Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling




Simple Summary
Abstract
1. Introduction
2. Antitumor Immunity Impairment: Role of Structural and Functional Abnormalities
3. Improving Immune–Vascular Crosstalk for Cancer Immunotherapy
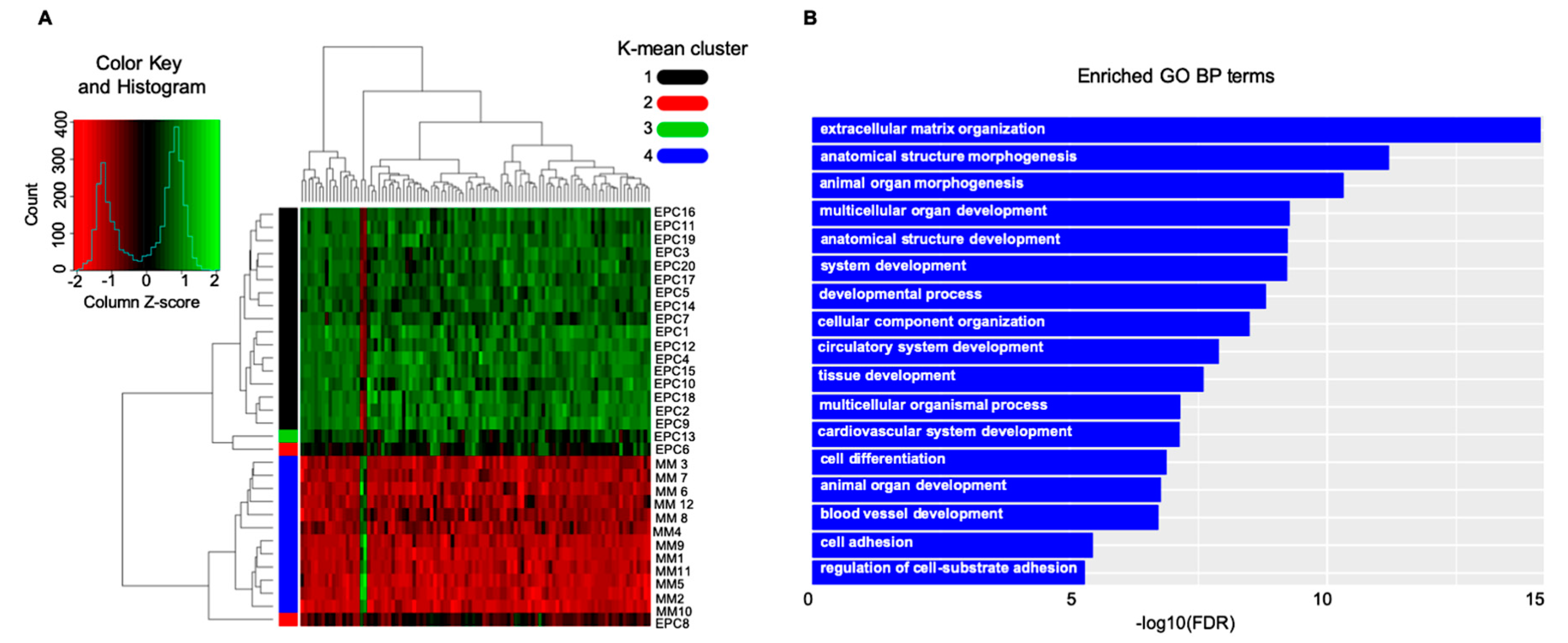
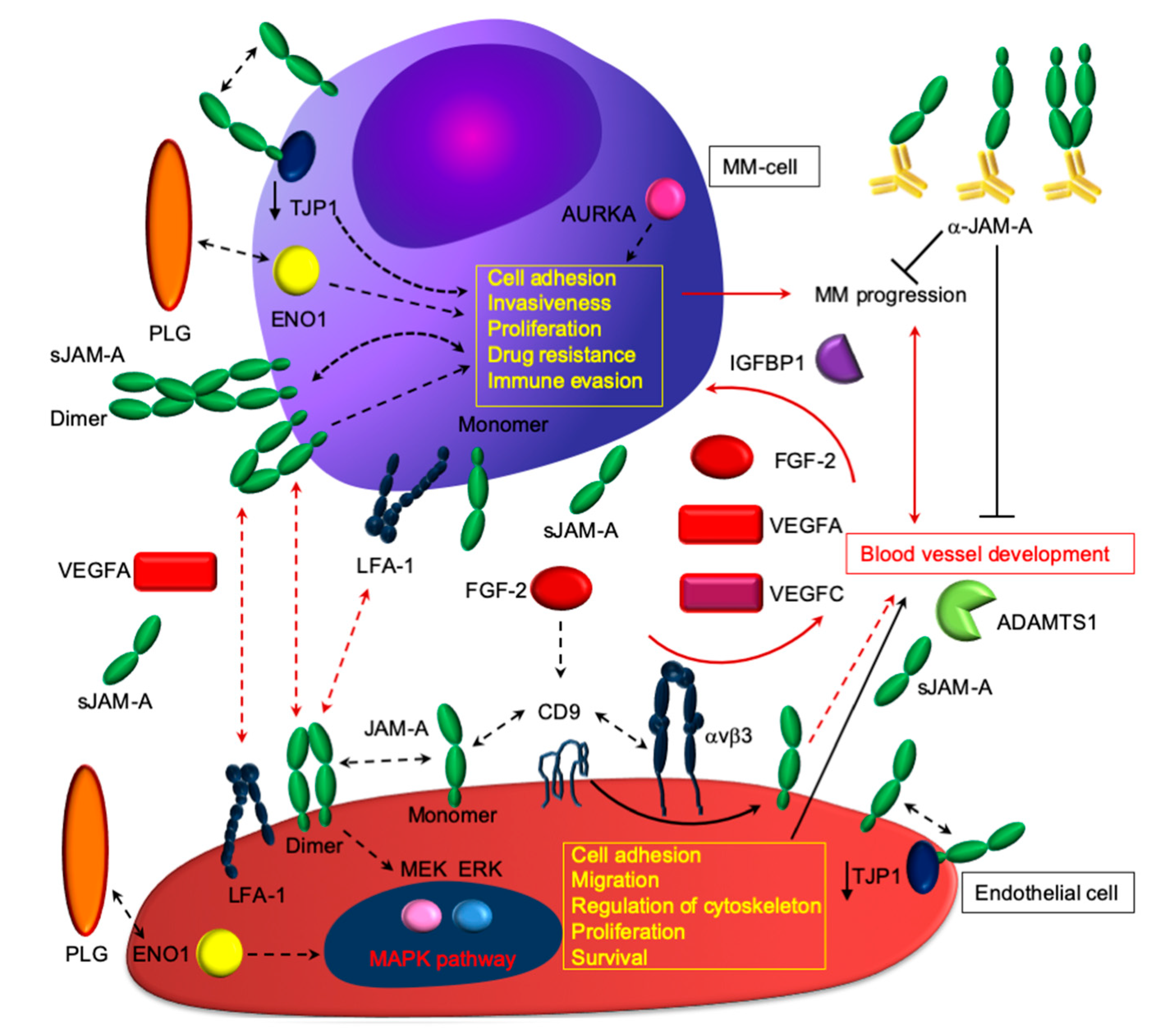
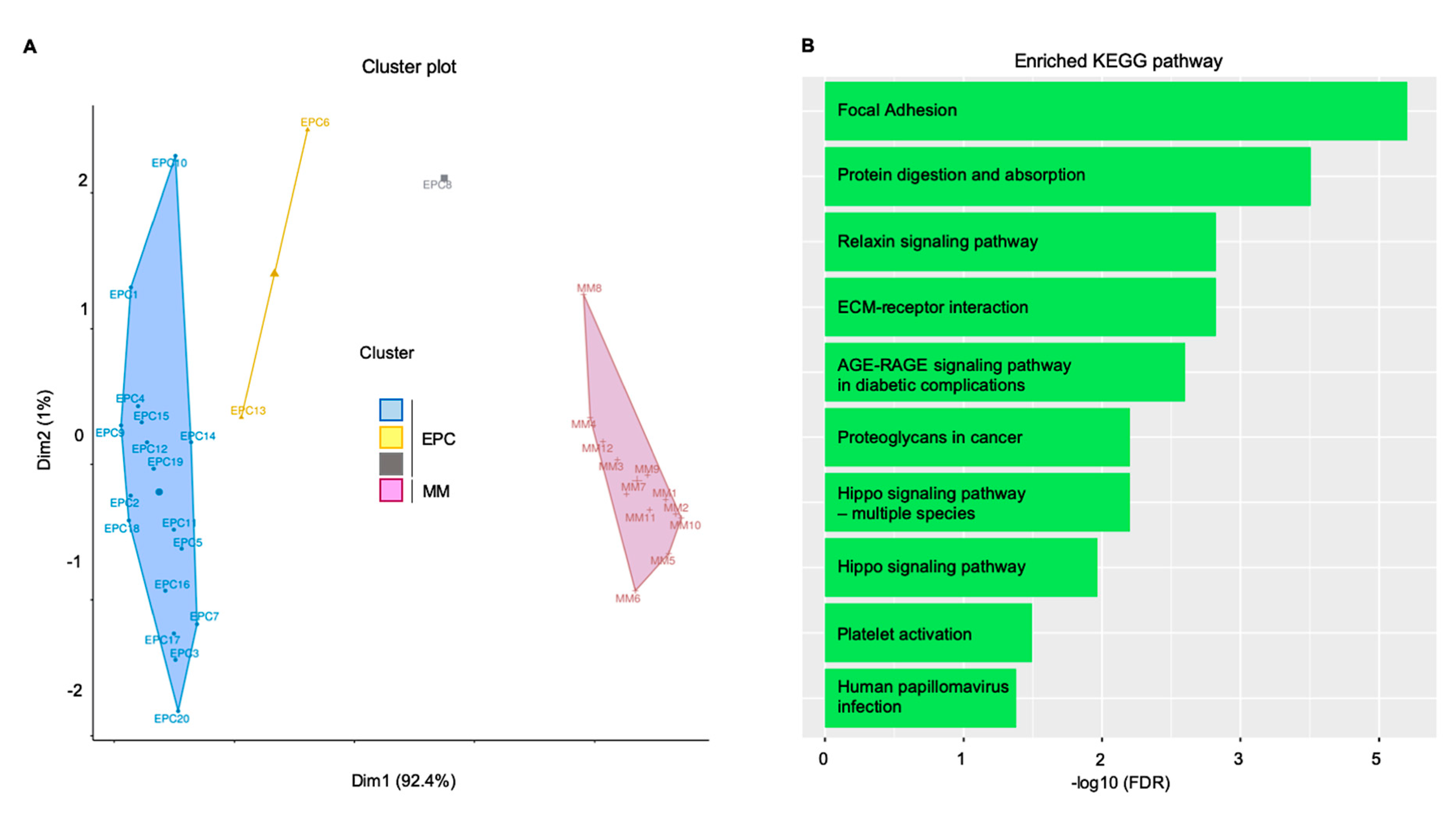
4. Multiple Myeloma (MM) as a Paradigm for Endothelial Gatekeeper Function within the Neoplastic Niche: In Silico Functional Enrichment Study Identifies Prognostic Relevant Gene Profiles in MM Bone Marrow Derived Endothelial Cells
5. Measuring T Cell Exit from Tumors: How Do Lymphatic Vessels Shape the Intratumoral Repertoire
6. Boosting Cancer Immunotherapy Using Anti-Angiogenics: Therapeutic Windows and Challenges Offered by the Visualization and Reprogramming of the Tumor Milieu
Hypoxia as a Key Factor for Angiogenesis and Immune Equilibrium
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCL | Chemokine ligand |
| CXCL | Chemokine ligand |
| ENO1 | Enolase1 |
| ESAM | Endothelial cell-selective adhesion molecule |
| FGF2 | Fibroblast growth factor 2 |
| ICAM | Intercellular adhesion molecule |
| IDO1 | Indoleamine 2,3-Dioxygenase 1 |
| HLA-E | Human leukocyte antigen E |
| IFNγ | Interferon gamma |
| JAMs | Junctional adhesion molecules |
| LFA1 | Lymphocyte function-associated antigen 1 (also known as αLβ2-integrin) |
| MHC | Major histocompatibility complex |
| NEU1 | Epidermal growth factor like domain 7 (Egfl7) |
| NO | Nitric oxide |
| PD-L1/2 | Programmed death-ligand 1/2 |
| PECAM1 | Platelet/endothelial-cell adhesion molecule 1 |
| TIM3 | T-cell immunoglobulin and mucin domain 3 |
| TNFα | Tumour necrosis factor alpha |
| VCAM | Vascular cell-adhesion molecule |
| VE-cadherin | Vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| VLA4 | Very late antigen 4 (also known as α4β1-integrin) |
References
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The history of the angiogenic switch concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Presta, M. The discovery of angiogenic factors. Gen. Pharmacol. Vasc. Syst. 2000, 35, 227–231. [Google Scholar] [CrossRef]
- Vermeulen, P.B.; Gasparini, G.; Fox, S.B.; Toi, M.; Martin, L.; Mcculloch, P.; Pezzella, F.; Viale, G.; Weidner, N.; Harris, A.L.; et al. Quantification of angiogenesis in solid human tumours: An international consensus on the methodology and criteria of evaluation. Eur. J. Cancer 1996, 32, 2474–2484. [Google Scholar] [CrossRef]
- Hasan, J.; Byers, R.; Jayson, G.C. Intra-tumoural microvessel density in human solid tumours. Br. J. Cancer 2002, 86, 1566–1577. [Google Scholar] [CrossRef]
- Leone, P.; Buonavoglia, A.; Fasano, R.; Solimando, A.G.; De Re, V.; Cicco, S.; Vacca, A.; Racanelli, V. Insights into the Regulation of Tumor Angiogenesis by Micro-RNAs. J. Clin. Med. 2019, 8, 2030. [Google Scholar] [CrossRef]
- Lamanuzzi, A.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Leone, P.; Racanelli, V.; Nico, B.; Ribatti, D.; Ditonno, P.; Prete, M.; et al. Inhibition of mTOR complex 2 restrains tumor angiogenesis in multiple myeloma. Oncotarget 2018, 9, 20563–20577. [Google Scholar] [CrossRef]
- Li, W.W.; Hutnik, M.; Gehr, G. Antiangiogenesis in haematological malignancies. Br. J. Haematol. 2008, 143, 622–631. [Google Scholar] [CrossRef]
- Hendrix, M.J.C.; Seftor, E.A.; Seftor, R.E.B.; Chao, J.-T.; Chien, D.-S.; Chu, Y.-W. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol. Ther. 2016, 159, 83–92. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis. In Biology of Endothelial Cells; Jaffe, E.A., Ed.; Developments in Cardiovascular Medicine; Springer: Boston, MA, USA, 1984; Volume 27, pp. 412–428. ISBN 978-1-4612-9786-4. [Google Scholar]
- Moserle, L.; Jimenez-Valerio, G.; Casanovas, O. Antiangiogenic Therapies: Going beyond Their Limits. Cancer Discov. 2014, 4, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; Solimando, A.G.; Krebs, M.; Leone, P.; Susca, N.; Brunetti, O.; Racanelli, V.; Vacca, A.; Silvestris, N. Anti-angiogenesis and Immunotherapy: Novel Paradigms to Envision Tailored Approaches in Renal Cell-Carcinoma. J. Clin. Med. 2020, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Sakariassen, P.O.; Prestegarden, L.; Wang, J.; Skaftnesmo, K.-O.; Mahesparan, R.; Molthoff, C.; Sminia, P.; Sundlisaeter, E.; Misra, A.; Tysnes, B.B.; et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16466–16471. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia Auckl. N. Z. 