SAHA Overcomes 5-FU Resistance in IFIT2-Depleted Oral Squamous Cell Carcinoma Cells

Abstract
Simple Summary
Abstract
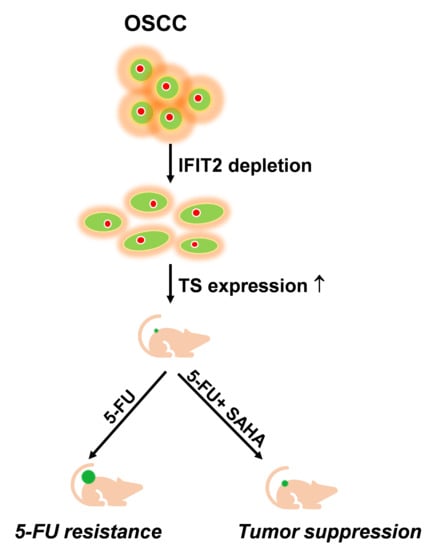
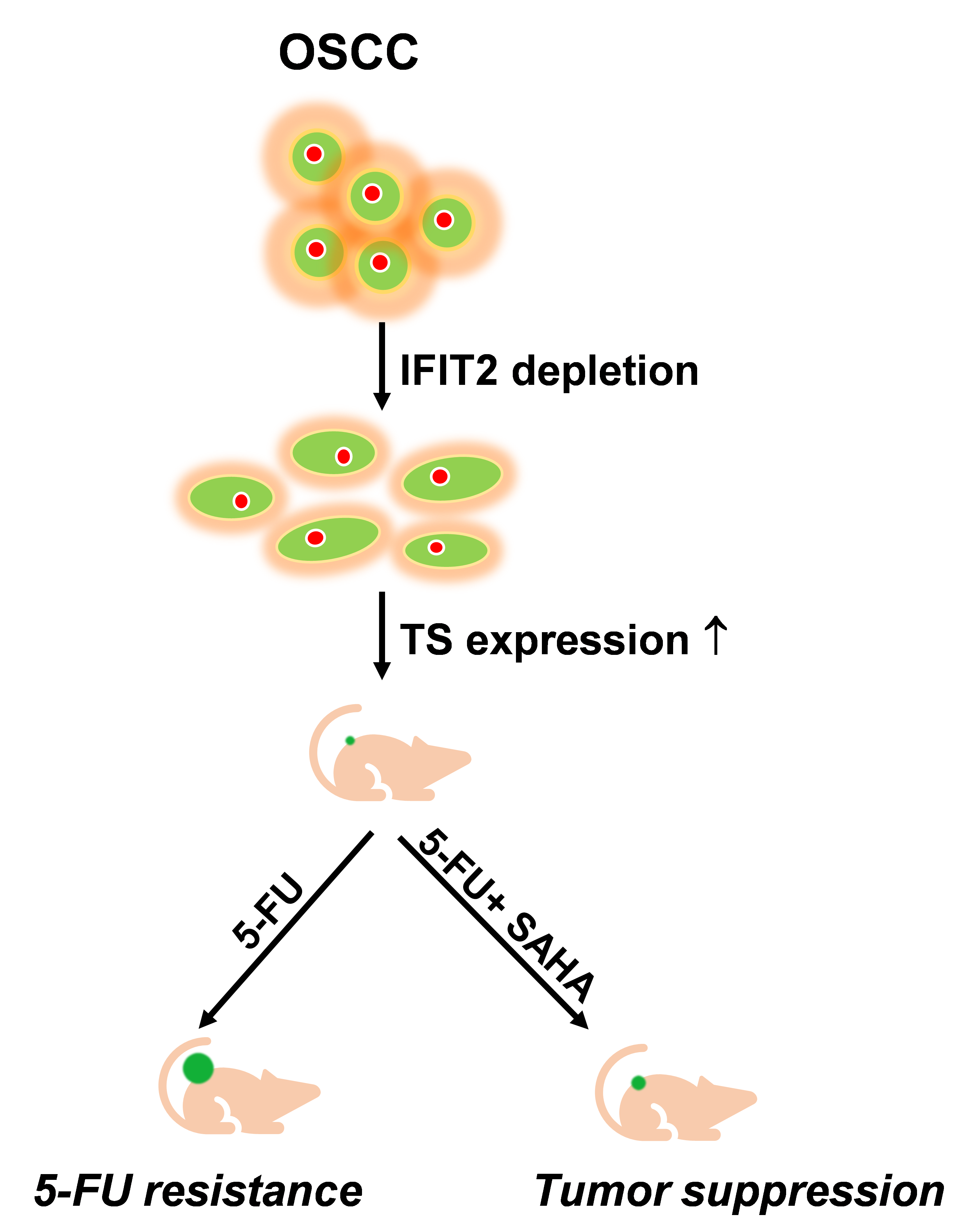
1. Introduction
2. Results
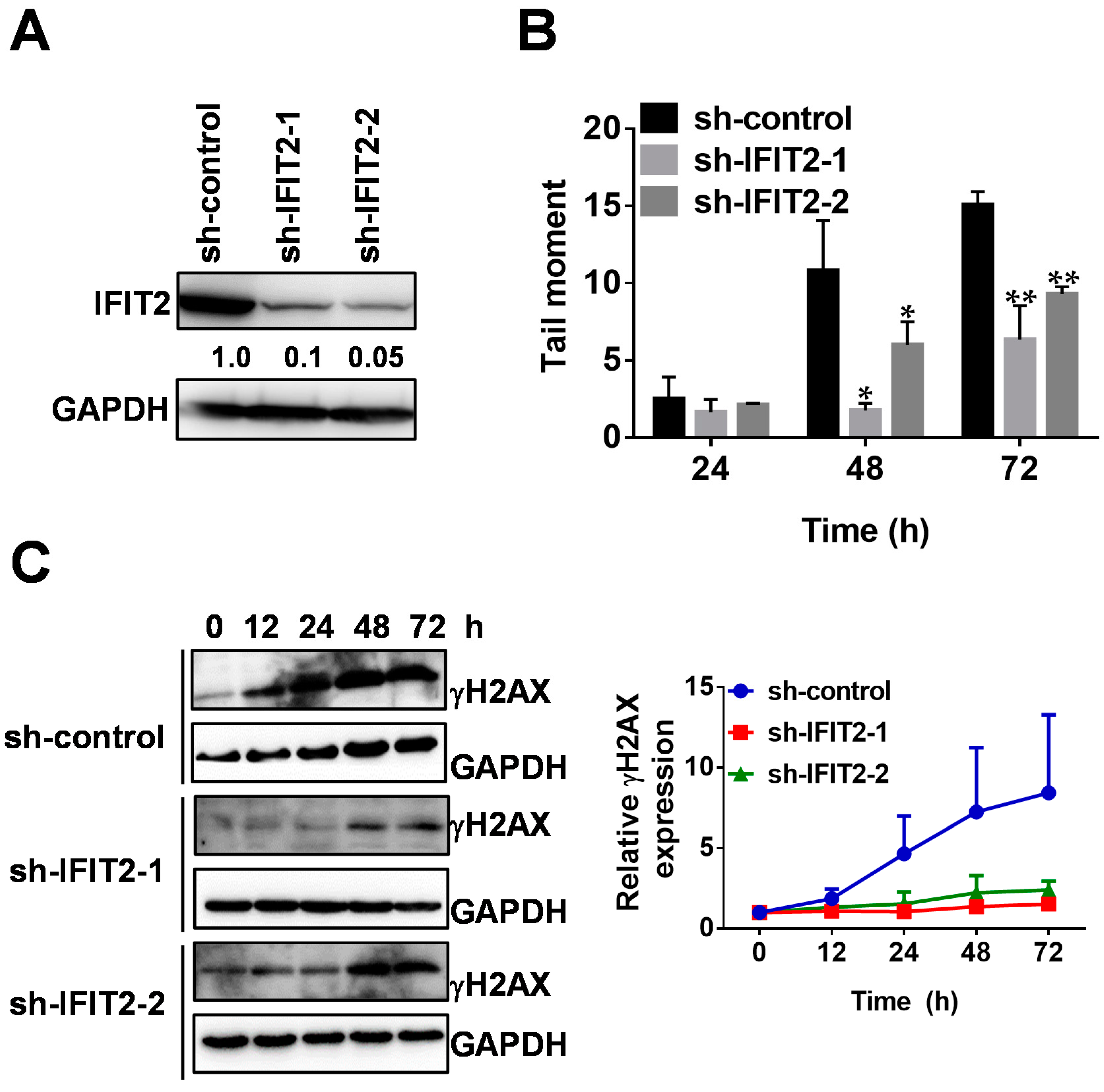
2.1. 5-FU Resistance in IFIT2-Knockdown Cells
2.2. Attenuated DNA Damage Activity of 5-FU in IFIT2-Knockdown Cells
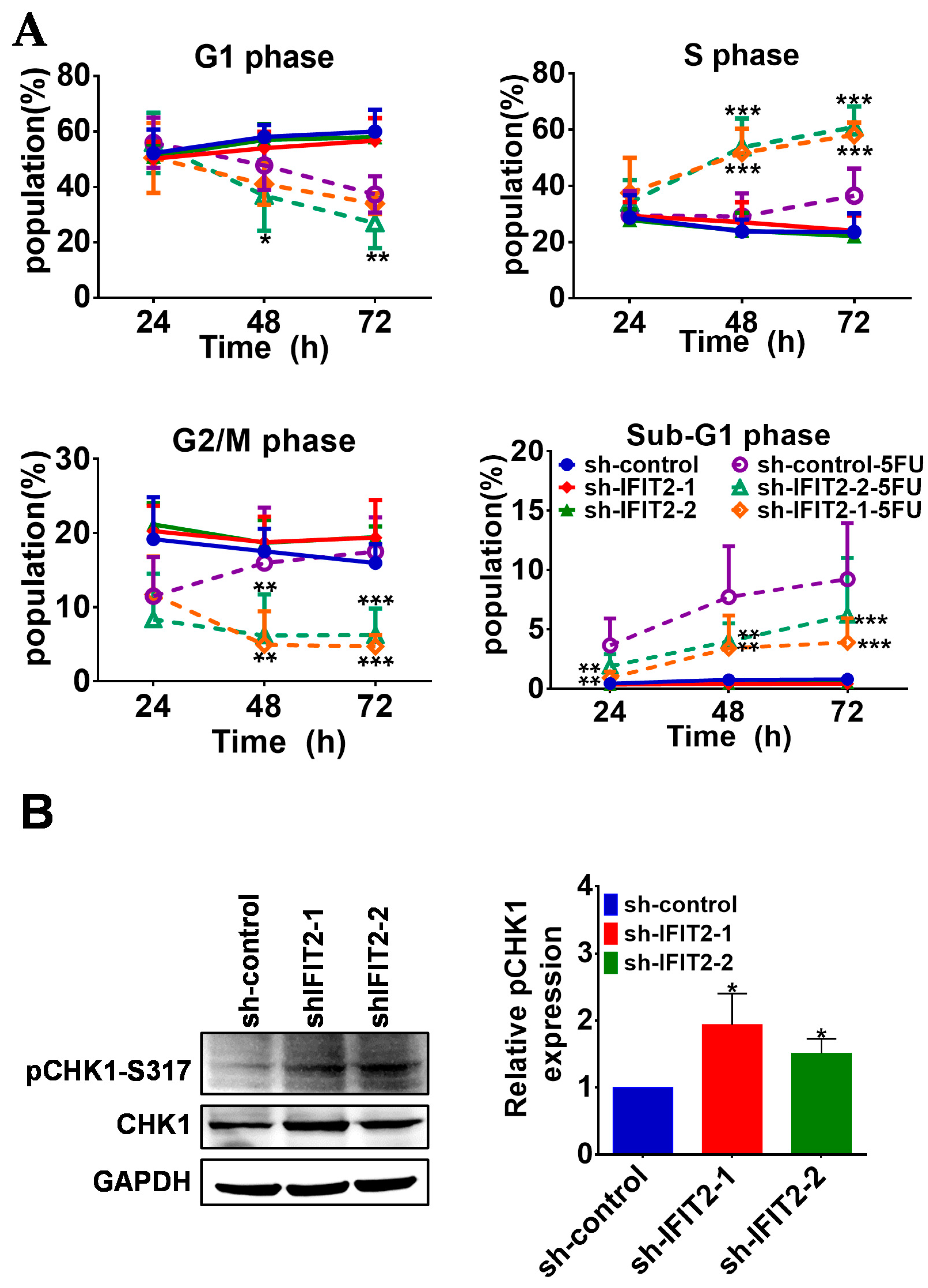
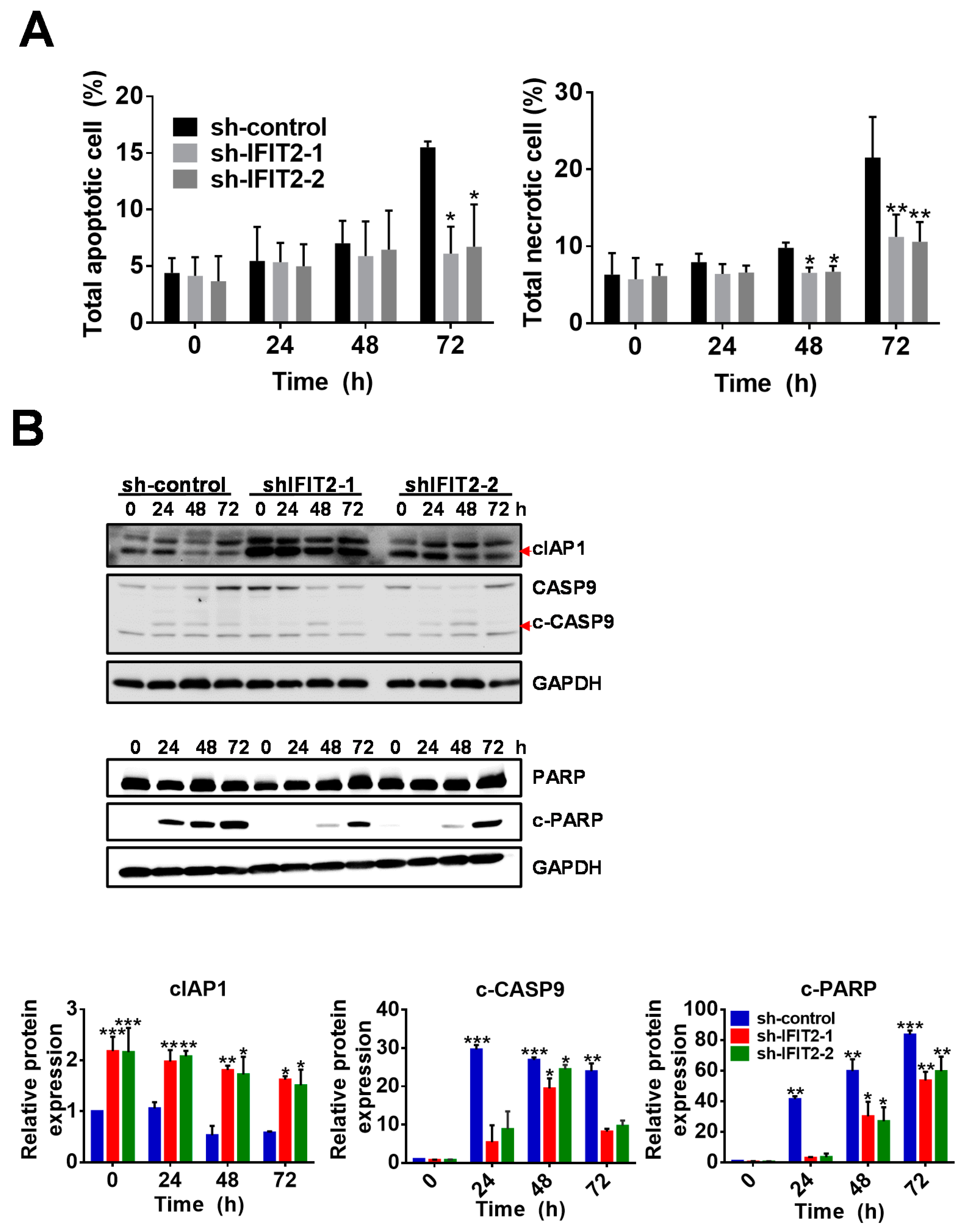
2.3. Effect of 5-FU on Cell Cycle Progression and Apoptosis in IFIT2-Knockdown Cells
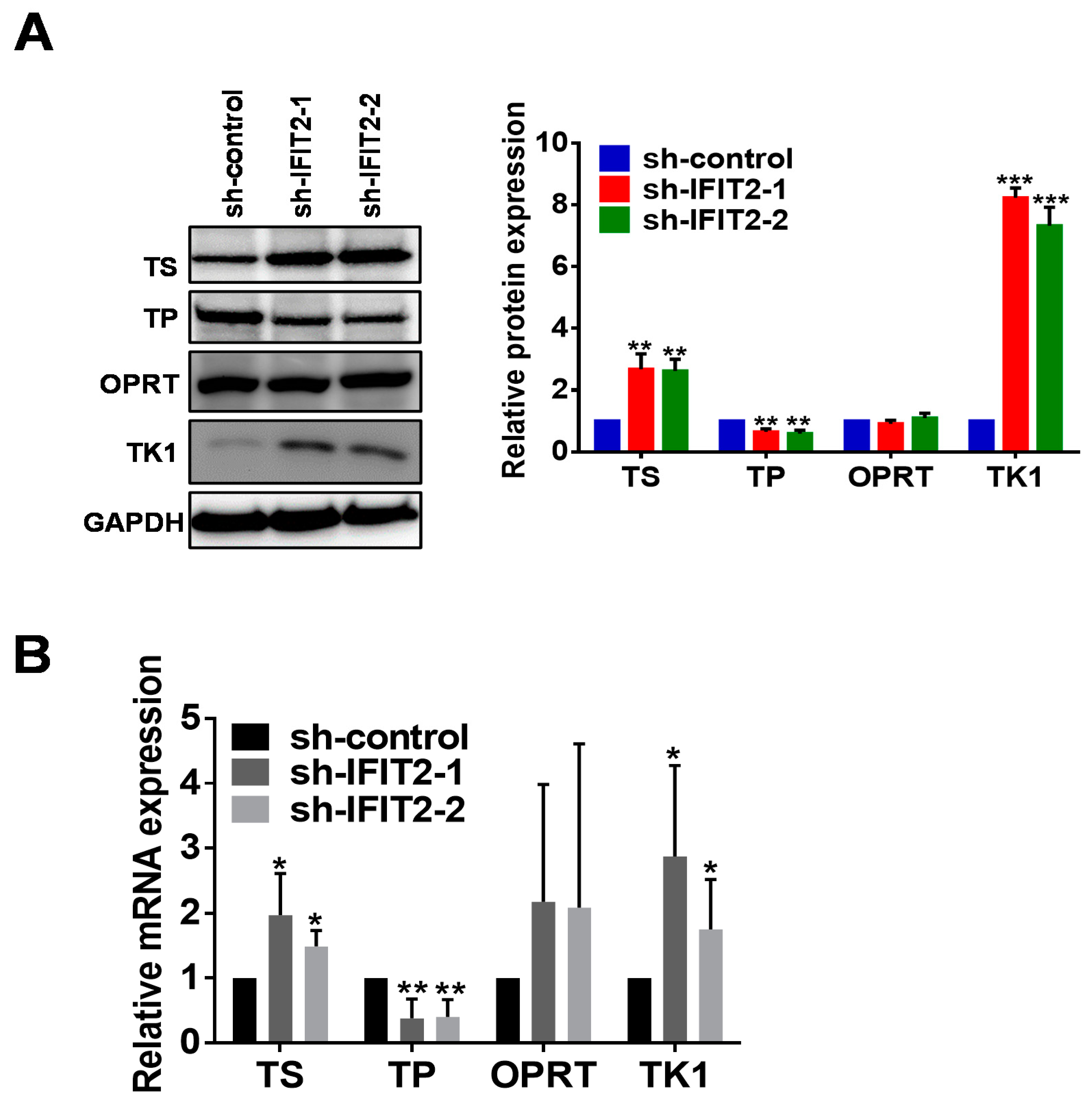
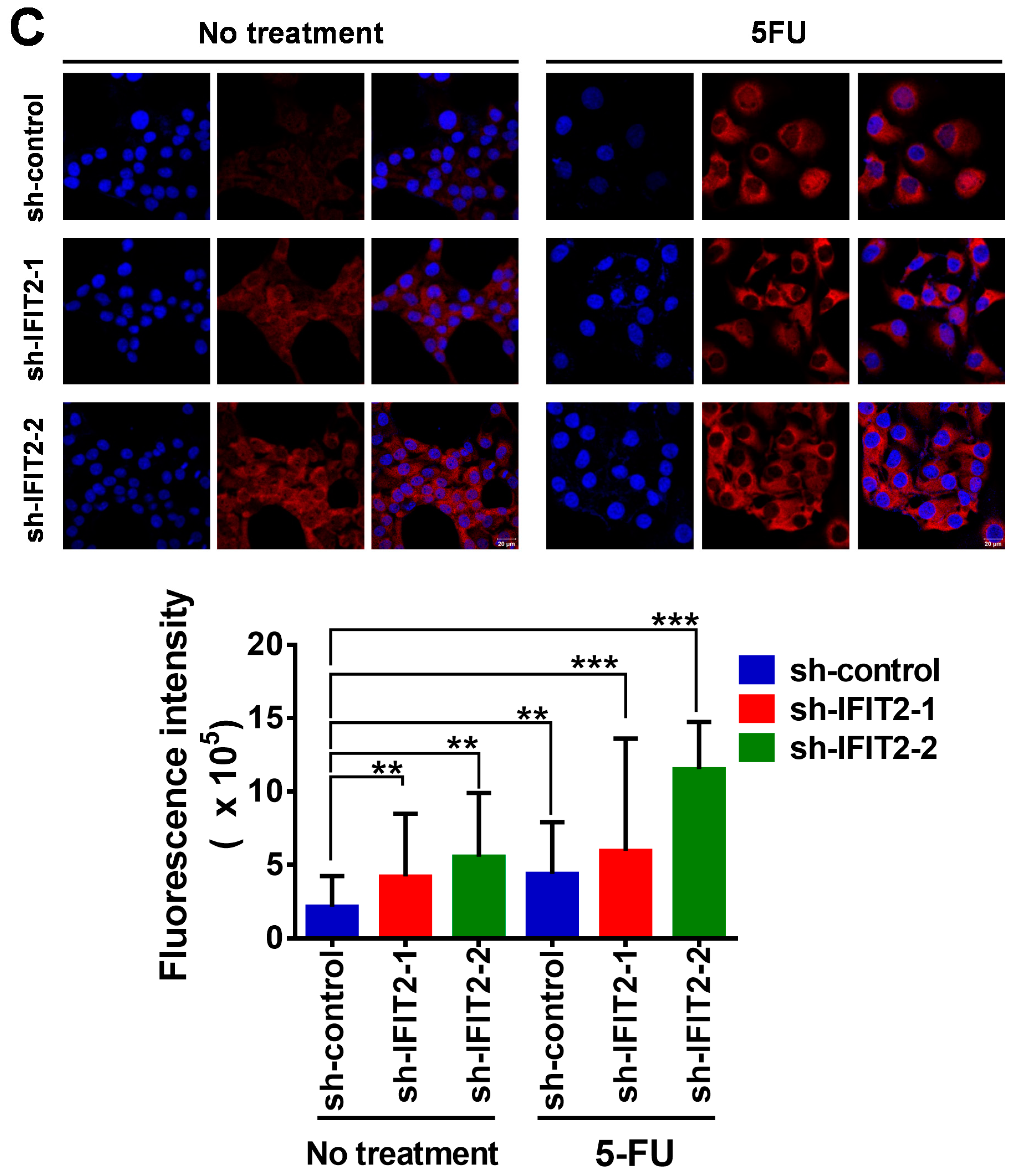
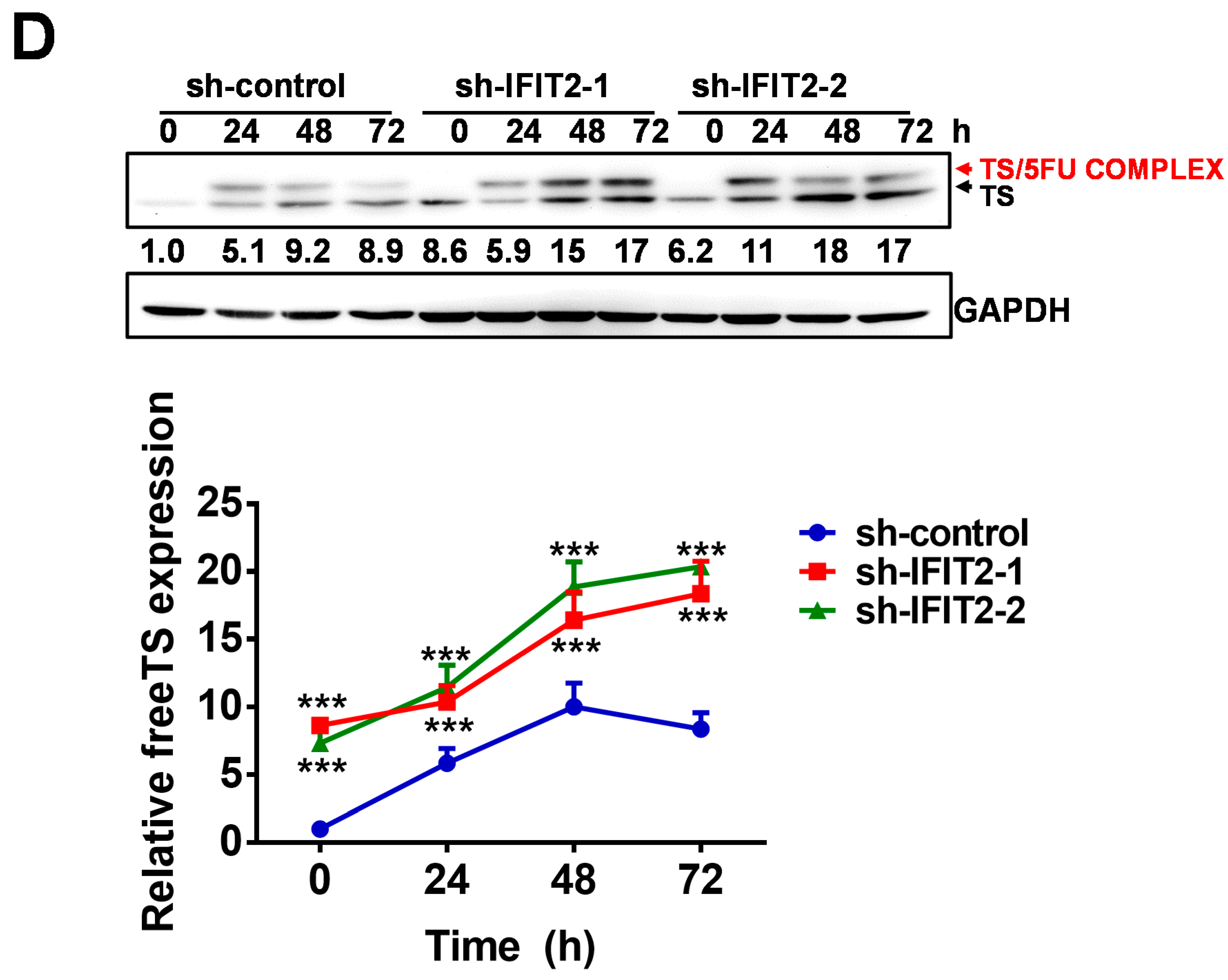
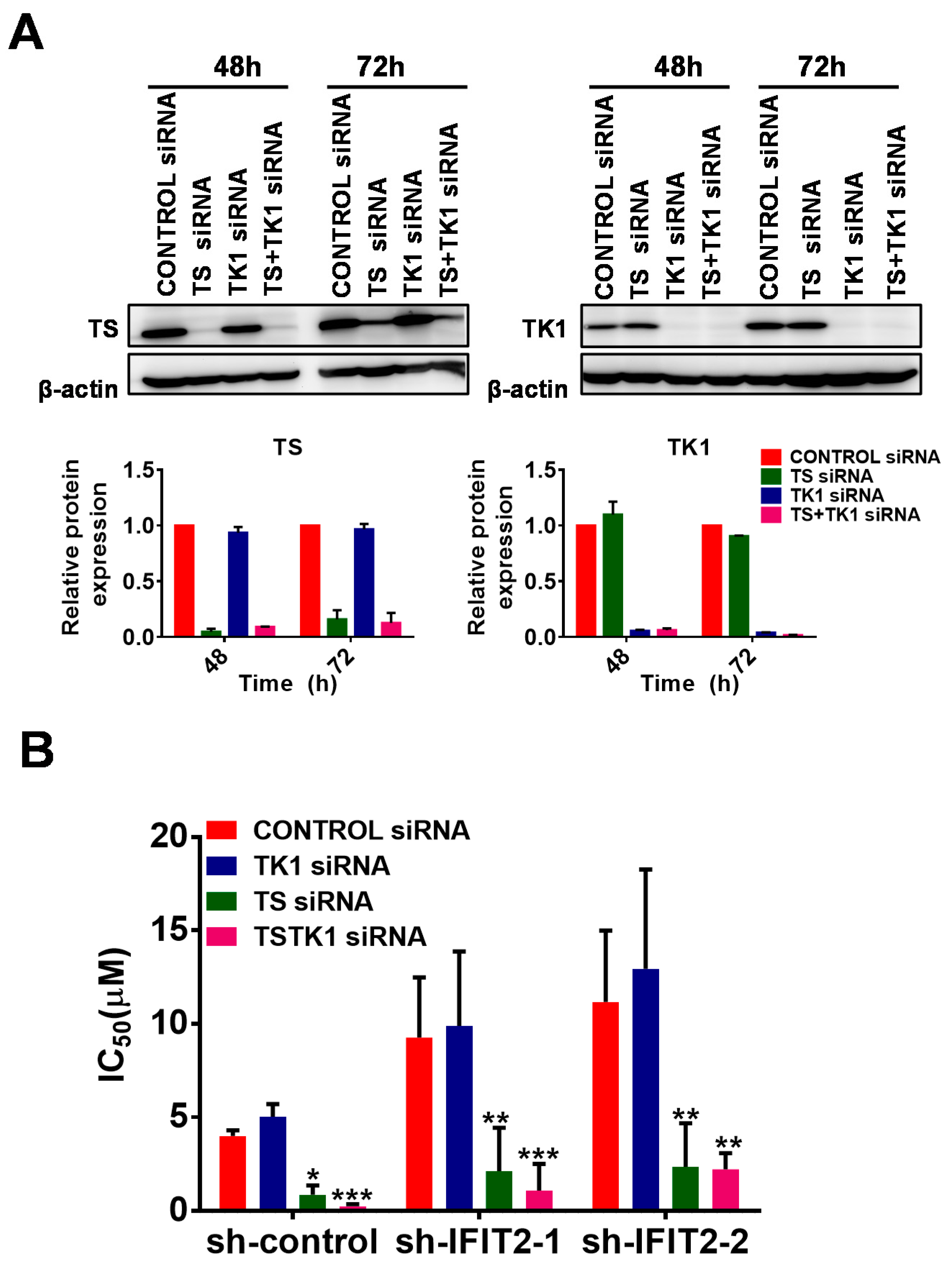
2.4. Altered Expression of 5-FU Metabolic Enzymes in IFIT2-Knockdown Cells
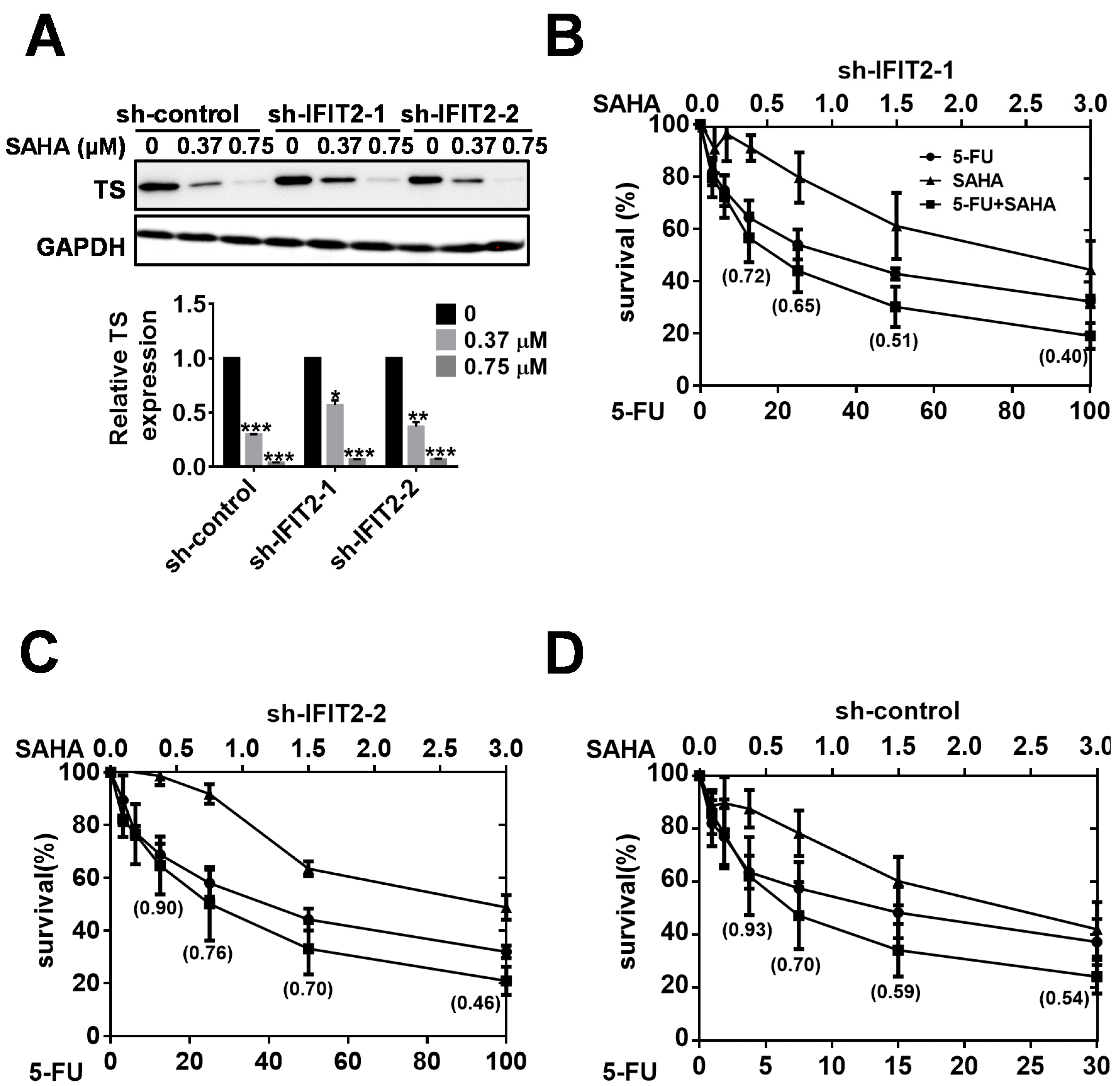
2.5. Synergistic Inhibition of Tumor Growth by the Combination of SAHA and 5-FU
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Cytotoxicity Assay
4.3. Comet Assay
4.4. Western Blot Analysis
4.5. Cell Cycle Analysis
4.6. Annexin V Staining Assay
4.7. Quantitative Real-Time PCR (Q-PCR)
4.8. Immunofluorescence Staining
4.9. siRNA Transfection
4.10. Xenograft Animal Model
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fensterl, V.; Sen, G.C. Interferon-induced ifit proteins: Their role in viral pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Pidugu, V.K.; Wu, M.M.; Yen, A.H.; Pidugu, H.B.; Chang, K.W.; Liu, C.J.; Lee, T.C. Ifit1 and ifit3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-egfr recycling. Oncogene 2019, 38, 3232–3247. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. The isg56/ifit1 gene family. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Gorna, M.W.; Superti-Furga, G. Ifits: Emerging roles as key anti-viral proteins. Front. Immunol. 2014, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.M.; Liu, C.J.; Lee, T.C. Emerging functions of human ifit proteins in cancer. Front. Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed]
- Lo, U.G.; Bao, J.; Cen, J.; Yeh, H.C.; Luo, J.; Tan, W.; Hsieh, J.T. Interferon-induced ifit5 promotes epithelial-to-mesenchymal transition leading to renal cancer invasion. Am. J. Clin. Exp. Urol. 2019, 7, 31–45. [Google Scholar] [PubMed]
- Zhao, Y.; Altendorf-Hofmann, A.; Pozios, I.; Camaj, P.; Daberitz, T.; Wang, X.; Niess, H.; Seeliger, H.; Popp, F.; Betzler, C.; et al. Elevated interferon-induced protein with tetratricopeptide repeats 3 (ifit3) is a poor prognostic marker in pancreatic ductal adenocarcinoma. J. Cancer Res. Clin. Oncol. 2017, 143, 1061–1068. [Google Scholar] [CrossRef]
