The Δ133p53 Isoforms, Tuners of the p53 Pathway



Abstract
Simple Summary
Abstract
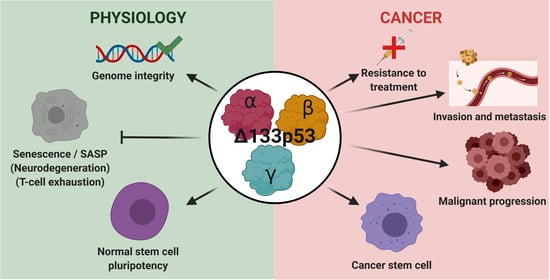
1. Introduction
2. Regulatory Mechanisms of the Δ133p53 Isoforms
2.1. RNA Level
2.2. Protein Level
3. The Δ133p53 Isoforms in Cancers
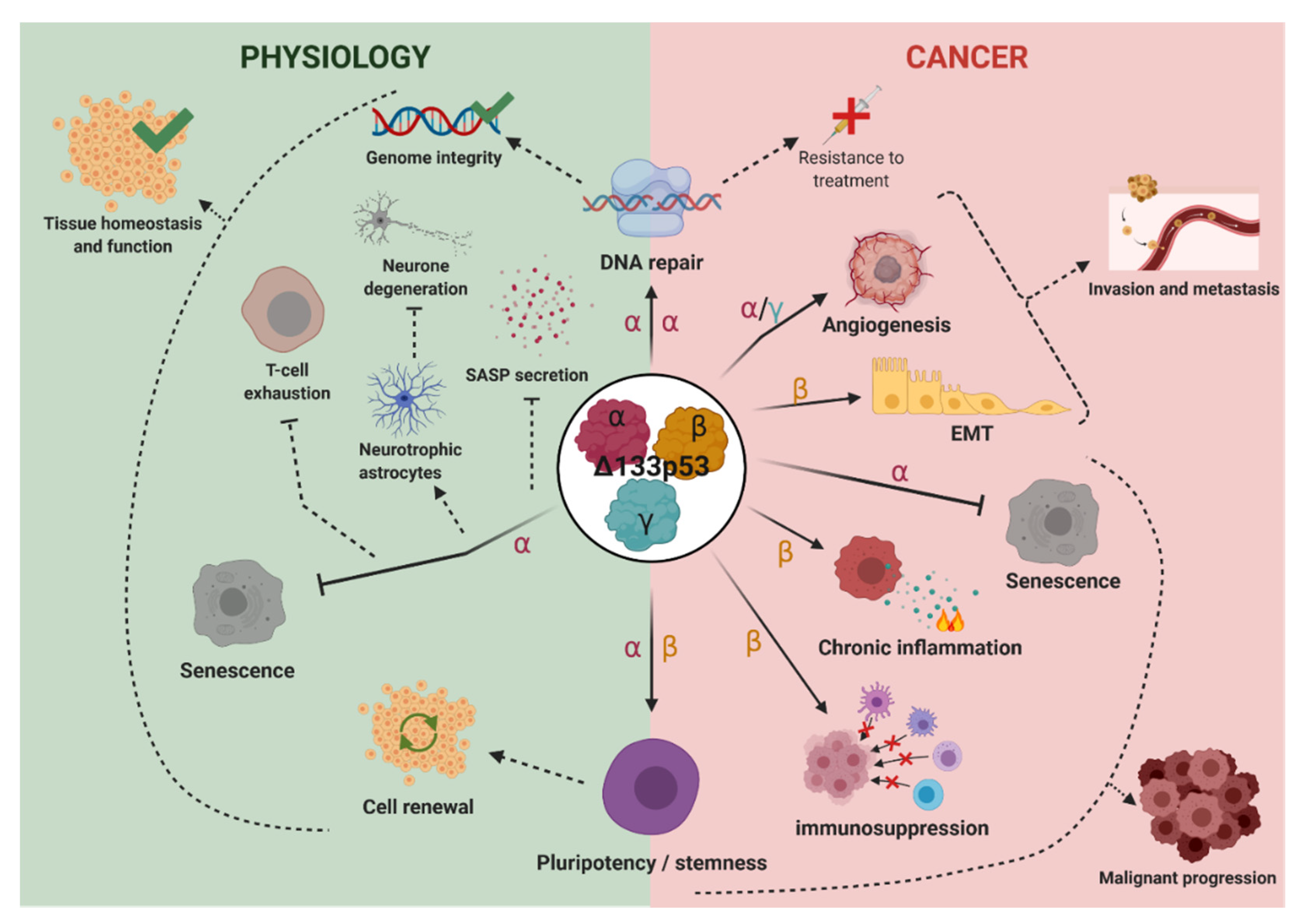
4. Biological Activities
4.1. Δ133p53α
4.1.1. Δ133p53α in Cellular Senescence and SASP
4.1.2. Δ133p53α in DNA Repair
4.1.3. Δ133p53α in Pluripotent Stem Cells Regulation
4.1.4. Δ133p53α in Cancer Biology
4.2. Δ133p53β
5. N-Terminally Truncated p53 Isoforms through Evolution: Models for Human Δ133p53 Isoforms?
5.1. Drosophila
5.2. Mouse
5.3. Zebrafish
6. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. P53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Levine, A.J. The p53 Protein: From Cell Regulation to Cancer; A Cold Spring Harbor Perspectives in Medicine Collection; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2016. [Google Scholar]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Bellini, I.; Pitto, L.; Marini, M.G.; Porcu, L.; Moi, P.; Garritano, S.; Boldrini, L.; Rainaldi, G.; Fontanini, G.; Chiarugi, M.; et al. DeltaN133p53 expression levels in relation to haplotypes of the TP53 internal promoter region. Hum. Mutat. 2010, 31, 456–465. [Google Scholar] [CrossRef]
- Marcel, V.; Petit, I.; Murray-Zmijewski, F.; Goullet de Rugy, T.; Fernandes, K.; Meuray, V.; Diot, A.; Lane, D.P.; Aberdam, D.; Bourdon, J.-C. Diverse p63 and p73 isoforms regulate Δ133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012, 19, 816–826. [Google Scholar] [CrossRef]
- Eiholzer, R.A.; Mehta, S.; Kazantseva, M.; Drummond, C.J.; McKinney, C.; Young, K.; Slater, D.; Morten, B.C.; Avery-Kiejda, K.A.; Lasham, A.; et al. Intronic TP53 Polymorphisms Are Associated with Increased Δ133TP53 Transcript, Immune Infiltration and Cancer Risk. Cancers 2020, 12, 2472. [Google Scholar] [CrossRef]
- Moore, H.C.; Jordan, L.B.; Bray, S.E.; Baker, L.; Quinlan, P.R.; Purdie, C.A.; Thompson, A.M.; Bourdon, J.-C.; Fuller-Pace, F.V. The RNA helicase p68 modulates expression and function of the Δ133 isoform(s) of p53, and is inversely associated with Δ133p53 expression in breast cancer. Oncogene 2010, 29, 6475–6484. [Google Scholar] [CrossRef]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.-C.; Lane, D.P.; Bourdon, J.-C. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.-M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.-C. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Crutchley, J.; Zhang, D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage-induced Alternative Splicing Pathway that Regulates p53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Chen, M.; Dai, R.; Zhang, Y.; Zhao, H.; Song, Z.; Zhang, L.; Li, Z.; Feng, Y.; Gao, H.; et al. SRSF1 promotes vascular smooth muscle cell proliferation through a Δ133p53/EGR1/KLF5 pathway. Nat. Commun. 2017, 8, 16016. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA. FASEB J. 2019, 33, 4225–4235. [Google Scholar] [CrossRef]
- Ma, J.; Martin, J.D.; Zhang, H.; Auger, K.R.; Ho, T.F.; Kirkpatrick, R.B.; Grooms, M.H.; Johanson, K.O.; Tummino, P.J.; Copeland, R.A.; et al. A second p53 binding site in the central domain of Mdm2 is essential for p53 ubiquitination. Biochemistry 2006, 45, 9238–9245. [Google Scholar] [CrossRef]
