; Ortiz Rivera, I.; Alvarez, C.; Husain, K.; DeLoach, D.; Krystal, G.; Hibbs, M.L.; Jiang, K.; Ghansah, T. Apigenin Increases SHIP-1 Expression, Promotes Tumoricidal Macrophages and Anti-Tumor Immune Responses in Murine Pancreatic Cancer. Cancers 2020, 12, 3631. https://doi.org/10.3390/cancers12123631
Villalobos-Ayala K, Ortiz Rivera I, Alvarez C, Husain K, DeLoach D, Krystal G, Hibbs ML, Jiang K, Ghansah T. Apigenin Increases SHIP-1 Expression, Promotes Tumoricidal Macrophages and Anti-Tumor Immune Responses in Murine Pancreatic Cancer. Cancers. 2020; 12(12):3631. https://doi.org/10.3390/cancers12123631
Chicago/Turabian StyleVillalobos-Ayala, Krystal, Ivannie Ortiz Rivera, Ciara Alvarez, Kazim Husain, DeVon DeLoach, Gerald Krystal, Margaret L. Hibbs, Kun Jiang, and Tomar Ghansah. 2020. "Apigenin Increases SHIP-1 Expression, Promotes Tumoricidal Macrophages and Anti-Tumor Immune Responses in Murine Pancreatic Cancer" Cancers 12, no. 12: 3631. https://doi.org/10.3390/cancers12123631
APA StyleVillalobos-Ayala, K., Ortiz Rivera, I., Alvarez, C., Husain, K., DeLoach, D., Krystal, G., Hibbs, M. L., Jiang, K., & Ghansah, T. (2020). Apigenin Increases SHIP-1 Expression, Promotes Tumoricidal Macrophages and Anti-Tumor Immune Responses in Murine Pancreatic Cancer. Cancers, 12(12), 3631. https://doi.org/10.3390/cancers12123631