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Rusnati, M.; Riboldi, E.; Mitola, S.; Presta, M. Dendritic cell-endothelial cell cross-talk in angiogenesis. Trends Immunol. 2007, 28, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Abdalla, A.M.E.; Xiao, L.; Ullah, M.W.; Yu, M.; Ouyang, C.; Yang, G. Current Challenges of Cancer Anti-angiogenic Therapy and the Promise of Nanotherapeutics. Theranostics 2018, 8, 533–548. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Fisher, D.T.; Muhitch, J.B.; Kim, M.; Doyen, K.C.; Bogner, P.N.; Evans, S.S.; Skitzki, J.J. Intraoperative intravital microscopy permits the study of human tumour vessels. Nat. Commun. 2016, 7, 10684. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Da Vià, M.C.; Solimando, A.G.; Garitano-Trojaola, A.; Barrio, S.; Munawar, U.; Strifler, S.; Haertle, L.; Rhodes, N.; Teufel, E.; Vogt, C.; et al. CIC Mutation as a Molecular Mechanism of Acquired Resistance to Combined BRAF-MEK Inhibition in Extramedullary Multiple Myeloma with Central Nervous System Involvement. Oncologist 2019. [Google Scholar] [CrossRef]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Solimando, A.G.; Brandl, A.; Mattenheimer, K.; Graf, C.; Ritz, M.; Ruckdeschel, A.; Stühmer, T.; Mokhtari, Z.; Rudelius, M.; Dotterweich, J.; et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia 2018, 32, 736–743. [Google Scholar] [CrossRef]
- Rudelius, M.; Rosenfeldt, M.T.; Leich, E.; Rauert-Wunderlich, H.; Solimando, A.G.; Beilhack, A.; Ott, G.; Rosenwald, A. Inhibition of focal adhesion kinase overcomes resistance of mantle cell lymphoma to ibrutinib in the bone marrow microenvironment. Haematologica 2018, 103, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.A. A hybrid mathematical model of solid tumour invasion: The importance of cell adhesion. Math. Med. Biol. J. IMA 2005, 22, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; Calabrese, A.; Solimando, A.G.; Notaristefano, A.; Panarelli, M.M.G.; Brunetti, O. Bone metastasis as primary presentation of pancreatic ductal adenocarcinoma: A case report and literature review. Clin. Case Rep. 2019, 7, 1972–1976. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking JAM-A on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica 2020. [Google Scholar] [CrossRef]
- Moghadam, A.R.; Patrad, E.; Tafsiri, E.; Peng, W.; Fangman, B.; Pluard, T.J.; Accurso, A.; Salacz, M.; Shah, K.; Ricke, B.; et al. Ral signaling pathway in health and cancer. Cancer Med. 2017, 6, 2998–3013. [Google Scholar] [CrossRef]
- Solimando, A.G.; Ribatti, D.; Vacca, A.; Einsele, H. Targeting B-cell non Hodgkin lymphoma: New and old tricks. Leuk. Res. 2016, 42, 93–104. [Google Scholar] [CrossRef]
- Farahani, E.; Patra, H.K.; Jangamreddy, J.R.; Rashedi, I.; Kawalec, M.; Rao Pariti, R.K.; Batakis, P.; Wiechec, E. Cell adhesion molecules and their relation to (cancer) cell stemness. Carcinogenesis 2014, 35, 747–759. [Google Scholar] [CrossRef]
- Seibold, M.; Stühmer, T.; Kremer, N.; Mottok, A.; Scholz, C.-J.; Schlosser, A.; Leich, E.; Holzgrabe, U.; Brünnert, D.; Barrio, S.; et al. RAL GTPases mediate multiple myeloma cell survival and are activated independently of oncogenic RAS. Haematologica 2019. [Google Scholar] [CrossRef]
- Wu, N.Z.; Klitzman, B.; Dodge, R.; Dewhirst, M.W. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res. 1992, 52, 4265–4268. [Google Scholar]
- Ganss, R.; Hanahan, D. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 1998, 58, 4673–4681. [Google Scholar]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Gröne, H.-J.; Hämmerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Cancer Immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Siu, L.L.; Ivy, S.P.; Dixon, E.L.; Gravell, A.E.; Reeves, S.A.; Rosner, G.L. Challenges and Opportunities in Adapting Clinical Trial Design for Immunotherapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4950–4958. [Google Scholar] [CrossRef]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Solimando, A.G.; Crudele, L.; Leone, P.; Argentiero, A.; Guarascio, M.; Silvestris, N.; Vacca, A.; Racanelli, V. Immune Checkpoint Inhibitor-Related Myositis: From Biology to Bedside. Int. J. Mol. Sci. 2020, 21, 3054. [Google Scholar] [CrossRef]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving cancer immunotherapy using nanomedicines: Progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef]