- Camus, S.; Ménendez, S.; Fernandes, K.; Kua, N.; Liu, G.; Xirodimas, D.P.; Lane, D.P.; Bourdon, J.-C. The p53 isoforms are differentially modified by Mdm2. Cell Cycle Georget. Tex 2012, 11, 1646–1655. [Google Scholar] [CrossRef][Green Version]
- Thut, C.J.; Goodrich, J.A.; Tjian, R. Repression of p53-mediated transcription by MDM2: A dual mechanism. Genes Dev. 1997, 11, 1974–1986. [Google Scholar] [CrossRef]
- Mirnezami, A.H.; Campbell, S.J.; Darley, M.; Primrose, J.N.; Johnson, P.W.M.; Blaydes, J.P. Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. Curr. Biol. CB 2003, 13, 1234–1239. [Google Scholar] [CrossRef]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257. [Google Scholar] [CrossRef]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- DeHart, C.J.; Chahal, J.S.; Flint, S.J.; Perlman, D.H. Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol. Cell. Proteom. MCP 2014, 13, 1–17. [Google Scholar] [CrossRef]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Anensen, N.; Oyan, A.M.; Bourdon, J.-C.; Kalland, K.H.; Bruserud, O.; Gjertsen, B.T. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 3985–3992. [Google Scholar] [CrossRef]
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545. [Google Scholar] [CrossRef]
- Hjelle, S.M.; Sulen, A.; Øye, O.K.; Jørgensen, K.; McCormack, E.; Hollund, B.E.; Gjertsen, B.T. Leukocyte p53 protein biosignature through standard-aligned two-dimensional immunoblotting. J. Proteom. 2012, 76, 69–78. [Google Scholar] [CrossRef]
- Kloster, M.M.; Naderi, E.H.; Haaland, I.; Gjertsen, B.T.; Blomhoff, H.K.; Naderi, S. cAMP signalling inhibits p53 acetylation and apoptosis via HDAC and SIRT deacetylases. Int. J. Oncol. 2013, 42, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.D.; Hicke, L. Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem. 2003, 278, 35857–35860. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Tsai, P.; Lasham, A.; Campbell, H.; Reddel, R.; Braithwaite, A.; Print, C. A Study of TP53 RNA Splicing Illustrates Pitfalls of RNA-seq Methodology. Cancer Res. 2016, 76, 7151–7159. [Google Scholar] [CrossRef] [PubMed]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 isoform Δ133p53β in glioblastomas: An alternative to mutant p53 in promoting tumor development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Δ133p53β isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis. 2019, 10, 631. [Google Scholar] [CrossRef]
- Lasham, A.; Tsai, P.; Fitzgerald, S.J.; Mehta, S.Y.; Knowlton, N.S.; Braithwaite, A.W.; Print, C.G. Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts. Cancers 2020, 12, 769. [Google Scholar] [CrossRef]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef]
- Arsic, N.; Ho-Pun-Cheung, A.; Evelyne, C.; Assenat, E.; Jarlier, M.; Anguille, C.; Colard, M.; Pezet, M.; Roux, P.; Gadea, G. The p53 isoform delta133p53ß regulates cancer cell apoptosis in a RhoB-dependent manner. PLoS ONE 2017, 12, e0172125. [Google Scholar] [CrossRef]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. ∆133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef]
