Correlating Radiomic Features of Heterogeneity on CT with Circulating Tumor DNA in Metastatic Melanoma

 ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Results
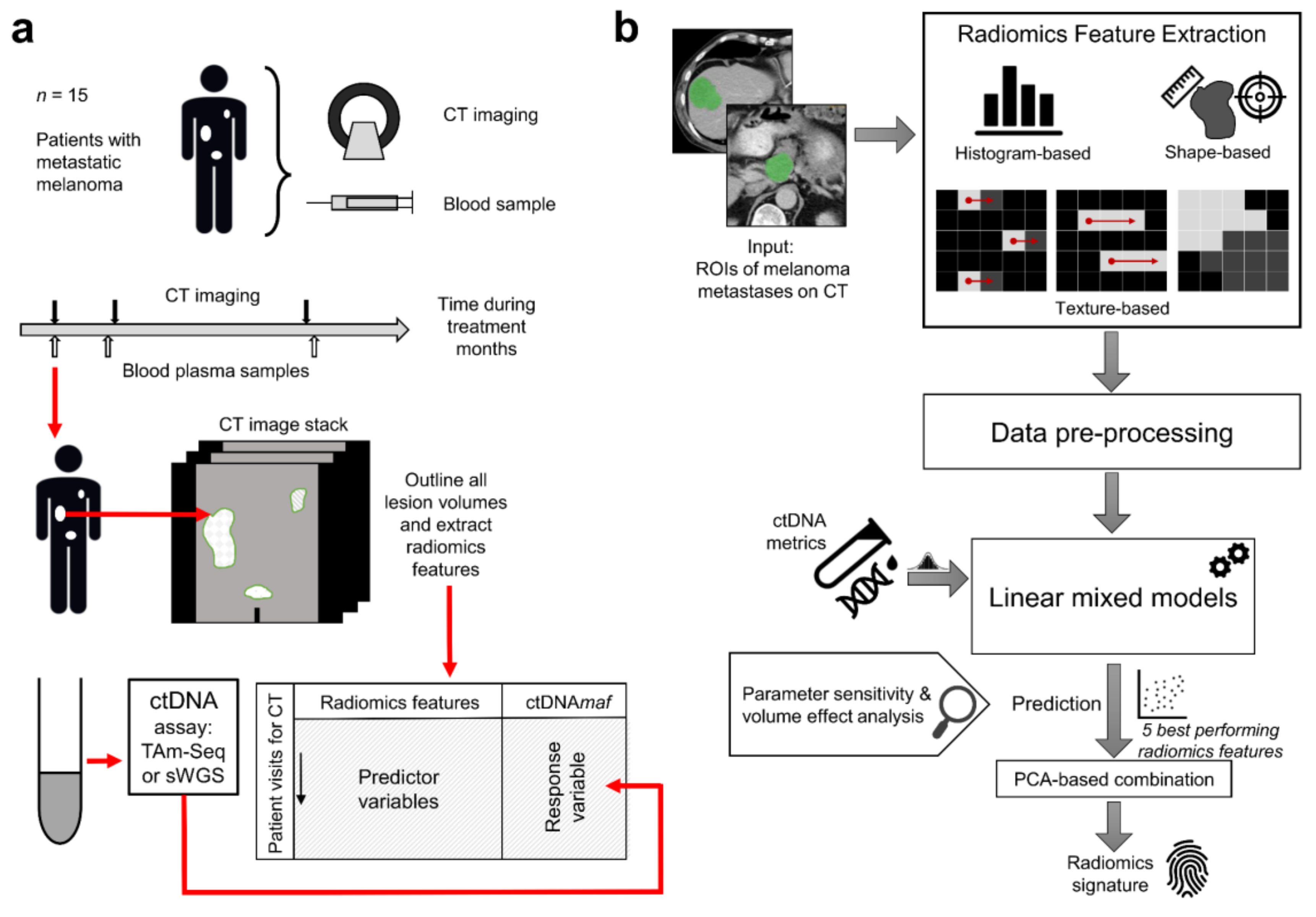
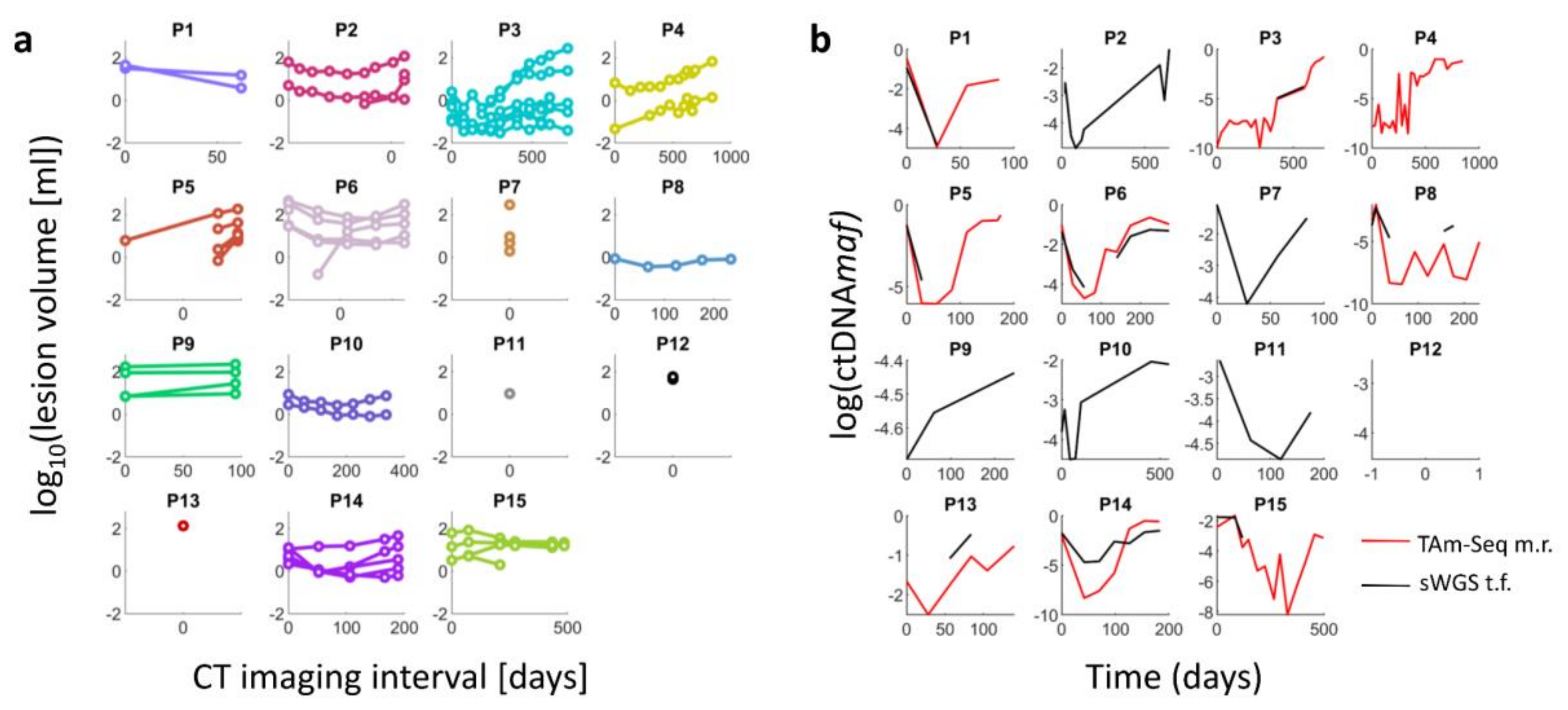
2.1. Change in the Volume of Lesions over Time and Radiomic Feature Extraction
2.2. Comparison of Radiomic Features and ctDNAmaf without Controlling for Lesion Volume
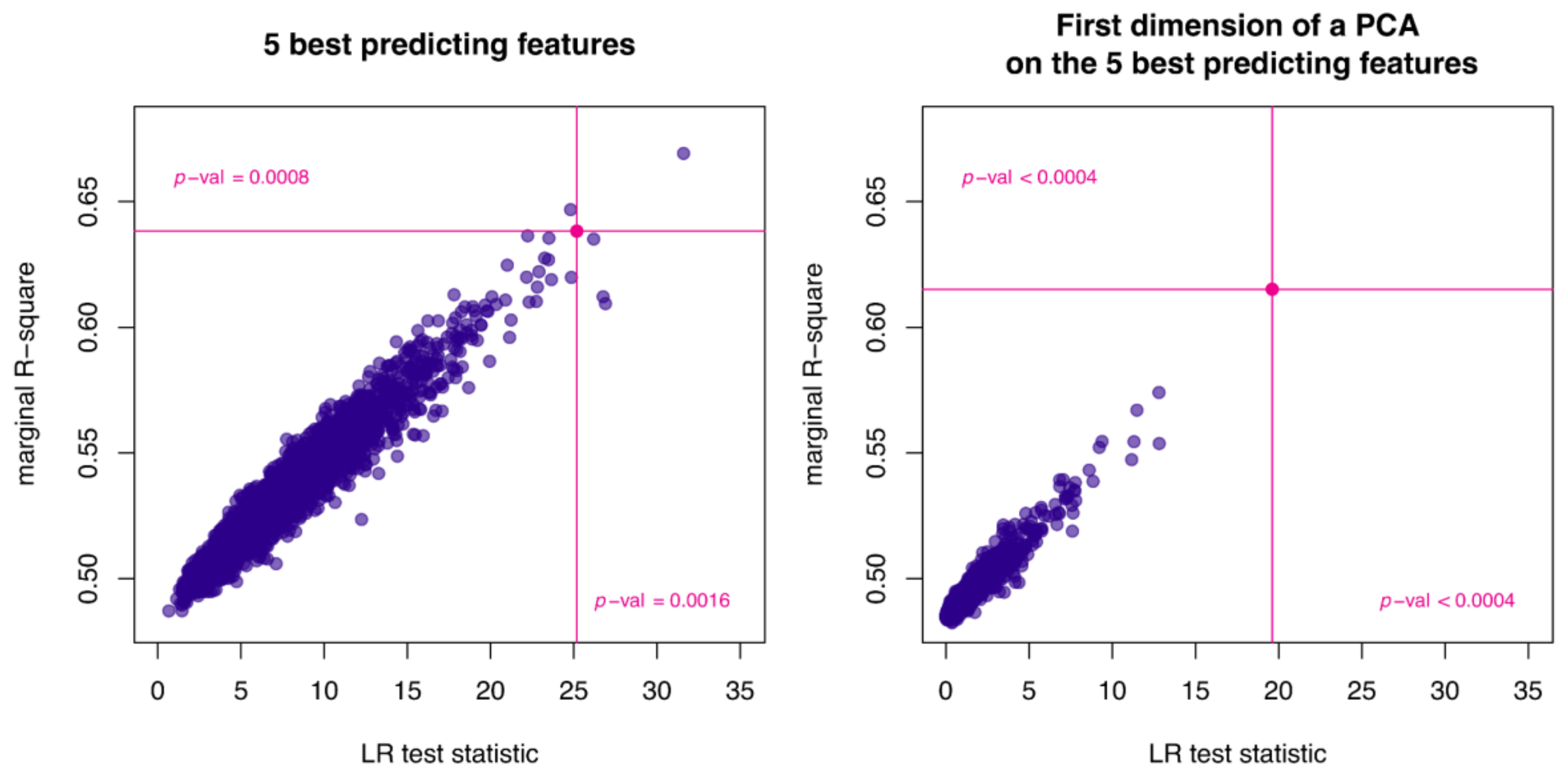
2.3. Comparison of Radiomic Features and ctDNAmaf Controlling for Lesion Volume
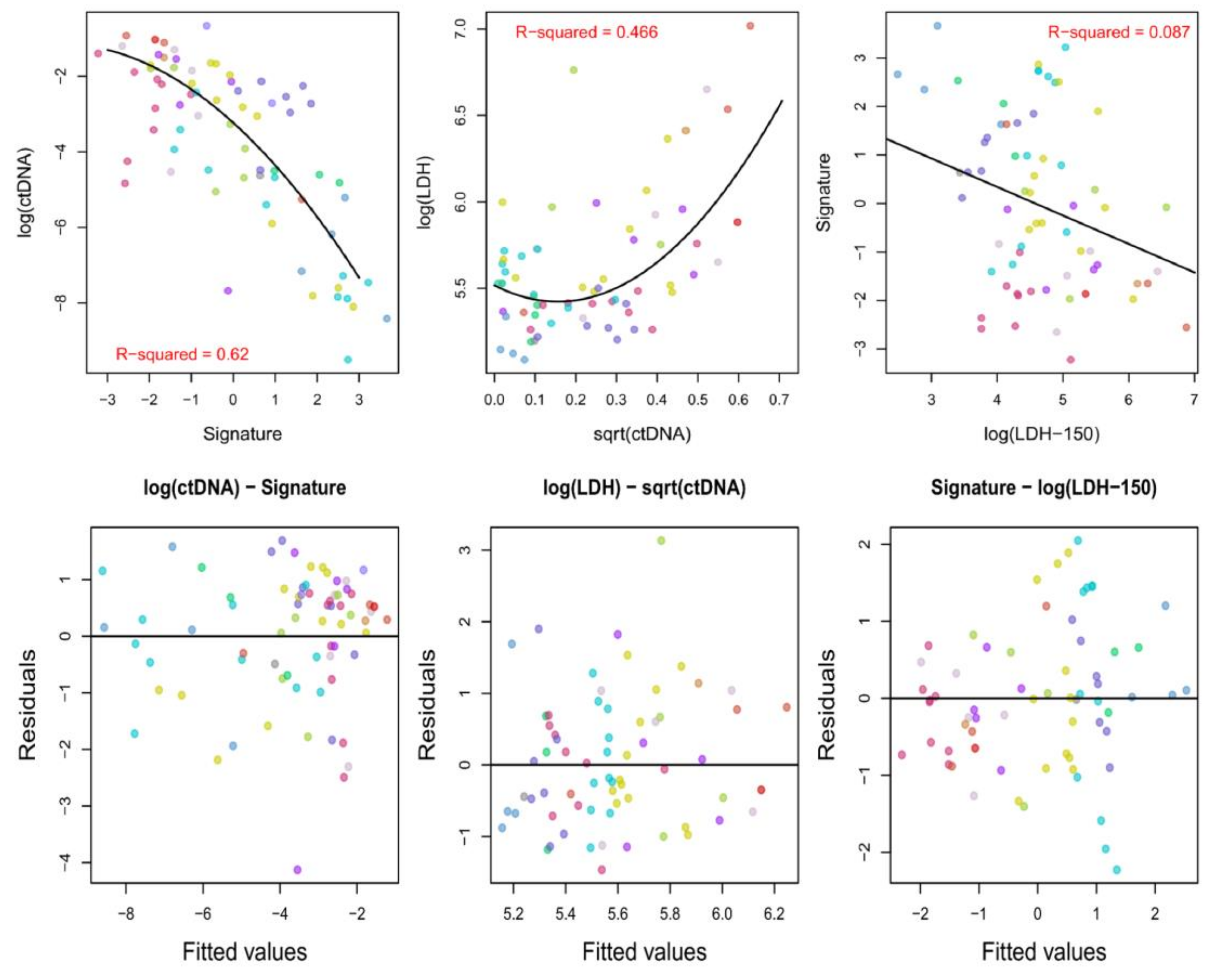
2.4. Analysis of the Associations between ctDNAmaf, the Derived Radiomics Signature, and Serum LDH Levels
3. Discussion
4. Materials and Methods
4.1. Patient Sample
4.2. Image Acquisition and Analysis
4.3. ctDNA Quantification
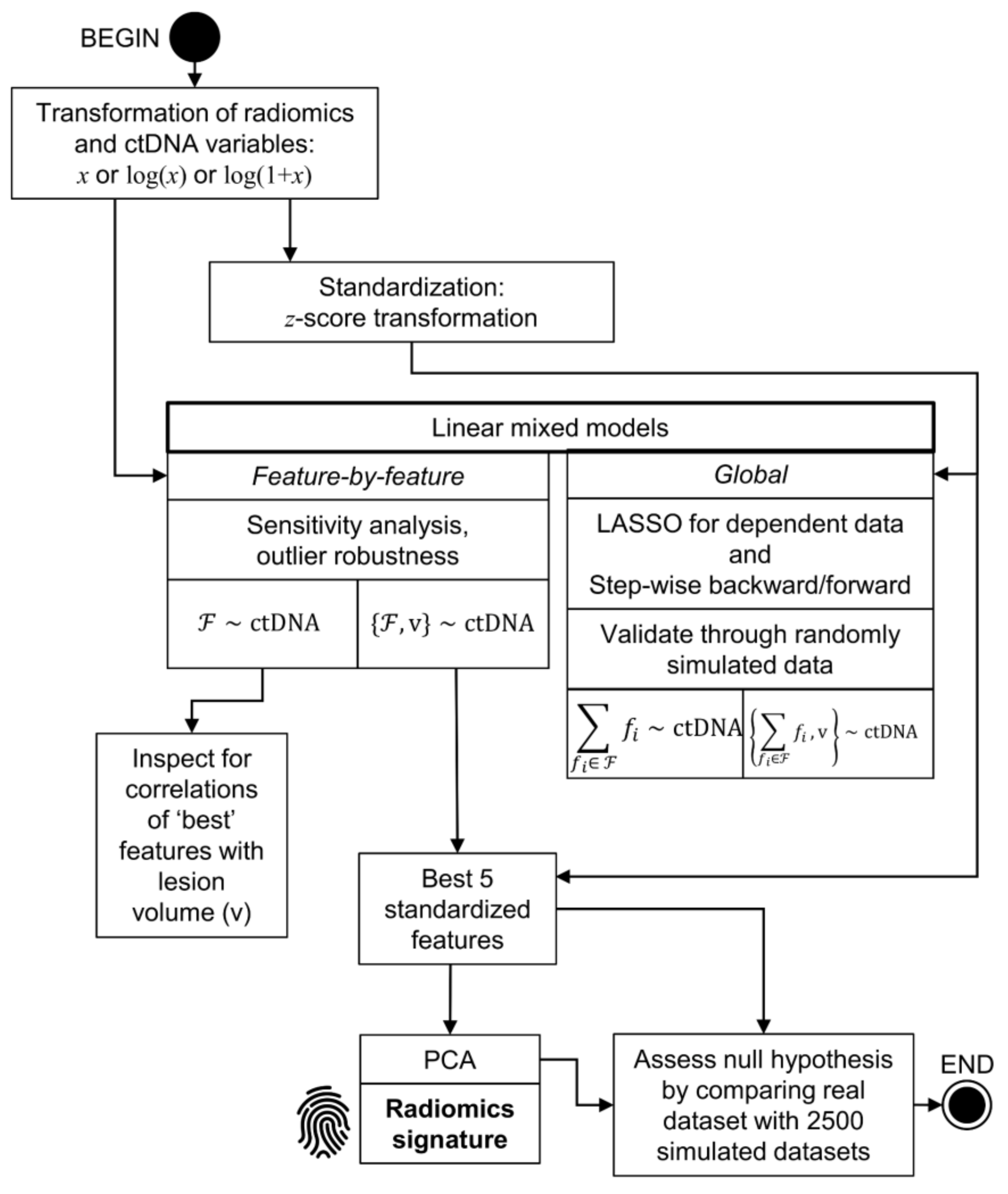
4.4. Statistical Analysis Methods
4.4.1. Descriptive Analyses and Data Transformation
4.4.2. Analysis without Controlling for Lesion Volume
4.4.3. Analysis Controlling for Lesion Volume
4.4.4. Analysis of the Associations between ctDNA, LDH Levels, and the Derived Radiomics Signature
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef] [Green Version]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients With Metastatic Melanoma Treated With Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.N.; Arshad, M.; Thornton, A.; Avesani, G.; Cunnea, P.; Curry, E.; Kanavati, F.; Liang, J.; Nixon, K.; Williams, S.T.; et al. A mathematical-descriptor of tumor-mesoscopic-structure from computed-tomography images annotates prognostic- and molecular-phenotypes of epithelial ovarian cancer. Nat. Commun. 2019, 10, 764. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Early change in circulating tumor DNA as a potential predictor of response to chemotherapy in patients with metastatic colorectal cancer. Sci. Rep. 2019, 9, 17358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.A.; Binkley, M.S.; Osmundson, E.C.; Alizadeh, A.A.; Diehn, M. Predicting Radiotherapy Responses and Treatment Outcomes Through Analysis of Circulating Tumor DNA. Semin. Radiat. Oncol. 2015, 25, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Lambin, P.; Rios-Velazquez, E.; Leijenaar, R.; Carvalho, S.; Van Stiphout, R.G.; Granton, P.; Zegers, C.M.; Gillies, R.; Boellard, R.; Dekker, A.; et al. Radiomics: Extracting more information from medical images using advanced feature analysis. Eur. J. Cancer 2012, 48, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larue, R.T.; Defraene, G.; De Ruysscher, D.; Lambin, P.; Van Elmpt, W. Quantitative radiomics studies for tissue characterization: A review of technology and methodological procedures. Br. J. Radiol. 2017, 90, 20160665. [Google Scholar] [CrossRef]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images Are More than Pictures, They Are Data. Radiology 2016, 278, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Aerts, H.J.; Velazquez, E.R.; Leijenaar, R.T.; Parmar, C.; Grossmann, P.; Carvalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef]