- Liu, Y.; Shaw, S.K.; Ma, S.; Yang, L.; Luscinskas, F.W.; Parkos, C.A. Regulation of Leukocyte Transmigration: Cell Surface Interactions and Signaling Events. J. Immunol. 2004, 172, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone marrow endothelial cells sustain a tumor-specific CD8+ T cell subset with suppressive function in myeloma patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef]
- Chae, Y.K.; Choi, W.M.; Bae, W.H.; Anker, J.; Davis, A.A.; Agte, S.; Iams, W.T.; Cruz, M.; Matsangou, M.; Giles, F.J. Overexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancer. Sci. Rep. 2018, 8, 1023. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers 2019, 11, 330. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis from Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Fang, Z.; Liu, X.; Deng, S.; Zhou, P.; Wang, X.; Zhang, C.; Yin, R.; Hu, H.; Chen, X.; et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Investig. 2018, 128, 2104–2115. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.J.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Goon, P.K.Y.; Lip, G.Y.H.; Boos, C.J.; Stonelake, P.S.; Blann, A.D. Circulating endothelial cells, endothelial progenitor cells, and endothelial microparticles in cancer. Neoplasia N. Y. 2006, 8, 79–88. [Google Scholar] [CrossRef]
- Gao, D.; Nolan, D.J.; Mellick, A.S.; Bambino, K.; McDonnell, K.; Mittal, V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 2008, 319, 195–198. [Google Scholar] [CrossRef]
- Moschetta, M.; Mishima, Y.; Kawano, Y.; Manier, S.; Paiva, B.; Palomera, L.; Aljawai, Y.; Calcinotto, A.; Unitt, C.; Sahin, I.; et al. Targeting vasculogenesis to prevent progression in multiple myeloma. Leukemia 2016, 30, 1103–1115. [Google Scholar] [CrossRef]
- Moschetta, M.; Mishima, Y.; Sahin, I.; Manier, S.; Glavey, S.; Vacca, A.; Roccaro, A.M.; Ghobrial, I.M. Role of endothelial progenitor cells in cancer progression. Biochim. Biophys. Acta 2014, 1846, 26–39. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Vacca, A.; Melaccio, A.; Sportelli, A.; Solimando, A.G.; Dammacco, F.; Ria, R. Subcutaneous immunoglobulins in patients with multiple myeloma and secondary hypogammaglobulinemia: A randomized trial. Clin. Immunol. 2018, 191, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Nucci, M.; Anaissie, E. Infections in Patients with Multiple Myeloma in the Era of High-Dose Therapy and Novel Agents. Clin. Infect. Dis. 2009, 49, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Reale, A.; Solimando, A.G.; Mangialardi, G.; Moschetta, M.; Gelao, L.; Iodice, G.; Vacca, A. Induction therapy and stem cell mobilization in patients with newly diagnosed multiple myeloma. Stem Cells Int. 2012, 2012, 607260. [Google Scholar] [CrossRef] [PubMed]
- Cicco, S.; Solimando, A.G.; Leone, P.; Battaglia, S.; Ria, R.; Vacca, A.; Racanelli, V. Suspected Pericardial Tuberculosis Revealed as an Amyloid Pericardial Mass. Case Rep. Hematol. 2018, 2018, 8606430. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Sportelli, A.; Troiano, T.; Demarinis, L.; Di Serio, F.; Ostuni, A.; Dammacco, F.; Vacca, A.; Ria, R. A multiple myeloma that progressed as type I cryoglobulinemia with skin ulcers and foot necrosis: A case report. Medicine 2018, 97, e12355. [Google Scholar] [CrossRef]
- Cicco, S.; Solimando, A.G.; Buono, R.; Susca, N.; Inglese, G.; Melaccio, A.; Prete, M.; Ria, R.; Racanelli, V.; Vacca, A. Right Heart Changes Impact on Clinical Phenotype of Amyloid Cardiac Involvement: A Single Centre Study. Life 2020, 10, 247. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef]
- Solimando, A.G.; Vacca, A.; Ribatti, D. A Comprehensive Biological and Clinical Perspective Can Drive a Patient-Tailored Approach to Multiple Myeloma: Bridging the Gaps between the Plasma Cell and the Neoplastic Niche. J. Oncol. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Noonan, K.; Borrello, I. The immune microenvironment of myeloma. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2011, 4, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggiò, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Desantis, V.; Di Marzo, L.; Craparotta, I.; Beltrame, L.; Marchini, S.; Annese, T.; Visino, F.; Arciuli, M.; Saltarella, I.; et al. Bone marrow fibroblasts overexpress miR-27b and miR-214 in step with multiple myeloma progression, dependent on tumour cell-derived exosomes: Myeloma cell-derived exosomes and fibroblast miRNA expression. J. Pathol. 2019, 247, 241–253. [Google Scholar] [CrossRef]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Melaccio, A.; Solimando, A.G.; Mariggiò, M.A.; Racanelli, V.; Paradiso, A.; Vacca, A.; Frassanito, M.A. MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance. Int. J. Mol. Sci. 2020, 21, 3084. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Via’, M.C.; Leone, P.; Croci, G.; Borrelli, P.; Tabares Gaviria, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Adhesion-Mediated Multiple Myeloma (MM) Disease Progression: Junctional Adhesion Molecule a Enhances Angiogenesis and Multiple Myeloma Dissemination and Predicts Poor Survival. Blood 2019, 134, 855. [Google Scholar] [CrossRef]