- Koh, S.Y.; Moon, J.Y.; Unno, T.; Cho, S.K. Baicalein suppresses stem cell-like characteristics in radio- and chemoresistant mda-mb-231 human breast cancer cells through up-regulation of ifit2. Nutrients 2019, 11, 624. [Google Scholar] [CrossRef]
- Chen, L.; Zhai, W.; Zheng, X.; Xie, Q.; Zhou, Q.; Tao, M.; Zhu, Y.; Wu, C.; Jiang, J. Decreased ifit2 expression promotes gastric cancer progression and predicts poor prognosis of the patients. Cell Physiol. Biochem. 2018, 45, 15–25. [Google Scholar] [CrossRef]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef]
- Reich, N.C. A death-promoting role for isg54/ifit2. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2013, 33, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Song, L.; Cong, Q.; Wang, J.; Xu, H.; Chu, Y.; Li, Q.; Zhang, Y.; Zou, X.; Zhang, C.; et al. The lim protein ajuba promotes colorectal cancer cell survival through suppression of jak1/stat1/ifit2 network. Oncogene 2017, 36, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Ohsugi, T.; Yamaguchi, K.; Zhu, C.; Ikenoue, T.; Furukawa, Y. Decreased expression of interferon-induced protein 2 (ifit2) by wnt/beta-catenin signaling confers anti-apoptotic properties to colorectal cancer cells. Oncotarget 2017, 8, 100176–100186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, Y.; Liu, S.; Zeng, L.; Wan, L.; Zhang, Z. Curcumin induces apoptosis in human leukemic cell lines through an ifit2-dependent pathway. Cancer Biol. Ther. 2017, 18, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, N.; Liu, S.; Jiang, M.; Wan, J.; Zhang, Y.; Wan, L.; Xie, C.; Le, A. Overexpression of ifit2 inhibits the proliferation of chronic myeloid leukemia cells by regulating the bcr-abl/akt/mtor pathway. Int. J. Mol. Med. 2020, 45, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.C.; Chang, K.W.; Liu, C.J.; Kao, S.Y.; Lee, T.C. Ifn-induced protein with tetratricopeptide repeats 2 inhibits migration activity and increases survival of oral squamous cell carcinoma. Mol. Cancer Res. MCR 2008, 6, 1431–1439. [Google Scholar] [CrossRef]
- Lai, K.C.; Liu, C.J.; Chang, K.W.; Lee, T.C. Depleting ifit2 mediates atypical pkc signaling to enhance the migration and metastatic activity of oral squamous cell carcinoma cells. Oncogene 2013, 32, 3686–3697. [Google Scholar] [CrossRef]