- Sala, E.; Mema, E.; Himoto, Y.; Veeraraghavan, H.; Brenton, J.D.; Snyder, A.; Weigelt, B.; Vargas, H.A. Unravelling tumour heterogeneity using next-generation imaging: Radiomics, radiogenomics, and habitat imaging. Clin. Radiol. 2017, 72, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S.B.; Patel, A.A. Monitoring immunotherapy outcomes with circulating tumor DNA. Immunotherapy 2018, 10, 1023–1025. [Google Scholar] [CrossRef]
- Thierry, A.R.; Mouliere, F.; Gongora, C.; Ollier, J.; Robert, B.; Ychou, M.; Del Rio, M.; Molina, F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010, 38, 6159–6175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, A.A.; Bischoff, F.Z.; Dang, D.; Baldwin, M.F.; Han, L.Y.; Lin, Y.G.; Merritt, W.M.; Landen, C.N., Jr.; Lu, C.; Gershenson, D.M.; et al. Circulating cell-free DNA: A novel biomarker for response to therapy in ovarian carcinoma. Cancer Biol. Ther. 2006, 5, 1369–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weissgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.I. A concordance correlation coefficient to evaluate reproducibility. Biometrics 1989, 45, 255–268. [Google Scholar] [CrossRef]
- Nioche, C.; Orlhac, F.; Boughdad, S.; Reuze, S.; Goya-Outi, J.; Robert, C.; Pellot-Barakat, C.; Soussan, M.; Frouin, F.; Buvat, I. LIFEx: A Freeware for Radiomic Feature Calculation in Multimodality Imaging to Accelerate Advances in the Characterization of Tumor Heterogeneity. Cancer Res. 2018, 78, 4786–4789. [Google Scholar] [CrossRef] [Green Version]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Yi, X.; Ma, J.; Guan, Y.; Chen, R.; Yang, L.; Xia, X. The feasibility of using mutation detection in ctDNA to assess tumor dynamics. Int. J. Cancer 2017, 140, 2642–2647. [Google Scholar] [CrossRef]
- Yip, S.S.; Aerts, H.J. Applications and limitations of radiomics. Phys. Med. Biol. 2016, 61, R150–R166. [Google Scholar] [CrossRef] [Green Version]
- Parekh, V.; Jacobs, M.A. Radiomics: A new application from established techniques. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 207–226. [Google Scholar] [CrossRef] [Green Version]
- Eton, O.; Legha, S.S.; Moon, T.E.; Buzaid, A.C.; Papadopoulos, N.E.; Plager, C.; Burgess, A.M.; Bedikian, A.Y.; Ring, S.; Dong, Q.; et al. Prognostic factors for survival of patients treated systemically for disseminated melanoma. J. Clin. Oncol. 1998, 16, 1103–1111. [Google Scholar] [CrossRef]