- Saadatmand, S.; de Kruijf, E.M.; Sajet, A.; Dekker-Ensink, N.G.; van Nes, J.G.H.; Putter, H.; Smit, V.T.H.B.M.; van de Velde, C.J.H.; Liefers, G.J.; Kuppen, P.J.K. Expression of cell adhesion molecules and prognosis in breast cancer. Br. J. Surg. 2013, 100, 252–260. [Google Scholar] [CrossRef]
- Moh, M.C.; Shen, S. The roles of cell adhesion molecules in tumor suppression and cell migration: A new paradox. Cell Adhes. Migr. 2009, 3, 334–336. [Google Scholar] [CrossRef]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275–41289. [Google Scholar] [CrossRef]
- Teoh, G.; Anderson, K.C. Interaction of Tumor and Host Cells With Adhesion and Extracellular Matrix Molecules in the Development of Multiple Myeloma. Hematol. Oncol. Clin. N. Am. 1997, 11, 27–42. [Google Scholar] [CrossRef]
- Braunstein, M.J.; Campagne, F.; Mukherjee, P.; Carrasco, D.R.; Sukhdeo, K.; Protopopov, A.; Anderson, K.C.; Batuman, O. Genome-Wide Profiling of Endothelial Progenitor Cells in Multiple Myeloma: Disease-Relevant Pathways and Overlaps with Common Cancer Biomarkers. Blood 2008, 112, 626. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gil Marques, F.; Poli, E.; Malaquias, J.; Carvalho, T.; Portêlo, A.; Ramires, A.; Aldeia, F.; Ribeiro, R.M.; Vitorino, E.; Diegues, I.; et al. Low doses of ionizing radiation activate endothelial cells and induce angiogenesis in peritumoral tissues. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2019, 141, 256–261. [Google Scholar] [CrossRef]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics (Oxf. Engl.) 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Zeileis, A.; Fisher, J.C.; Hornik, K.; Ihaka, R.; McWhite, C.D.; Murrell, P.; Stauffer, R.; Wilke, C.O. Colorspace: A Toolbox for Manipulating and Assessing Colors and Palettes. arXiv 2019, arXiv:190306490. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Use R! Springer: Dordrecht, The Netherlands, 2009; ISBN 978-0-387-98141-3. [Google Scholar]
- Abe, M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int. J. Hematol. 2011, 94, 334–343. [Google Scholar] [CrossRef]
- Leich, E.; Weißbach, S.; Klein, H.-U.; Grieb, T.; Pischimarov, J.; Stühmer, T.; Chatterjee, M.; Steinbrunn, T.; Langer, C.; Eilers, M.; et al. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion- and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013, 3, e102. [Google Scholar] [CrossRef]
- Jridi, I.; Catacchio, I.; Majdoub, H.; Shahbazzadeh, D.; El Ayeb, M.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Vacca, A.; Borchani, L. The small subunit of Hemilipin2, a new heterodimeric phospholipase A2 from Hemiscorpius lepturus scorpion venom, mediates the antiangiogenic effect of the whole protein. Toxicon Off. J. Int. Soc. Toxinology 2017, 126, 38–46. [Google Scholar] [CrossRef]
- Flati, V.; Pastore, L.I.; Griffioen, A.W.; Satijn, S.; Toniato, E.; D’Alimonte, I.; Laglia, E.; Marchetti, P.; Gulino, A.; Martinotti, S. Endothelial cell anergy is mediated by bFGF through the sustained activation of p38-MAPK and NF-kappaB inhibition. Int. J. Immunopathol. Pharmacol. 2006, 19, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Peddibhotla, S.S.D.; Brinkmann, B.F.; Kummer, D.; Tuncay, H.; Nakayama, M.; Adams, R.H.; Gerke, V.; Ebnet, K. Tetraspanin CD9 links junctional adhesion molecule-A to αvβ3 integrin to mediate basic fibroblast growth factor-specific angiogenic signaling. Mol. Biol. Cell 2013, 24, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Damen, C.A.; Mayo, K.H.; Barendsz-Janson, A.F.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int. J. Cancer 1999, 80, 315–319. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Oude Egbrink, M.G.A.; Kuijpers, M.J.E.; van der Niet, S.T.; Heijnen, V.V.T.; Bouma-ter Steege, J.C.A.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Cook-Mills, J.M.; Deem, T.L. Active participation of endothelial cells in inflammation. J. Leukoc. Biol. 2005, 77, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Lauko, A.; Mu, Z.; Gutmann, D.H.; Naik, U.P.; Lathia, J.D. Junctional Adhesion Molecules in Cancer: A Paradigm for the Diverse Functions of Cell–Cell Interactions in Tumor Progression. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef]