- Lai, K.C.; Liu, C.J.; Lin, T.J.; Mar, A.C.; Wang, H.H.; Chen, C.W.; Hong, Z.X.; Lee, T.C. Blocking tnf-alpha inhibits angiogenesis and growth of ifit2-depleted metastatic oral squamous cell carcinoma cells. Cancer Lett. 2016, 370, 207–215. [Google Scholar] [CrossRef]
- Shield, K.D.; Ferlay, J.; Jemal, A.; Sankaranarayanan, R.; Chaturvedi, A.K.; Bray, F.; Soerjomataram, I. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J. Clin. 2017, 67, 51–64. [Google Scholar] [CrossRef]
- Rivera, C.; Venegas, B. Histological and molecular aspects of oral squamous cell carcinoma (review). Oncol. Lett. 2014, 8, 7–11. [Google Scholar] [CrossRef]
- Shingaki, S.; Takada, M.; Sasai, K.; Bibi, R.; Kobayashi, T.; Nomura, T.; Saito, C. Impact of lymph node metastasis on the pattern of failure and survival in oral carcinomas. Am. J. Surg. 2003, 185, 278–284. [Google Scholar] [CrossRef]
- Jerjes, W.; Upile, T.; Petrie, A.; Riskalla, A.; Hamdoon, Z.; Vourvachis, M.; Karavidas, K.; Jay, A.; Sandison, A.; Thomas, G.J.; et al. Clinicopathological parameters, recurrence, locoregional and distant metastasis in 115 t1-t2 oral squamous cell carcinoma patients. Head Neck Oncol. 2010, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.A.; Graboyes, E.M.; Wahlquist, A.E.; Neskey, D.M.; Kaczmar, J.M.; Schopper, H.K.; Sharma, A.K.; Morgan, P.F.; Nguyen, S.A.; Day, T.A. Primary surgery vs radiotherapy for early stage oral cavity cancer. Otolaryngol. Head Neck Surg. Off. J. Am. Acad. Otolaryngol. Head Neck Surg. 2018, 158, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.G.; Tan, D.S.; Tan, V.K.; Wang, W.; Hwang, J.; Tan, N.C.; Sivanandan, R.; Tan, H.K.; Lim, W.T.; Ang, M.K.; et al. Randomized trial comparing surgery and adjuvant radiotherapy versus concurrent chemoradiotherapy in patients with advanced, nonmetastatic squamous cell carcinoma of the head and neck: 10-year update and subset analysis. Cancer 2015, 121, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, K.B.; Gupta, N. Clinicopathological prognostic implicators of oral squamous cell carcinoma: Need to understand and revise. N. Am. J. Med. Sci. 2013, 5, 671–679. [Google Scholar] [CrossRef]
- Almangush, A.; Mäkitie, A.A.; Triantafyllou, A.; de Bree, R.; Strojan, P.; Rinaldo, A.; Hernandez-Prera, J.C.; Suárez, C.; Kowalski, L.P.; Ferlito, A.; et al. Staging and grading of oral squamous cell carcinoma: An update. Oral Oncol. 2020, 107, 104799. [Google Scholar] [CrossRef]