- Nutthasirikul, N.; Limpaiboon, T.; Leelayuwat, C.; Patrakitkomjorn, S.; Jearanaikoon, P. Ratio disruption of the ∆133p53 and TAp53 isoform equilibrium correlates with poor clinical outcome in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2013, 42, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Nutthasirikul, N.; Hahnvajanawong, C.; Techasen, A.; Limpaiboon, T.; Leelayuwat, C.; Chau-In, S.; Jearanaikoon, P. Targeting the ∆133p53 isoform can restore chemosensitivity in 5-fluorouracil-resistant cholangiocarcinoma cells. Int. J. Oncol. 2015, 47, 2153–2164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fragou, A.; Tzimagiorgis, G.; Karageorgopoulos, C.; Barbetakis, N.; Lazopoulos, A.; Papaioannou, M.; Haitoglou, C.; Kouidou, S. Increased Δ133p53 mRNA in lung carcinoma corresponds with reduction of p21 expression. Mol. Med. Rep. 2017, 15, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Gong, H.; Yuan, C.; Liu, G.; Huang, J.; Li, Z.; Luo, J. Δ133p53/FLp53 Predicts Poor Clinical Outcome in Esophageal Squamous Cell Carcinoma. Cancer Manag Res. 2020, 12, 7405–7417. [Google Scholar] [CrossRef]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef]
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of p53 Isoform Expression on Survival in High-Grade Serous Ovarian Cancers. Sci. Rep. 2019, 9, 5244. [Google Scholar] [CrossRef]
- Knezović Florijan, M.; Ozretić, P.; Bujak, M.; Pezzè, L.; Ciribilli, Y.; Kaštelan, Ž.; Slade, N.; Hudolin, T. The role of p53 isoforms’ expression and p53 mutation status in renal cell cancer prognosis. Urol. Oncol. 2019, 37, 578.e1–578.e10. [Google Scholar] [CrossRef]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Δ133p53β variant. ELife 2016, 5, e14734. [Google Scholar] [CrossRef]
- Milićević, Z.; Bajić, V.; Živković, L.; Kasapović, J.; Andjelković, U.; Spremo-Potparević, B. Identification of p53 and its isoforms in human breast carcinoma cells. Sci. World J. 2014, 2014, 618698. [Google Scholar] [CrossRef]
- Ozretić, P.; Hanžić, N.; Proust, B.; Sabol, M.; Trnski, D.; Radić, M.; Musani, V.; Ciribilli, Y.; Milas, I.; Puljiz, Z.; et al. Expression profiles of p53/p73, NME and GLI families in metastatic melanoma tissue and cell lines. Sci. Rep. 2019, 9, 12470. [Google Scholar] [CrossRef]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Pan, X.; Chen, H.; Rao, L.; Zeng, Y.; Hang, H.; Peng, J.; Xiao, L.; Chen, J. p53 isoform Δ133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep. 2016, 6, 37281. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Park, K.-Y.; Isogaya, K.; Hiyoshi, Y.; Li, H.; Anami, K.; Robles, A.I.; Mondal, A.M.; Fujita, K.; Serrano, M.; et al. Δ133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017, 24, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Oh, L.; Hainaut, P.; Blanchet, S.; Ariffin, H. Expression of p53 N-terminal isoforms in B-cell precursor acute lymphoblastic leukemia and its correlation with clinicopathological profiles. BMC Cancer 2020, 20, 110. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Noto, J.; Zaika, E.; Romero-Gallo, J.; Correa, P.; El-Rifai, W.; Peek, R.M.; Zaika, A. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc. Natl. Acad. Sci. USA 2012, 109, E2543–E2550. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Sang, X.-G.; Wang, Y.-Z.; Cui, C.; Zhang, L.; Ji, W.-S. Role of Δ133p53 isoform in NF-κB inhibitor PDTC-mediated growth inhibition of MKN45 gastric cancer cells. World J. Gastroenterol. 2017, 23, 2716–2722. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.K.; Martinez, J.D. The significance of p53 isoform expression in serous ovarian cancer. Future Oncol. Lond. Engl. 2012, 8, 683–686. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef]