- Georganaki, M.; van Hooren, L.; Dimberg, A. Vascular Targeting to Increase the Efficiency of Immune Checkpoint Blockade in Cancer. Front. Immunol. 2018, 9, 3081. [Google Scholar] [CrossRef]
- Salik, B.; Smyth, M.J.; Nakamura, K. Targeting immune checkpoints in hematological malignancies. J. Hematol. Oncol. J Hematol. Oncol. 2020, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Georganaki, M.; Ramachandran, M.; Tuit, S.; Núñez, N.G.; Karampatzakis, A.; Fotaki, G.; van Hooren, L.; Huang, H.; Lugano, R.; Ulas, T.; et al. Tumor endothelial cell up-regulation of IDO1 is an immunosuppressive feed-back mechanism that reduces the response to CD40-stimulating immunotherapy. Oncoimmunology 2020, 9, 1730538. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Kirkiles-Smith, N.C.; Shepherd, B.R.; McNiff, J.M.; Carr, E.J.; Pober, J.S. Human effector memory CD4+ T cells directly recognize allogeneic endothelial cells in vitro and in vivo. J. Immunol. Baltim. Md 1950 2007, 179, 4397–4404. [Google Scholar] [CrossRef] [PubMed]
- Kochan, G.; Escors, D.; Breckpot, K.; Guerrero-Setas, D. Role of non-classical MHC class I molecules in cancer immunosuppression. Oncoimmunology 2013, 2, e26491. [Google Scholar] [CrossRef]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.-C.; Pircher, A.; Geldhof, V.; de Rooij, L.P.M.H.; Kalucka, J.; Sokol, L.; et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]
- Barthel, S.R.; Gavino, J.D.; Descheny, L.; Dimitroff, C.J. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin. Ther. Targets 2007, 11, 1473–1491. [Google Scholar] [CrossRef]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef]
- Delfortrie, S.; Pinte, S.; Mattot, V.; Samson, C.; Villain, G.; Caetano, B.; Lauridant-Philippin, G.; Baranzelli, M.-C.; Bonneterre, J.; Trottein, F.; et al. Egfl7 promotes tumor escape from immunity by repressing endothelial cell activation. Cancer Res. 2011, 71, 7176–7186. [Google Scholar] [CrossRef]
- Pannier, D.; Philippin-Lauridant, G.; Baranzelli, M.-C.; Bertin, D.; Bogart, E.; Delprat, V.; Villain, G.; Mattot, V.; Bonneterre, J.; Soncin, F. High expression levels of egfl7 correlate with low endothelial cell activation in peritumoral vessels of human breast cancer. Oncol. Lett. 2016, 12, 1422–1428. [Google Scholar] [CrossRef]
- Salama, Y.; Heida, A.H.; Yokoyama, K.; Takahashi, S.; Hattori, K.; Heissig, B. The EGFL7-ITGB3-KLF2 axis enhances survival of multiple myeloma in preclinical models. Blood Adv. 2020, 4, 1021–1037. [Google Scholar] [CrossRef]
- De Sanctis, F.; Ugel, S.; Facciponte, J.; Facciabene, A. The dark side of tumor-associated endothelial cells. Semin. Immunol. 2018, 35, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Karrison, T.; Rowley, D.A.; Schreiber, H. IFN-γ– and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J. Clin. Investig. 2008, 118, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Enzler, T.; Gillessen, S.; Manis, J.P.; Ferguson, D.; Fleming, J.; Alt, F.W.; Mihm, M.; Dranoff, G. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J. Exp. Med. 2003, 197, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- List, A.F. Vascular endothelial growth factor signaling pathway as an emerging target in hematologic malignancies. Oncologist 2001, 6 (Suppl. 5), 24–31. [Google Scholar] [CrossRef][Green Version]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 2018, 16, 242. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Du, T.; Chauhan, D.; Anderson, K.C. Preclinical validation of Alpha-Enolase (ENO1) as a novel immunometabolic target in multiple myeloma. Oncogene 2020, 39, 2786–2796. [Google Scholar] [CrossRef]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.-I.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef]
- Lund, A.W.; Wagner, M.; Fankhauser, M.; Steinskog, E.S.; Broggi, M.A.; Spranger, S.; Gajewski, T.F.; Alitalo, K.; Eikesdal, H.P.; Wiig, H.; et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Investig. 2016, 126, 3389–3402. [Google Scholar] [CrossRef]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef]