- Troiano, G.; Rubini, C.; Togni, L.; Caponio, V.C.A.; Zhurakivska, K.; Santarelli, A.; Cirillo, N.; Lo Muzio, L.; Mascitti, M. The immune phenotype of tongue squamous cell carcinoma predicts early relapse and poor prognosis. Cancer Med. 2020, 9, 8333–8344. [Google Scholar] [CrossRef]
- Chang, W.C.; Chang, C.F.; Li, Y.H.; Yang, C.Y.; Su, R.Y.; Lin, C.K.; Chen, Y.W. A histopathological evaluation and potential prognostic implications of oral squamous cell carcinoma with adverse features. Oral Oncol. 2019, 95, 65–73. [Google Scholar] [CrossRef]
- Mattavelli, D.; Lombardi, D.; Missale, F.; Calza, S.; Battocchio, S.; Paderno, A.; Bozzola, A.; Bossi, P.; Vermi, W.; Piazza, C.; et al. Prognostic nomograms in oral squamous cell carcinoma: The negative impact of low neutrophil to lymphocyte ratio. Front. Oncol. 2019, 9, 339. [Google Scholar] [CrossRef]
- Sim, Y.C.; Hwang, J.H.; Ahn, K.M. Overall and disease-specific survival outcomes following primary surgery for oral squamous cell carcinoma: Analysis of consecutive 67 patients. J. Korean Assoc. Oral Maxillofac. Surg. 2019, 45, 83–90. [Google Scholar] [CrossRef]
- Nor, J.E.; Gutkind, J.S. Head and neck cancer in the new era of precision medicine. J. Dent. Res. 2018, 97, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Ketabat, F.; Pundir, M.; Mohabatpour, F.; Lobanova, L.; Koutsopoulos, S.; Hadjiiski, L.; Chen, X.; Papagerakis, P.; Papagerakis, S. Controlled drug delivery systems for oral cancer treatment-current status and future perspectives. Pharmaceutics 2019, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Lorch, J.H.; Goloubeva, O.; Haddad, R.I.; Cullen, K.; Sarlis, N.; Tishler, R.; Tan, M.; Fasciano, J.; Sammartino, D.E.; Posner, M.R.; et al. Induction chemotherapy with cisplatin and fluorouracil alone or in combination with docetaxel in locally advanced squamous-cell cancer of the head and neck: Long-term results of the tax 324 randomised phase 3 trial. Lancet Oncol. 2011, 12, 153–159. [Google Scholar] [CrossRef]
- Shin, D.M.; Khuri, F.R. Advances in the management of recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck 2013, 35, 443–453. [Google Scholar] [CrossRef]
- Ferrari, D.; Codeca, C.; Fiore, J.; Luciani, A.; Foa, P. A review on the treatment of relapsed/metastatic head and neck cancer. Expert Opin. Pharmacother. 2009, 10, 2625–2632. [Google Scholar] [CrossRef]