- Gong, H.; Zhang, Y.; Jiang, K.; Ye, S.; Chen, S.; Zhang, Q.; Peng, J.; Chen, J. p73 coordinates with Δ133p53 to promote DNA double-strand break repair. Cell Death Differ. 2018, 25, 1063–1079. [Google Scholar] [CrossRef]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro-Oncology 2019, 21, 474–485. [Google Scholar] [CrossRef]
- von Muhlinen, N.; Horikawa, I.; Alam, F.; Isogaya, K.; Lissa, D.; Vojtesek, B.; Lane, D.P.; Harris, C.C. p53 isoforms regulate premature aging in human cells. Oncogene 2018, 37, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Pan, X.; Yuan, Z.-M.; Peng, J.; Chen, J. p53 coordinates with Δ133p53 isoform to promote cell survival under low-level oxidative stress. J. Mol. Cell Biol. 2016, 8, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.M.; Zhou, H.; Horikawa, I.; Suprynowicz, F.A.; Li, G.; Dakic, A.; Rosenthal, B.; Ye, L.; Harris, C.C.; Schlegel, R.; et al. Δ133p53α, a natural p53 isoform, contributes to conditional reprogramming and long-term proliferation of primary epithelial cells. Cell Death Dis. 2018, 9, 750. [Google Scholar] [CrossRef]
- Arsic, N.; Gadea, G.; Lagerqvist, E.L.; Busson, M.; Cahuzac, N.; Brock, C.; Hollande, F.; Gire, V.; Pannequin, J.; Roux, P. The p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell Rep. 2015, 4, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.-C. The p53 isoform, Δ133p53α, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Dey, S. Induction of p73, Δ133p53, Δ160p53, pAKT lead to neuroprotection via DNA repair by 5-LOX inhibition. Mol. Biol. Rep. 2020, 47, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Roth, I.; Campbell, H.; Rubio, C.; Vennin, C.; Wilson, M.; Wiles, A.; Williams, G.; Woolley, A.; Timpson, P.; Berridge, M.V.; et al. The Δ133p53 isoform and its mouse analogue Δ122p53 promote invasion and metastasis involving pro-inflammatory molecules interleukin-6 and CCL2. Oncogene 2016, 35, 4981–4989. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C.H. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef]
- Beck, J.; Turnquist, C.; Horikawa, I.; Harris, C. Targeting cellular senescence in cancer and aging: Roles of p53 and its isoforms. Carcinogenesis 2020, 41, 1017–1029. [Google Scholar] [CrossRef]
- Romanova, L.Y.; Willers, H.; Blagosklonny, M.V.; Powell, S.N. The interaction of p53 with replication protein A mediates suppression of homologous recombination. Oncogene 2004, 23, 9025–9033. [Google Scholar] [CrossRef] [PubMed]
- Keimling, M.; Wiesmüller, L. DNA double-strand break repair activities in mammary epithelial cells--influence of endogenous p53 variants. Carcinogenesis 2009, 30, 1260–1268. [Google Scholar] [CrossRef]