- Weide, B.; Elsasser, M.; Buttner, P.; Pflugfelder, A.; Leiter, U.; Eigentler, T.K.; Bauer, J.; Witte, M.; Meier, F.; Garbe, C. Serum markers lactate dehydrogenase and S100B predict independently disease outcome in melanoma patients with distant metastasis. Br. J. Cancer 2012, 107, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Butler, T.M.; Spellman, P.T.; Gray, J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr. Opin. Genet. Dev. 2017, 42, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Heider, K.; Gale, D.; Murphy, S.; Fisher, E.; Mouliere, F.; Ruiz-Valdepenas, A.; Santonja, A.; Morris, J.; Chandrananda, D.; et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Durot, C.; Mule, S.; Soyer, P.; Marchal, A.; Grange, F.; Hoeffel, C. Metastatic melanoma: Pretreatment contrast-enhanced CT texture parameters as predictive biomarkers of survival in patients treated with pembrolizumab. Eur. Radiol. 2019, 29, 3183–3191. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.G.; Madsen, A.T.; Gammelgaard, K.R.; Aggerholm-Pedersen, N.; Sorensen, B.S.; Ollegaard, T.H.; Jakobsen, M.R. Inflammatory Cytokines and ctDNA Are Biomarkers for Progression in Advanced-Stage Melanoma Patients Receiving Checkpoint Inhibitors. Cancers 2020, 12, 1414. [Google Scholar] [CrossRef] [PubMed]
- Herbreteau, G.; Vallee, A.; Knol, A.C.; Theoleyre, S.; Quereux, G.; Frenard, C.; Varey, E.; Hofman, P.; Khammari, A.; Dreno, B.; et al. Circulating Tumour DNA Is an Independent Prognostic Biomarker for Survival in Metastatic BRAF or NRAS-Mutated Melanoma Patients. Cancers 2020, 12, 1871. [Google Scholar] [CrossRef]
- Ahlborn, L.B.; Tuxen, I.V.; Mouliere, F.; Kinalis, S.; Schmidt, A.Y.; Rohrberg, K.S.; Santoni-Rugiu, E.; Nielsen, F.C.; Lassen, U.; Yde, C.W.; et al. Circulating tumor DNA as a marker of treatment response in BRAF V600E mutated non-melanoma solid tumors. Oncotarget 2018, 9, 32570–32579. [Google Scholar] [CrossRef] [Green Version]
- Hesketh, R.L.; Zhu, A.X.; Oklu, R. Radiomics and circulating tumor cells: Personalized care in hepatocellular carcinoma? Diagn. Interv. Radiol. 2015, 21, 78–84. [Google Scholar] [CrossRef]
- Neri, E.; Del Re, M.; Paiar, F.; Erba, P.; Cocuzza, P.; Regge, D.; Danesi, R. Radiomics and liquid biopsy in oncology: The holons of systems medicine. Insights Imaging 2018, 9, 915–924. [Google Scholar] [CrossRef] [Green Version]
- Efron, B. Nonparametric estimates of standard error: The jackknife, the bootstrap and other methods. Biometrika 1981, 68, 589–599. [Google Scholar] [CrossRef]