- Clavel, M.; Vermorken, J.B.; Cognetti, F.; Cappelaere, P.; de Mulder, P.H.; Schornagel, J.H.; Tueni, E.A.; Verweij, J.; Wildiers, J.; Clerico, M.; et al. Randomized comparison of cisplatin, methotrexate, bleomycin and vincristine (cabo) versus cisplatin and 5-fluorouracil (cf) versus cisplatin (c) in recurrent or metastatic squamous cell carcinoma of the head and neck. A phase iii study of the eortc head and neck cancer cooperative group. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 1994, 5, 521–526. [Google Scholar]
- Shin, D.M.; Khuri, F.R.; Glisson, B.S.; Ginsberg, L.; Papadimitrakopoulou, V.M.; Clayman, G.; Lee, J.J.; Ang, K.K.; Lippman, S.M.; Hong, W.K. Phase ii study of paclitaxel, ifosfamide, and carboplatin in patients with recurrent or metastatic head and neck squamous cell carcinoma. Cancer 2001, 91, 1316–1323. [Google Scholar] [CrossRef]
- Bernier, J. Cetuximab in the treatment of head and neck cancer. Expert Rev. Anticancer Ther. 2006, 6, 1539–1552. [Google Scholar] [CrossRef]
- Muratori, L.; La Salvia, A.; Sperone, P.; Di Maio, M. Target therapies in recurrent or metastatic head and neck cancer: State of the art and novel perspectives. A systematic review. Crit. Rev. Oncol./Hematol. 2019, 139, 41–52. [Google Scholar] [CrossRef]
- Du, F.; Liu, H.; Lu, Y.; Zhao, X.; Fan, D. Epithelial-to-mesenchymal transition: Liaison between cancer metastasis and drug resistance. Crit. Rev. Oncog. 2017, 22, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. Emt: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Niess, H.; Camaj, P.; Renner, A.; Ischenko, I.; Zhao, Y.; Krebs, S.; Mysliwietz, J.; Jackel, C.; Nelson, P.J.; Blum, H.; et al. Side population cells of pancreatic cancer show characteristics of cancer stem cells responsible for resistance and metastasis. Target. Oncol. 2015, 10, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Brockstein, B.E. Management of recurrent head and neck cancer: Recent progress and future directions. Drugs 2011, 71, 1551–1559. [Google Scholar] [CrossRef]
- Houghton, J.A.; Houghton, P.J. Elucidation of pathways of 5-fluorouracil metabolism in xenografts of human colorectal adenocarcinoma. Eur. J. Cancer Clin. Oncol. 1983, 19, 807–815. [Google Scholar] [CrossRef]
- Noro, R.; Miyanaga, A.; Minegishi, Y.; Okano, T.; Seike, M.; Soeno, C.; Kataoka, K.; Matsuda, K.; Yoshimura, A.; Gemma, A. Histone deacetylase inhibitor enhances sensitivity of non-small-cell lung cancer cells to 5-fu/s-1 via down-regulation of thymidylate synthase expression and up-regulation of p21(waf1/cip1) expression. Cancer Sci. 2010, 101, 1424–1430. [Google Scholar] [CrossRef]