- Coppedè, F.; Migliore, L. DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Joshi, Y.B.; Praticò, D. The 5-Lipoxygenase Pathway: Oxidative and Inflammatory Contributions to the Alzheimer’s Disease Phenotype. Available online: https://pubmed.ncbi.nlm.nih.gov/25642165/ (accessed on 16 September 2020).
- Zorić, A.; Horvat, A.; Slade, N. Differential effects of diverse p53 isoforms on TAp73 transcriptional activity and Apoptosis. Carcinogenesis 2013, 34, 522–529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-J.; Amatruda, J.F.; Abrams, J.M. p53 ancestry: Gazing through an evolutionary lens. Nat. Rev. Cancer 2009, 9, 758–762. [Google Scholar] [CrossRef]
- Chen, J.; Ruan, H.; Ng, S.M.; Gao, C.; Soo, H.M.; Wu, W.; Zhang, Z.; Wen, Z.; Lane, D.P.; Peng, J. Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 2005, 19, 2900–2911. [Google Scholar] [CrossRef]
- Storer, N.Y.; Zon, L.I. Zebrafish models of p53 functions. Cold Spring Harb. Perspect. Biol. 2010, 2, a001123. [Google Scholar] [CrossRef]
- Sulak, M.; Fong, L.; Mika, K.; Chigurupati, S.; Yon, L.; Mongan, N.P.; Emes, R.D.; Lynch, V.J. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. eLife 2016, 5, e11994. [Google Scholar] [CrossRef]
- Slatter, T.L.; Hung, N.; Campbell, H.; Rubio, C.; Mehta, R.; Renshaw, P.; Williams, G.; Wilson, M.; Engelmann, A.; Jeffs, A.; et al. Hyperproliferation, cancer, and inflammation in mice expressing a Δ133p53-like isoform. Blood 2011, 117, 5166–5177. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Yang, Q.; Tong, W.M.; Hergenhahn, M.; Wang, Z.Q.; Hollstein, M. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: A new biomedical research tool. Oncogene 2001, 20, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Vijayakumar, V.; Fernández-Cuesta, L.; Hafsi, H.; Sagne, C.; Hautefeuille, A.; Olivier, M.; Hainaut, P. p53 regulates the transcription of its Delta133p53 isoform through specific response elements contained within the TP53 P2 internal promoter. Oncogene 2010, 29, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Yin, L.; Chang, C.; Peng, J.; Chen, J. Protein interaction between p53 and Δ113p53 is required for the anti-apoptotic function of Δ113p53. J. Genet. Genom. Yi Chuan Xue Bao 2014, 41, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ng, S.M.; Chang, C.; Zhang, Z.; Bourdon, J.-C.; Lane, D.P.; Peng, J. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 2009, 23, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.R.; Kari, C.; Ren, Q.; Daroczi, B.; Dicker, A.P.; Rodeck, U. Differential regulation of p53 function by the N-terminal ΔNp53 and Δ113p53 isoforms in zebrafish embryos. BMC Dev. Biol. 2010, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- McElderry, J.; Carrington, B.; Bishop, K.; Kim, E.; Pei, W.; Chen, Z.; Ramanagoudr-Bhojappa, R.; Prakash, A.; Burgess, S.M.; Liu, P.P.; et al. Splicing factor DHX15 affects tp53 and mdm2 expression via alternate splicing and promoter usage. Hum. Mol. Genet. 2019, 28, 4173–4185. [Google Scholar] [CrossRef]