- Lu, L.; Liang, Y.; Schwartz, L.H.; Zhao, B. Reliability of Radiomic Features Across Multiple Abdominal CT Image Acquisition Settings: A Pilot Study Using ACR CT Phantom. Tomography 2019, 5, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Shafiq-Ul-Hassan, M.; Latifi, K.; Zhang, G.; Ullah, G.; Gillies, R.; Moros, E. Voxel size and gray level normalization of CT radiomic features in lung cancer. Sci. Rep. 2018, 8, 10545. [Google Scholar] [CrossRef] [PubMed]
- Mackin, D.; Fave, X.; Zhang, L.; Fried, D.; Yang, J.; Taylor, B.; Rodriguez-Rivera, E.; Dodge, C.; Jones, A.K.; Court, L. Measuring Computed Tomography Scanner Variability of Radiomics Features. Investig. Radiol. 2015, 50, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Menzies, A.M.; Carlino, M.S.; McEvoy, A.C.; Sandhu, S.; Weppler, A.M.; Diefenbach, R.J.; Dawson, S.J.; Kefford, R.F.; Millward, M.J.; et al. Longitudinal Monitoring of ctDNA in Patients with Melanoma and Brain Metastases Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2020, 26, 4064–4071. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.W.; Ashburner, J.; Breman, H.; Fissell, K.; Haselgrove, C.; Holmes, C.J.; Lancaster, J.L.; Rex, D.E.; Smith, S.M.; Woodward, J.B.; et al. A (sort of) new image data format standard: NIfTI-1. In Proceedings of the 10th Annual Meeting of the Organization for Human Brain Mapping, Budapest, Hungary, 13–17 June 2004. [Google Scholar]
- Zwanenburg, A.; Vallieres, M.; Abdalah, M.A.; Aerts, H.; Andrearczyk, V.; Apte, A.; Ashrafinia, S.; Bakas, S.; Beukinga, R.J.; Boellaard, R.; et al. The Image Biomarker Standardization Initiative: Standardized Quantitative Radiomics for High-Throughput Image-based Phenotyping. Radiology 2020, 295, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Haralick, R.M. Statistical and Structural Approaches to Texture. Proc. IEEE 1979, 67, 786–804. [Google Scholar] [CrossRef]
- Haralick, R.M.; Shanmugam, K.; Dinstein, I. Textural Features for Image Classification. IEEE Trans. Syst. Man Cybern. 1973, Smc3, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Amadasun, M.; King, R. Textural Features Corresponding to Textural Properties. IEEE Trans. Syst. Man Cybern. 1989, 19, 1264–1274. [Google Scholar] [CrossRef]
- Galloway, M.M. Texture analysis using gray level run lengths. Comput. Graph. Image Process. 1975, 4, 172–179. [Google Scholar] [CrossRef]
- Thibault, G.; Fertil, B.; Navarro, C.; Pereira, S.; Cau, P.; Levy, N.; Sequeira, J.; Mari, J.L. Shape and Texture Indexes Application to Cell Nuclei Classification. Int. J. Pattern Recogn. 2013, 27, 1357002. [Google Scholar] [CrossRef]
- Van Griethuysen, J.J.M.; Fedorov, A.; Parmar, C.; Hosny, A.; Aucoin, N.; Narayan, V.; Beets-Tan, R.G.H.; Fillion-Robin, J.C.; Pieper, S.; Aerts, H. Computational Radiomics System to Decode the Radiographic Phenotype. Cancer Res. 2017, 77, e104–e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.C.M. Monitoring Trace Levels of ctDNA Using Integration of Variant Reads. Ph.D. Thesis, University of Cambridge, Cambridge, UK, May 2019. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Pinheiro, J.C.; Bates, D.M. Mixed-Effects Models in S and S-PLUS; Springer: New York, NY, USA, 2000. [Google Scholar]