- Ocker, M.; Alajati, A.; Ganslmayer, M.; Zopf, S.; Luders, M.; Neureiter, D.; Hahn, E.G.; Schuppan, D.; Herold, C. The histone-deacetylase inhibitor saha potentiates proapoptotic effects of 5-fluorouracil and irinotecan in hepatoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 385–394. [Google Scholar] [CrossRef]
- Hernando-Cubero, J.; Matos-Garcia, I.; Alonso-Orduna, V.; Capdevila, J. The role of fluoropirimidines in gastrointestinal tumours: From the bench to the bed. J. Gastrointest. Cancer 2017, 48, 135–147. [Google Scholar] [CrossRef]
- Nishijima, T.F.; Suzuki, M.; Muss, H.B. A comparison of toxicity profiles between the lower and standard dose capecitabine in breast cancer: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2016, 156, 227–236. [Google Scholar] [CrossRef]
- Leveque, D.; Becker, G. The role of therapeutic drug monitoring in the management of safety of anticancer agents: A focus on 3 cytotoxics. Expert Opin. Drug Saf. 2019, 18, 1009–1015. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Glei, M.; Schneider, T.; Schlormann, W. Comet assay: An essential tool in toxicological research. Arch. Toxicol. 2016, 90, 2315–2336. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. Gammah2ax and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Guo, X.; Goessl, E.; Jin, G.; Collie-Duguid, E.S.; Cassidy, J.; Wang, W.; O’Brien, V. Cell cycle perturbation and acquired 5-fluorouracil chemoresistance. Anticancer Res. 2008, 28, 9–14. [Google Scholar]
- Ijichi, K.; Adachi, M.; Ogawa, T.; Hasegawa, Y.; Murakami, S. Cell-cycle distribution and thymidilate synthatase (ts) expression correlate with 5-fu resistance in head and neck carcinoma cells. Anticancer Res. 2014, 34, 2907–2911. [Google Scholar]
- Xiao, Z.; Xue, J.; Sowin, T.J.; Rosenberg, S.H.; Zhang, H. A novel mechanism of checkpoint abrogation conferred by chk1 downregulation. Oncogene 2005, 24, 1403–1411. [Google Scholar] [CrossRef][Green Version]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Peters, G.J.; van der Wilt, C.L.; van Triest, B.; Codacci-Pisanelli, G.; Johnston, P.G.; van Groeningen, C.J.; Pinedo, H.M. Thymidylate synthase and drug resistance. Eur. J. Cancer 1995, 31, 1299–1305. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Lee, J.S.; Min, H.Y.; Lee, H.Y. Acquired resistance to 5-fluorouracil via hsp90/src-mediated increase in thymidylate synthase expression in colon cancer. Oncotarget 2015, 6, 32622–32633. [Google Scholar] [CrossRef]