- Ye, S.; Zhao, T.; Zhang, W.; Tang, Z.; Gao, C.; Ma, Z.; Xiong, J.W.; Peng, J.; Tan, W.Q.; Chen, J. p53 isoform Δ113p53 promotes zebrafish heart regeneration by maintaining redox homeostasis. Cell Death Dis. 2020, 11, 568. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. The Paradox of p53: What, How, and Why? Cold Spring Harb. Perspect. Med. 2016, 6, a026328. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.P.; Effros, R.B. T cell replicative senescence in human aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar] [CrossRef] [PubMed]
- Prieto, L.I.; Baker, D.J. Cellular Senescence and the Immune System in Cancer. Gerontology 2019, 65, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 322–336.e8. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Campbell, H.G.; Slatter, T.L.; Jeffs, A.; Mehta, R.; Rubio, C.; Baird, M.; Braithwaite, A.W. Does Δ133p53 isoform trigger inflammation and autoimmunity? Cell Cycle Georget. Tex 2012, 11, 446–450. [Google Scholar] [CrossRef]
- Sawhney, S.; Hood, K.; Shaw, A.; Braithwaite, A.W.; Stubbs, R.; Hung, N.A.; Royds, J.A.; Slatter, T.L. Alpha-enolase is upregulated on the cell surface and responds to plasminogen activation in mice expressing a ∆133p53α mimic. PLoS ONE 2015, 10, e0116270. [Google Scholar] [CrossRef]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef]
- Costantini, C.; Scrable, H.; Puglielli, L. An Aging Pathway Controls the TrkA to p75NTR Receptor Switch and Amyloid Beta-Peptide Generation. EMBO J. 2006, 25, 1997–2006. [Google Scholar] [CrossRef]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; Gil del Alcazar, C.; Burger, C.; Scrable, H.; Puglielli, L. Altered Longevity-Assurance Activity of p53:p44 in the Mouse Causes Memory Loss, Neurodegeneration and Premature Death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. P44, the ‘Longevity-Assurance’ Isoform of P53, Regulates Tau Phosphorylation and Is Activated in an Age-Dependent Fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef]
- Gambino, V.; De Michele, G.; Venezia, O.; Migliaccio, P.; Dall’Olio, V.; Bernard, L.; Paolo Minardi, S.P.; Della Fazia, M.A.; Bartoli, D.; Servillo, G.; et al. Oxidative Stress Activates a Specific p53 Transcriptional Response That Regulates Cellular Senescence and Aging. Aging Cell 2013, 12, 435–445. [Google Scholar] [CrossRef] [PubMed]
- IARC TP53 Search. Available online: https://p53.iarc.fr/TP53SomaticMutations.aspx (accessed on 11 June 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Total Δ133p53 mRNAs (α, β and γ) Association with Disease | Ref. |
|---|---|---|
| Colon cancer | Total Δ133p53 mRNAs are downregulated in adenoma vs. normal tissue, but overexpressed in carcinoma vs. adenoma or normal tissue | [39] |
| Total Δ133p53 mRNAs expression is correlated with higher risks of recurrence | [40] | |
| Elevation of total Δ133p53 mRNAs is associated with shorter disease-free survival | [41] | |
| Cholangiocarcinoma | Total Δ133p53 mRNAs upregulation, and decreased TA mRNAs, is associated with shortened overall survival | [42] |
| Total Δ133p53 mRNAs reduction sensitizes to 5-Fluorouracil treatment | [43] | |
| Lung carcinoma | Total Δ133p53 mRNAs are overexpressed in tumor as compared to adjacent non-cancerous tissue | [44] |
| Esophageal Squamous Cell Carcinoma | High tissue or serum total Δ133p53/TAp53 ratio is associated with poor overall and progression-free survival. | [45] |