- Koller, M. robustlmm: An R Package for Robust Estimation of Linear Mixed-Effects Models. J. Stat. Softw. 2016, 75, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Nakagawa, S.; Johnson, P.C.D.; Schielzeth, H. The coefficient of determination R-2 and intra-class correlation coefficient from generalized linear mixed-effects models revisited and expanded. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Schelldorfer, J.; Buhlmann, P.; Van De Geer, S. Estimation for High-Dimensional Linear Mixed-Effects Models Using l(1)-Penalization. Scand. J. Stat. 2011, 38, 197–214. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. Roy. Stat. Soc. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Campbell, N.A.; Lopuhaa, H.P.; Rousseeuw, P.J. On the calculation of a robust S-estimator of a covariance matrix. Stat. Med. 1998, 17, 2685–2695. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender | Lesion Analyzed | Intra-Axial Brain Lesions 1 | AJCC Stage | BRAF Mutation Status | Baseline Serum LDH (UL) | Treatment | CT Visit Number | RECIST 1.1 Response Assessment | Target Lesion: Descriptive Volume Changes 2 | Lesion Volume at Baseline (mL) | Target Lesion: Fractional Volume at Baseline (%) 3 | Number of Lesions (>1 cm3) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Left axillary lymph node metastasis | N | IV | + | - | Vemurafenib | i | Progressive Disease | Baseline | 45 | 58 | 2 |
| ii | Partial Response | Large decrease | |||||||||||
| 2 | M | Para-aortic lymph node metastasis | N | IV | + | 261 | Vemurafenib | i | Partial Response | Baseline | 65 | 93 | 4 |
| ii | Partial Response | Large decrease | |||||||||||
| iii | Partial Response | Large decrease | |||||||||||
| iv | Partial Response | Large decrease | |||||||||||
| v | Partial Response | Large decrease | |||||||||||
| vi | Progressive Disease | Small increase | |||||||||||
| vii | Stable Disease | Large increase (borderline) | |||||||||||
| viii | Progressive Disease | Large increase | |||||||||||
| ix | Progressive Disease | Large increase | |||||||||||
| 3 | M | Axillary lymph node metastasis left | Y | IV | + | 252 | Vemurafenib | i | Progressive Disease | Baseline | 3 | 63 | 7 |
| ii | Partial Response | Large decrease | |||||||||||
| iii | Stable Disease | Small decrease | |||||||||||
| iv | Stable Disease | Small decrease | |||||||||||
| v | Stable Disease | Small decrease | |||||||||||