- Aschele, C.; Lonardi, S.; Monfardini, S. Thymidylate synthase expression as a predictor of clinical response to fluoropyrimidine-based chemotherapy in advanced colorectal cancer. Cancer Treat. Rev. 2002, 28, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Allegra, C.J. Regulation of thymidylate synthase in human colon cancer cells treated with 5-fluorouracil and interferon-gamma. Adv. Exp. Med. Biol. 1993, 339, 143–151, discussion 147–165. [Google Scholar] [PubMed]
- Fanciullino, R.; Evrard, A.; Cuq, P.; Giacometti, S.; Peillard, L.; Mercier, C.; Aubert, C.; Milano, G.; Ciccolini, J. Genetic and biochemical modulation of 5-fluorouracil through the overexpression of thymidine kinase: An in-vitro study. Anticancer Drugs 2006, 17, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Park, S.; Park, J.K.; Kim, Y.; Kang, Y.; Yoo, Y.D. Establishment and characterization of 5-fluorouracil-resistant gastric cancer cells. Cancer Lett. 2000, 159, 95–101. [Google Scholar] [CrossRef]
- Di Gennaro, E.; Bruzzese, F.; Pepe, S.; Leone, A.; Delrio, P.; Subbarayan, P.R.; Avallone, A.; Budillon, A. Modulation of thymidilate synthase and p53 expression by hdac inhibitor vorinostat resulted in synergistic antitumor effect in combination with 5fu or raltitrexed. Cancer Biol. Ther. 2009, 8, 782–791. [Google Scholar] [CrossRef]
- Di Gennaro, E.; Piro, G.; Chianese, M.I.; Franco, R.; Di Cintio, A.; Moccia, T.; Luciano, A.; de Ruggiero, I.; Bruzzese, F.; Avallone, A.; et al. Vorinostat synergises with capecitabine through upregulation of thymidine phosphorylase. Br. J. Cancer 2010, 103, 1680–1691. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, J.H.; Jung, Y.; Kim, J.H.; Jong, H.S.; Kim, T.Y.; Bang, Y.J. Histone deacetylase inhibitor enhances 5-fluorouracil cytotoxicity by down-regulating thymidylate synthase in human cancer cells. Mol. Cancer Ther. 2006, 5, 3085–3095. [Google Scholar] [CrossRef]
- Fazzone, W.; Wilson, P.M.; Labonte, M.J.; Lenz, H.J.; Ladner, R.D. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int. J. Cancer 2009, 125, 463–473. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Lee, P.C.; Kakadiya, R.; Su, T.L.; Lee, T.C. Combination of bifunctional alkylating agent and arsenic trioxide synergistically suppresses the growth of drug-resistant tumor cells. Neoplasia 2010, 12, 376–387. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nature Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Chaniyara, R.; Tala, S.; Chen, C.W.; Zang, X.; Kakadiya, R.; Lin, L.F.; Chen, C.H.; Chien, S.I.; Chou, T.C.; Tsai, T.H.; et al. Novel antitumor indolizino[6–b]indoles with multiple modes of action: DNA cross-linking and topoisomerase i and ii inhibition. J. Med. Chem. 2013, 56, 1544–1563. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent | sh-Control | sh-IFIT2-1 | sh-IFIT2-2 |
|---|---|---|---|
| ANTIMETABOLITE | |||
| 5-Fluorouracil | 3.03 ± 0.83 | 13.37 ± 1.66 (4.4) b,*** | 11.08 ± 1.31 (3.7) *** |
| Cytarabine | 0.67 ± 0.24 | 0.91 ± 0.13 (1.4) *** | 1.19 ± 0.22 (1.8) *** |
| Gemcitabine | 0.011 ± 0.000 | 0.018 ± 0.001 (1.7) ** | 0.014 ± 0.001 (1.3) * |
| Raltitrexed | 0.105 ± 0.003 | 0.126 ± 0.001 (1.2) ** | 0.123 ± 0.002 (1.2) * |
| TOPOISOMERASE INHIBITOR | |||
| Irinotecan (I) | 3.88 ± 1.04 | 3.94 ± 0.55 (1.0) | 4.02 ± 0.64 (1.0) |
| Doxorubicin (II) | 0.08 ± 0.02 | 0.10 ± 0.02 (1.3) * | 0.08 ± 0.01 (1.0) |
| Etoposide (II) | 0.94 ± 0.35 | 1.04 ± 0.29 (1.1) | 0.97 ± 0.38 (1.0) |
| Mitoxantrone (II) | 0.003 ± 0.001 | 0.003 ± 0.000 (1.0) | 0.003 ± 0.001 (1.0) |
| PLATINUM BASED | |||
| Cisplatin | 3.38 ± 0.80 | 4.15 ± 0.96 (1.2) | 3.65 ± 1.17 (1.1) |
| Carboplatin | 31.36 ± 0.99 | 34.45 ± 4.03 (1.1) | 29.96 ± 7.67 (1.0) |
| Oxaliplatin | 6.11 ± 2.58 | 10.36 ± 3.98 (1.7) ** | 8.53 ± 1.41 (1.4) *** |
| TYROSINE KINASE INHIBITOR | |||
| Gefitinib | 5.10 ± 1.2 | 8.27 ± 2.54 (1.6) * | 9.74 ± 1.42 (1.9) ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regmi, P.; Lai, K.-C.; Liu, C.-J.; Lee, T.-C. SAHA Overcomes 5-FU Resistance in IFIT2-Depleted Oral Squamous Cell Carcinoma Cells. Cancers 2020, 12, 3527. https://doi.org/10.3390/cancers12123527
Regmi P, Lai K-C, Liu C-J, Lee T-C. SAHA Overcomes 5-FU Resistance in IFIT2-Depleted Oral Squamous Cell Carcinoma Cells. Cancers. 2020; 12(12):3527. https://doi.org/10.3390/cancers12123527
Chicago/Turabian StyleRegmi, Prabha, Kuo-Chu Lai, Chung-Ji Liu, and Te-Chang Lee. 2020. "SAHA Overcomes 5-FU Resistance in IFIT2-Depleted Oral Squamous Cell Carcinoma Cells" Cancers 12, no. 12: 3527. https://doi.org/10.3390/cancers12123527
APA StyleRegmi, P., Lai, K.-C., Liu, C.-J., & Lee, T.-C. (2020). SAHA Overcomes 5-FU Resistance in IFIT2-Depleted Oral Squamous Cell Carcinoma Cells. Cancers, 12(12), 3527. https://doi.org/10.3390/cancers12123527