- Secchiero, P.; Gonelli, A.; Celeghini, C.; Mirandola, P.; Guidotti, L.; Visani, G.; Capitani, S.; Zauli, G. Activation of the nitric oxide synthase pathway represents a key component of tumor necrosis factor–related apoptosis-inducing ligand–mediated cytotoxicity on hematologic malignancies. Blood 2001, 98, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef]
- Gooden, M.; Lampen, M.; Jordanova, E.S.; Leffers, N.; Trimbos, J.B.; van der Burg, S.H.; Nijman, H.; van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10656–10661. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.; Bernabeu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Stalin, J.; Nollet, M.; Garigue, P.; Fernandez, S.; Vivancos, L.; Essaadi, A.; Muller, A.; Bachelier, R.; Foucault-Bertaud, A.; Fugazza, L.; et al. Targeting soluble CD146 with a neutralizing antibody inhibits vascularization, growth and survival of CD146-positive tumors. Oncogene 2016, 35, 5489–5500. [Google Scholar] [CrossRef]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Scavelli, C.; Weber, E.; Aglianò, M.; Cirulli, T.; Nico, B.; Vacca, A.; Ribatti, D. Lymphatics at the crossroads of angiogenesis and lymphangiogenesis. J. Anat. 2004, 204, 433–449. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Crivellato, E.; Vacca, A. The structure of the vascular network of tumors. Cancer Lett. 2007, 248, 18–23. [Google Scholar] [CrossRef]
- Hunter, M.C.; Teijeira, A.; Halin, C. T Cell Trafficking through Lymphatic Vessels. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Eklund, L.; Bry, M.; Alitalo, K. Mouse models for studying angiogenesis and lymphangiogenesis in cancer. Mol. Oncol. 2013, 7, 259–282. [Google Scholar] [CrossRef]
- Tomura, M.; Yoshida, N.; Tanaka, J.; Karasawa, S.; Miwa, Y.; Miyawaki, A.; Kanagawa, O. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 10871–10876. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.M.; Churchill, M.J.; Breazeale, A.P.; Lane, R.S.; Nelson, N.A.; Lund, A.W. Quantifying Leukocyte Egress via Lymphatic Vessels from Murine Skin and Tumors. J. Vis. Exp. 2019, 58704. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Biasi, S.; Porta, C.; Lo Tartaro, D.; Depenni, R.; Pellacani, G.; Sabbatini, R.; Cossarizza, A. Single-Cell Approaches to Profile the Response to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 490. [Google Scholar] [CrossRef]
- Progatzky, F.; Dallman, M.J.; Lo Celso, C. From seeing to believing: Labelling strategies for in vivo cell-tracking experiments. Interface Focus 2013, 3, 20130001. [Google Scholar] [CrossRef]
- Ito, K.; Morimoto, J.; Kihara, A.; Matsui, Y.; Kurotaki, D.; Kanayama, M.; Simmons, S.; Ishii, M.; Sheppard, D.; Takaoka, A.; et al. Integrin α9 on lymphatic endothelial cells regulates lymphocyte egress. Proc. Natl. Acad. Sci. USA 2014, 111, 3080–3085. [Google Scholar] [CrossRef]
- Schwab, S.R.; Cyster, J.G. Finding a way out: Lymphocyte egress from lymphoid organs. Nat. Immunol. 2007, 8, 1295–1301. [Google Scholar] [CrossRef]
- Torcellan, T.; Hampton, H.R.; Bailey, J.; Tomura, M.; Brink, R.; Chtanova, T. In vivo photolabeling of tumor-infiltrating cells reveals highly regulated egress of T-cell subsets from tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 5677–5682. [Google Scholar] [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef]
- Condeelis, J.; Weissleder, R. In vivo imaging in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003848. [Google Scholar] [CrossRef]
- Vakoc, B.J.; Lanning, R.M.; Tyrrell, J.A.; Padera, T.P.; Bartlett, L.A.; Stylianopoulos, T.; Munn, L.L.; Tearney, G.J.; Fukumura, D.; Jain, R.K.; et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat. Med. 2009, 15, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Antonio, G.; Oronzo, B.; Vito, L.; Angela, C.; Antonel-la, A.; Roberto, C.; Giovanni, S.A.; Antonella, L. Immune system and bone microenvironment: Rationale for targeted cancer therapies. Oncotarget 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Heishi, T.; Khan, O.F.; Kowalski, P.S.; Incio, J.; Rahbari, N.N.; Chung, E.; Clark, J.W.; Willett, C.G.; Luster, A.D.; et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J. Clin. Investig. 2017, 127, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Bruns, O.T.; Bischof, T.S.; Harris, D.K.; Franke, D.; Shi, Y.; Riedemann, L.; Bartelt, A.; Jaworski, F.B.; Carr, J.A.; Rowlands, C.J.; et al. Next-generation in vivo optical imaging with short-wave infrared quantum dots. Nat. Biomed. Eng. 2017, 1, 56. [Google Scholar] [CrossRef]
- Brown, E.B.; Campbell, R.B.; Tsuzuki, Y.; Xu, L.; Carmeliet, P.; Fukumura, D.; Jain, R.K. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat. Med. 2001, 7, 864–868. [Google Scholar] [CrossRef]