| Ipilimumab | vi | Progressive Disease | Large increase | ||||||||||
| vii | Progressive Disease | Large increase | |||||||||||
| Pazopanib | viii | Progressive Disease | Large increase | ||||||||||
| ix | Stable Disease | Small increase | |||||||||||
| Dabrafenib/Trametinib | x | Progressive Disease | Small increase | ||||||||||
| xi | Progressive Disease | Large increase | |||||||||||
| 4 | F | Iliac lymph node metastasis right | N | IV | + | 246 | Vemurafenib | i | Stable Disease | Baseline | 7 | 99 | 2 |
| ii | Stable Disease | Small decrease | |||||||||||
| iii | Stable Disease | Small decrease | |||||||||||
| iv | Stable Disease | Small decrease | |||||||||||
| v | Stable Disease | Small decrease | |||||||||||
| vi | Stable Disease | Large increase | |||||||||||
| vii | Stable Disease | Large increase | |||||||||||
| viii | Progressive Disease | Large increase | |||||||||||
| ix | Progressive Disease | Large increase | |||||||||||
| pan-RAF inhibitor | x | Stable Disease | Small decrease | ||||||||||
| xi | Stable Disease | Small increase | |||||||||||
| xii | Progressive Disease | Large increase | |||||||||||
| 5 | M | Omental metastasis | U | IV | + | 258 | Vemurafenib | i | Partial Response | Baseline | 6 | 100 | 6 |
| ii | Progressive Disease | Large increase | |||||||||||
| Ipilimumab | iii | Progressive Disease | Small increase | ||||||||||
| 6 | M | Omental metastasis | N | IV | + | 773 | Dabrafenib/Trametinib | i | Progressive Disease | Baseline | 461 | 44 | 6 |
| ii | Partial Response | Large decrease | |||||||||||
| iii | Partial Response | Large decrease | |||||||||||
| iv | Progressive Disease | Large decrease (borderline) | |||||||||||
| v | Progressive Disease | Large increase | |||||||||||
| 7 | M | Right axillary lymph node | Y | IV | + | 699 | Vemurafenib | i | Progressive Disease | Baseline | 296 | 95 | 4 |
| 8 | F | Femoral, subcutaneous metastasis right | Y | IV | + | 175 | Vemurafenib | i | Progressive Disease | Baseline | 1 | 100 | 1 |
| ii | Stable Disease | Small decrease | |||||||||||
| iii | Stable Disease | Small increase | |||||||||||
| iv | Stable Disease | Small decrease | |||||||||||
| v | Progressive Disease | Large increase | |||||||||||
| 9 | F | Dorsal subcutaneous metastasis abutting the iliac crest | N | IV | + | 201 | Dabrafenib | i | n/a | Baseline | 2 | 93 | 4 |
| ii | n/a | Large increase | |||||||||||
| iii | n/a | Large increase | |||||||||||
| 10 | M | Right external iliac lymph node metastasis | Y | IV | + | 254 | Dabrafenib/Trametinib | i | Partial Response | Baseline | 9 | 75 | 2 |
| ii | Partial Response | Small decrease | |||||||||||
| iii | Partial Response | Small decrease | |||||||||||
| iv | Partial Response | Large decrease | |||||||||||