| Ovarian cancer | Total Δ133p53 mRNAs expression in mutant TP53 patients is associated with longer overall survival and disease-free survival | [46] |
| Total Δ133p53 mRNAs are associated with increased overall survival (and borderline disease-free survival) independently of TP53 mutation status | [47] | |
| Renal cell carcinoma | Wild-type tumors correlate with worse overall survival. Total Δ133p53 mRNAs are downregulated in WT tumors compared to mutant tumors and normal adjacent tissue | [48] |
| Cancer Type | Single Specific Δ133p53 Isoform Association with Disease | Ref. |
| Breast cancer | Δ133p53α mRNA is detected in tumor but not in normal tissue | [6] |
| Δ133p53β mRNA detection is associated with worse overall and disease-free survival | [49] | |
| Δ133p53β protein is overexpressed in invasive tumor as compared to non-invasive ones | [50] | |
| Glioblastoma | Δ133p53β is elevated in glioblastoma malignant cells and is associated with immunosuppressive and chemoresistant environment | [36] |
| Prostate cancer | Δ133p53β mRNA is elevated in tumor vs. non-neoplastic tissue and is associated with shorter progression-free survival | [37] |
| Melanoma | Increased Δ133p53β mRNA expression is associated with poorer overall survival | [51] |
| Δ133p53α is | Because Δ133p53α | Ref(s) |
|---|---|---|
| Non-mutagenic | Enhances DNA double-strand break repair | [37,42,51,52,59,60] |
| Does not induce chromosomal and microsatellite repeats abnormalities | [52] | |
| Does not cause increased mutation rate, unlike canonical p53α knockdown | [52] | |
| Non-oncogenic | Does not prevent p53α-dependent apoptosis of severely damaged cells | [52] |
| Increases replicative lifespan without immortalizing cells | [23,24,25,37,50,51,52] | |
| Does not induce malignant transformation | [23,24,25,37,50,51,52] |
| Biological Function | Δ133p53α | Δ133p53β | Δ133p53γ | |||
|---|---|---|---|---|---|---|
| Regulation | Ref(s) | Regulation | Ref(s) | Regulation | Ref(s) | |
| Proliferation |  | [6,15,23,24,25,39,53,54,61,62,63,64] | | [36,40,41,65] | | [41,49,66] |
| Normal stem cells pluripotency | | [53,54,64] | ND | - | ND | - |
| Cancer stemness | ND | - | | [65] | ND | - |
| Cellular senescence |  | [23,24,25,39,54,61,62] | ND | - | ND | - |
| DNA repair | | [39,52,54,60,62,67] | ND | - | ND | - |
| Angiogenesis | | [66] |  | [66] | | [66] |
| Motility/Invasion | | [41,49,66,68] | | [41,49] | | [41,49,66] |
| Immune cells infiltration | ND | - | | [36,37] | ND | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joruiz, S.M.; Beck, J.A.; Horikawa, I.; Harris, C.C. The Δ133p53 Isoforms, Tuners of the p53 Pathway. Cancers 2020, 12, 3422. https://doi.org/10.3390/cancers12113422
Joruiz SM, Beck JA, Horikawa I, Harris CC. The Δ133p53 Isoforms, Tuners of the p53 Pathway. Cancers. 2020; 12(11):3422. https://doi.org/10.3390/cancers12113422
Chicago/Turabian StyleJoruiz, Sebastien M., Jessica A. Beck, Izumi Horikawa, and Curtis C. Harris. 2020. "The Δ133p53 Isoforms, Tuners of the p53 Pathway" Cancers 12, no. 11: 3422. https://doi.org/10.3390/cancers12113422
APA StyleJoruiz, S. M., Beck, J. A., Horikawa, I., & Harris, C. C. (2020). The Δ133p53 Isoforms, Tuners of the p53 Pathway. Cancers, 12(11), 3422. https://doi.org/10.3390/cancers12113422