- Kirkpatrick, N.D.; Chung, E.; Cook, D.C.; Han, X.; Gruionu, G.; Liao, S.; Munn, L.L.; Padera, T.P.; Fukumura, D.; Jain, R.K. Video-rate resonant scanning multiphoton microscopy: An emerging technique for intravital imaging of the tumor microenvironment. Intravital 2012, 1. [Google Scholar] [CrossRef]
- Yuan, F.; Salehi, H.A.; Boucher, Y.; Vasthare, U.S.; Tuma, R.F.; Jain, R.K. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994, 54, 4564–4568. [Google Scholar]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Li, J.; Chekkoury, A.; Prakash, J.; Glasl, S.; Vetschera, P.; Koberstein-Schwarz, B.; Olefir, I.; Gujrati, V.; Omar, M.; Ntziachristos, V. Spatial heterogeneity of oxygenation and haemodynamics in breast cancer resolved in vivo by conical multispectral optoacoustic mesoscopy. Light Sci. Appl. 2020, 9, 57. [Google Scholar] [CrossRef]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.-L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, G.; Li, X.; Zhang, Y.; Jiang, Y.; Shen, J.; Liu, J.; Wang, Q.; Zhu, J.; Feng, X.; et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1α in hepatocellular carcinoma. BMC Cancer 2013, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomedicine 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- Brunetti, O.; Gnoni, A.; Licchetta, A.; Longo, V.; Calabrese, A.; Argentiero, A.; Delcuratolo, S.; Solimando, A.G.; Casadei-Gardini, A.; Silvestris, N. Predictive and Prognostic Factors in HCC Patients Treated with Sorafenib. Med. Kaunas Lith. 2019, 55, 707. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef]
- Longo, V.; Brunetti, O.; Gnoni, A.; Licchetta, A.; Delcuratolo, S.; Memeo, R.; Solimando, A.G.; Argentiero, A. Emerging role of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Med. Kaunas Lith. 2019, 55, 698. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef]
- Feng, J.; Byrne, N.M.; Al Jamal, W.; Coulter, J.A. Exploiting Current Understanding of Hypoxia Mediated Tumour Progression for Nanotherapeutic Development. Cancers 2019, 11, 1989. [Google Scholar] [CrossRef]
- Koumenis, C.; Wouters, B.G. “Translating” tumor hypoxia: Unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol. Cancer Res. MCR 2006, 4, 423–436. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Samples, J.; Willis, M.; Klauber-Demore, N. Targeting angiogenesis and the tumor microenvironment. Surg. Oncol. Clin. N. Am. 2013, 22, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Frassanito, M.A.; Tamma, R.; Saltarella, I.; Di Marzo, L.; Lamanuzzi, A.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; et al. Rhu-Epo down-regulates pro-tumorigenic activity of cancer-associated fibroblasts in multiple myeloma. Ann. Hematol. 2018, 97, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Rao, L.; Giannico, D.; Leone, P.; Solimando, A.G.; Maiorano, E.; Caporusso, C.; Duda, L.; Tamma, R.; Mallamaci, R.; Susca, N.; et al. HB-EGF-EGFR Signaling in Bone Marrow Endothelial Cells Mediates Angiogenesis Associated with Multiple Myeloma. Cancers 2020, 12, 173. [Google Scholar] [CrossRef]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.-S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Gerald, D.; Chintharlapalli, S.; Augustin, H.G.; Benjamin, L.E. Angiopoietin-2: An Attractive Target for Improved Antiangiogenic Tumor Therapy. Cancer Res. 2013, 73, 1649–1657. [Google Scholar] [CrossRef]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef]
- Chae, S.-S.; Kamoun, W.S.; Farrar, C.T.; Kirkpatrick, N.D.; Niemeyer, E.; de Graaf, A.M.A.; Sorensen, A.G.; Munn, L.L.; Jain, R.K.; Fukumura, D. Angiopoietin-2 Interferes with Anti-VEGFR2-Induced Vessel Normalization and Survival Benefit in Mice Bearing Gliomas. Clin. Cancer Res. 2010, 16, 3618–3627. [Google Scholar] [CrossRef]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Workman, C.J.; Vignali, D.A.A. Targeting regulatory T cells in tumors. FEBS J. 2016, 283, 2731–2748. [Google Scholar] [CrossRef] [PubMed]
- Maenhout, S.K.; Thielemans, K.; Aerts, J.L. Location, location, location: Functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology 2014, 3, e956579. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e5. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef]