| v | Stable Disease | Small increase | |||||||||||
| vi | Stable Disease | Large increase (borderline) | |||||||||||
| vii | Progressive Disease | Large increase | |||||||||||
| 11 | M | Left supraclavicular lymph node metastasis | Y | IV | + | 169 | Vemurafenib | i | Stable Disease | Baseline | 9 | 100 | 1 |
| 12 | F | Splenic lymph node metastasis | U | IV | + | 233 | No treatment | i | Progressive Disease | Baseline | 58 | 56 | 2 |
| 13 | M | Left axillary lymph node metastasis | U | IV | + | 492 | Vemurafenib | i | Progressive Disease | Baseline | 136 | 100 | 1 |
| 14 | M | Left iliac lymph node metastasis | Y | IV | + | 351 | Dabrafenib/Trametinib | i | Progressive Disease | Baseline | 11 | 31 | 5 |
| ii | Partial Response | Small increase | |||||||||||
| iii | Progressive Disease | Small increase | |||||||||||
| iv | Progressive Disease | Large increase | |||||||||||
| Ipilimumab | v | Progressive Disease | Small increase | ||||||||||
| 15 | F | Left inguinal lymph node metastasis | Y | IV | − | 392 | pan-RAF inhibitor | i | Progressive Disease | Baseline | 63 | 78 | 4 |
| ii | Progressive Disease | Small increase | |||||||||||
| Ipilimumab | iii | Stable Disease | Small decrease | ||||||||||
| Pembrolizumab | iv | Stable Disease | Small decrease | ||||||||||
| v | Stable Disease | Small decrease | |||||||||||
| vi | Stable Disease | Small increase |
| 1 Feature | t-Value | p-Value | Adj. p-Value | 2 Sig. | R2 |
|---|---|---|---|---|---|
| GLNUz | 8.263 | 1.11 × 10-11 | 1.54 × 10-9 | *** | 0.517 |
| GLNUr | 7.531 | 7.49 × 10-10 | 2.80 × 10-8 | *** | 0.496 |
| Coarseness | −7.527 | 6.86 × 10-10 | 2.80 × 10-8 | *** | 0.495 |
| ZLNU | 7.273 | 8.06 × 10-10 | 2.80 × 10-8 | *** | 0.458 |
| RLNU | 7.327 | 1.43 × 10-9 | 3.98 × 10-8 | *** | 0.477 |
| Volume | 7.295 | 1.79 × 10-9 | 4.16 × 10-8 | *** | 0.476 |
| Busyness | 6.608 | 1.57 × 10-8 | 3.12 × 10-7 | *** | 0.428 |
| Contrast | −6.143 | 2.81 × 10-7 | 4.89 × 10-6 | *** | 0.434 |
| SRLGE | −5.973 | 3.75 × 10-7 | 5.80 × 10-6 | *** | 0.411 |
| LGRE | −5.673 | 7.99 × 10-7 | 1.11 × 10-5 | *** | 0.376 |
| StdDev | −5.137 | 2.59 × 10-6 | 3.27 × 10-5 | *** | 0.295 |
| Mean | 5.218 | 4.24 × 10-6 | 4.92 × 10-5 | *** | 0.331 |
| ZP | −5.069 | 4.67 × 10-6 | 5.00 × 10-5 | *** | 0.298 |
| HGRE | 5.075 | 6.81 × 10-6 | 6.76 × 10-5 | *** | 0.321 |
| Entropy_h | −4.850 | 8.19 × 10-6 | 7.59 × 10-5 | *** | 0.285 |
| LRHGE | 4.703 | 3.04 × 10-5 | 2.65 × 10-4 | *** | 0.306 |
| Energy | 4.494 | 3.30 × 10-5 | 2.70 × 10-4 | *** | 0.262 |
| Correlation | −3.676 | 4.80 × 10-4 | 3.51 × 10-3 | ** | 0.191 |
| SRHGE | 3.699 | 4.75 × 10-4 | 3.51 × 10-3 | ** | 0.192 |
| LGZE | −3.740 | 5.23 × 10-4 | 3.64 × 10-3 | ** | 0.217 |
| Sphericity | −3.859 | 5.84 × 10-4 | 3.87 × 10-3 | ** | 0.230 |