- Hendry, S.A.; Farnsworth, R.H.; Solomon, B.; Achen, M.G.; Stacker, S.A.; Fox, S.B. The Role of the Tumor Vasculature in the Host Immune Response: Implications for Therapeutic Strategies Targeting the Tumor Microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Kowanetz, M.; Wu, X.; Zhuang, G.; Ngu, H.; Finkle, D.; Komuves, L.; Peale, F.; Ferrara, N. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J. Pathol. 2012, 227, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Lorgis, V.; Maura, G.; Coppa, G.; Hassani, K.; Taillandier, L.; Chauffert, B.; Apetoh, L.; Ladoire, S.; Ghiringhelli, F. Relation between bevacizumab dose intensity and high-grade glioma survival: A retrospective study in two large cohorts. J. Neurooncol. 2012, 107, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef]
- Gnoni, A.; Licchetta, A.; Memeo, R.; Argentiero, A.; Solimando, A.G.; Longo, V.; Delcuratolo, S.; Brunetti, O. Role of BRAF in Hepatocellular Carcinoma: A Rationale for Future Targeted Cancer Therapies. Medicina 2019, 55, 754. [Google Scholar] [CrossRef]
- Krebs, M.; Solimando, A.G.; Kalogirou, C.; Marquardt, A.; Frank, T.; Sokolakis, I.; Hatzichristodoulou, G.; Kneitz, S.; Bargou, R.; Kübler, H.; et al. miR-221-3p Regulates VEGFR2 Expression in High-Risk Prostate Cancer and Represents an Escape Mechanism from Sunitinib In Vitro. J. Clin. Med. 2020, 9, 670. [Google Scholar] [CrossRef]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D.; et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Proangiogenic Molecules * | Soluble Factors * | Immune Checkpoints | Major Histocompatibility Complex (MHC) | Adhesion Molecules * |
|---|---|---|---|---|
| FGF2 Modulate selective up- and down-regulation [56,103,104] of adhesion molecules (ICAM [30,105], VCAM [106], JAMs [35,107,108]) | Chemokines (CCL2/18, CXCL10/11, CXCL4) Deregulated chemokines, halting immune effector surveillance and attracting immune tolerogenic cells [27,43,105,109,110] | PD-L1/2 Cancer endothelium, also express immune checkpoints: a cross talk between aberrant vasculature, immune, and cancer cells creates an immune permissive tumor milieu [58,101,111,112,113,114] | MHC I Often overexpressed within the tumor niche, where the cancer associated endothelium is characterized by a lack of co-stimulatory molecules (B7.1–and B7.2) [58,115,116,117] | Selectin-mediated leukocyte rolling E-selectin/P-selectin Orchestrate leukocyte recruitment. [79,118,119] |
| NEU1 Induces a decreased adhesion molecule expression and boosts angiogenesis via NOTCH pathway [120,121,122] | Cytokines (IFNγ,TNFα) unresponsiveness and anergy along with PD-L1 overexpression [112,123,124,125,126,127] | ENO-1 acts both as a glycolytic enzyme and a plasminogen receptor expressed on the cell surface of tumor cells. Surface ENO1 plays a crucial role in cancer metabolism, tumor invasion and immune suppression in the cancer immune-microenvironment [35,128] | MHC II Can be decreased on tumor infiltrating vessels, thus contributing to an immune tolerogenic niche [117] | Integrin-mediated leukocyte rolling: ICAM1 binds to LFA1 (αLβ2 integrin); VCAM1 binds to VLA4 (α4β1 integrin) Multiple functions [27] ** Function |
| VEGF-A/C Vascular endothelial growth factors induce cell phenotype changes, recruiting immune suppressive cells [58,62,129,130] | NO Directly and indirectly affect effective immune response by altering leukocyte infiltration and suppressing CD8+ T cells [131,132,133,134] | IDO1 and TIM3 Immune regulatory checkpoints overexpress in cancer endothelial cells upon cytokines stimulation (i.e., IFNγ) able to induce T cells programmed cell death and cell cycle arrest, respectively [114] | HLA-E CD8+T cells infiltration in ovarian cancer correlated with improved better survival when HLA-E expression is decreased [135] | VE-cadherin and intracellular membrane compartments, containing PECAM1, JAMs, ESAM, ICAM2, and CD99 promote paracellular migration [27,136,137,138] # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solimando, A.G.; Summa, S.D.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. https://doi.org/10.3390/cancers12113380
Solimando AG, Summa SD, Vacca A, Ribatti D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers. 2020; 12(11):3380. https://doi.org/10.3390/cancers12113380
Chicago/Turabian StyleSolimando, Antonio Giovanni, Simona De Summa, Angelo Vacca, and Domenico Ribatti. 2020. "Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling" Cancers 12, no. 11: 3380. https://doi.org/10.3390/cancers12113380
APA StyleSolimando, A. G., Summa, S. D., Vacca, A., & Ribatti, D. (2020). Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers, 12(11), 3380. https://doi.org/10.3390/cancers12113380