| Kurtosis | 3.586 | 6.84 × 10-4 | 4.32 × 10-3 | ** | 0.198 |
| LRE | 3.522 | 1.04 × 10-3 | 6.26 × 10-3 | ** | 0.194 |
| SRE | −3.456 | 1.26 × 10-3 | 7.29 × 10-3 | ** | 0.191 |
| RP | −3.428 | 1.35 × 10-3 | 7.53 × 10-3 | ** | 0.187 |
| Uniformity | 3.189 | 2.23 × 10-3 | 1.19 × 10-2 | * | 0.146 |
| [random35] | 2.975 | 4.09 × 10-3 | 2.11 × 10-2 | * | 0.095 |
| Feature | t-Value | p-Value | Adjusted p-Value | R2 |
|---|---|---|---|---|
| Correlation | −3.227 | 0.0020 | 0.1708 | 0.536 |
| GLNUz | 3.151 | 0.0025 | 0.1708 | 0.515 |
| StdDev | −2.800 | 0.0067 | 0.2992 | 0.531 |
| LGRE | −2.700 | 0.0105 | 0.2992 | 0.536 |
| [random78] | −2.506 | 0.0149 | 0.2992 | 0.514 |
| Coarseness | −2.512 | 0.0150 | 0.2992 | 0.515 |
| [random45] | 2.497 | 0.0152 | 0.2992 | 0.508 |
| Mean | 2.419 | 0.0206 | 0.3060 | 0.523 |
| SRHGE | 2.359 | 0.0230 | 0.3060 | 0.518 |
| [random83] | 2.322 | 0.0234 | 0.3060 | 0.506 |
| LRLGE | −2.300 | 0.0259 | 0.3060 | 0.506 |
| HGRE | 2.308 | 0.0266 | 0.3060 | 0.520 |
| [random77] | 2.135 | 0.0367 | 0.3670 | 0.501 |
| [random80] | −2.126 | 0.0372 | 0.3670 | 0.498 |
| SRLGE | −2.131 | 0.0399 | 0.3672 | 0.519 |
| GLNUr | 2.034 | 0.0459 | 0.3736 | 0.509 |
| [random91] | −2.034 | 0.0460 | 0.3736 | 0.496 |
| [random79] | −2.005 | 0.0490 | 0.3758 | 0.502 |
| [random35} | 1.851 | 0.0687 | 0.4853 | 0.488 |
| Entropy_h | −1.839 | 0.0703 | 0.4853 | 0.504 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gill, A.B.; Rundo, L.; Wan, J.C.M.; Lau, D.; Zawaideh, J.P.; Woitek, R.; Zaccagna, F.; Beer, L.; Gale, D.; Sala, E.; et al. Correlating Radiomic Features of Heterogeneity on CT with Circulating Tumor DNA in Metastatic Melanoma. Cancers 2020, 12, 3493. https://doi.org/10.3390/cancers12123493
Gill AB, Rundo L, Wan JCM, Lau D, Zawaideh JP, Woitek R, Zaccagna F, Beer L, Gale D, Sala E, et al. Correlating Radiomic Features of Heterogeneity on CT with Circulating Tumor DNA in Metastatic Melanoma. Cancers. 2020; 12(12):3493. https://doi.org/10.3390/cancers12123493
Chicago/Turabian StyleGill, Andrew B, Leonardo Rundo, Jonathan C. M. Wan, Doreen Lau, Jeries P. Zawaideh, Ramona Woitek, Fulvio Zaccagna, Lucian Beer, Davina Gale, Evis Sala, and et al. 2020. "Correlating Radiomic Features of Heterogeneity on CT with Circulating Tumor DNA in Metastatic Melanoma" Cancers 12, no. 12: 3493. https://doi.org/10.3390/cancers12123493
APA StyleGill, A. B., Rundo, L., Wan, J. C. M., Lau, D., Zawaideh, J. P., Woitek, R., Zaccagna, F., Beer, L., Gale, D., Sala, E., Couturier, D.-L., Corrie, P. G., Rosenfeld, N., & Gallagher, F. A. (2020). Correlating Radiomic Features of Heterogeneity on CT with Circulating Tumor DNA in Metastatic Melanoma. Cancers, 12(12), 3493. https://doi.org/10.3390/cancers12123493