Impact of Genetic Ancestry on Prognostic Biomarkers in Uveal Melanoma

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Baseline Patient Characteristics
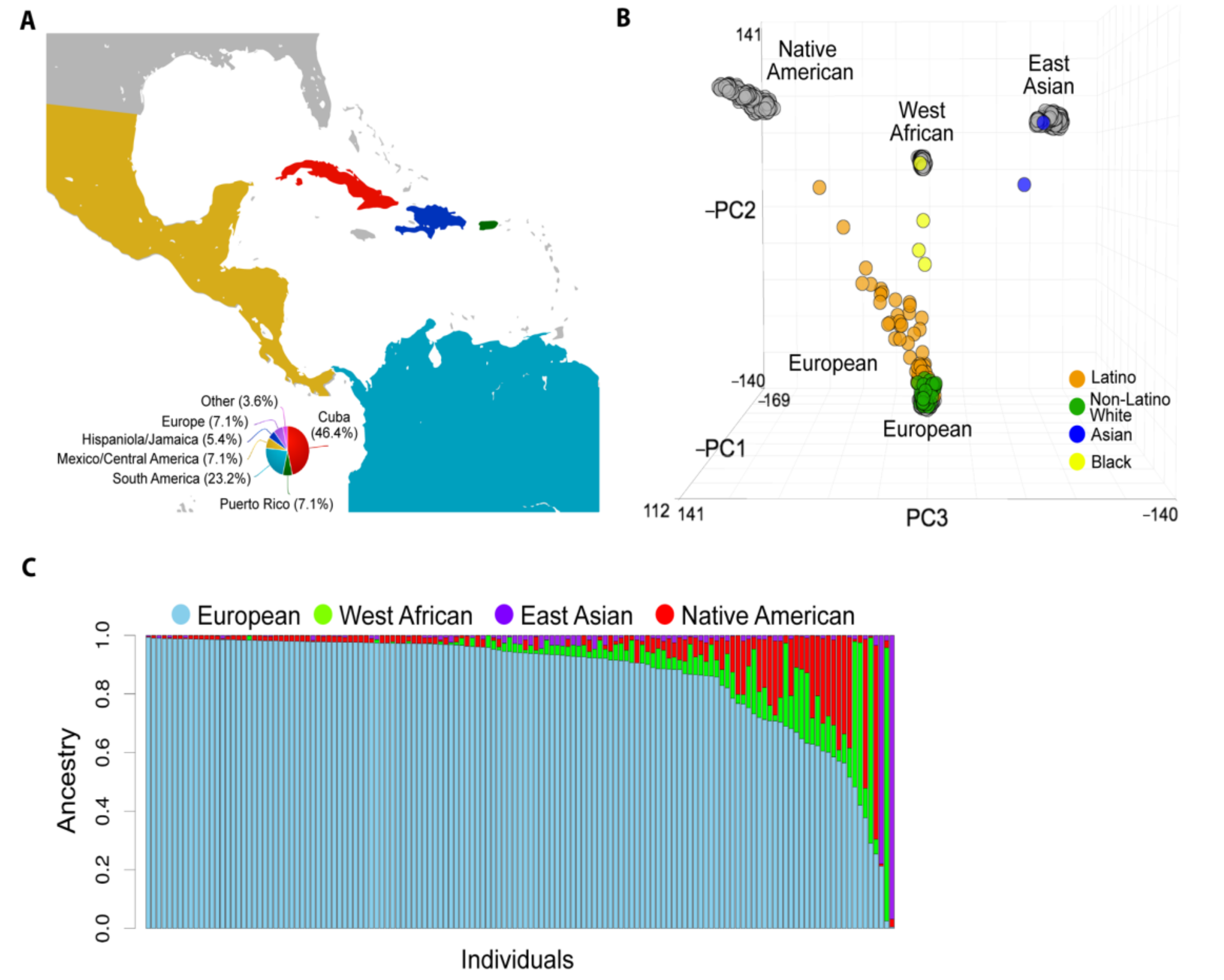
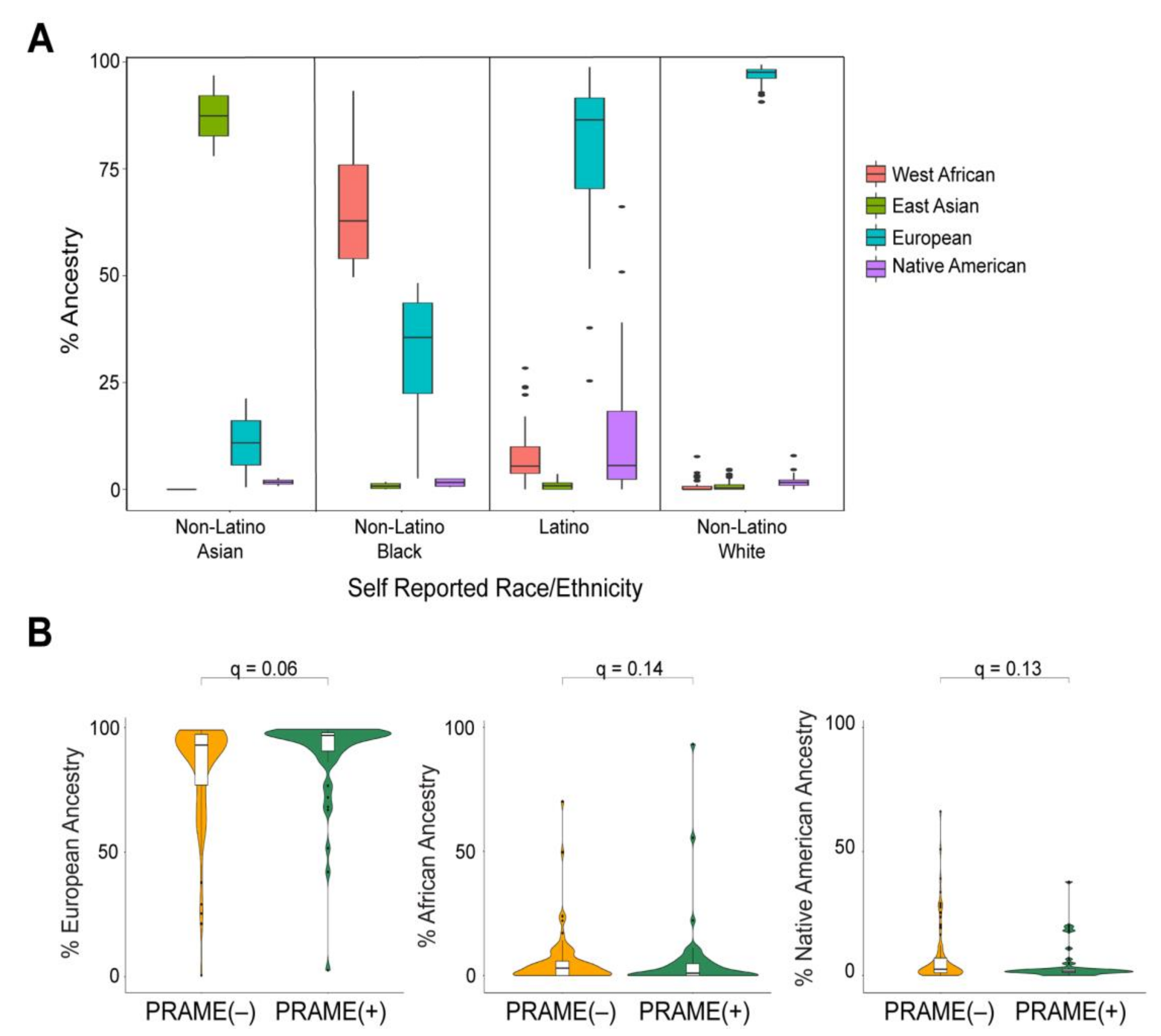
2.2. Global Genetic Ancestry and Association with Prognosis
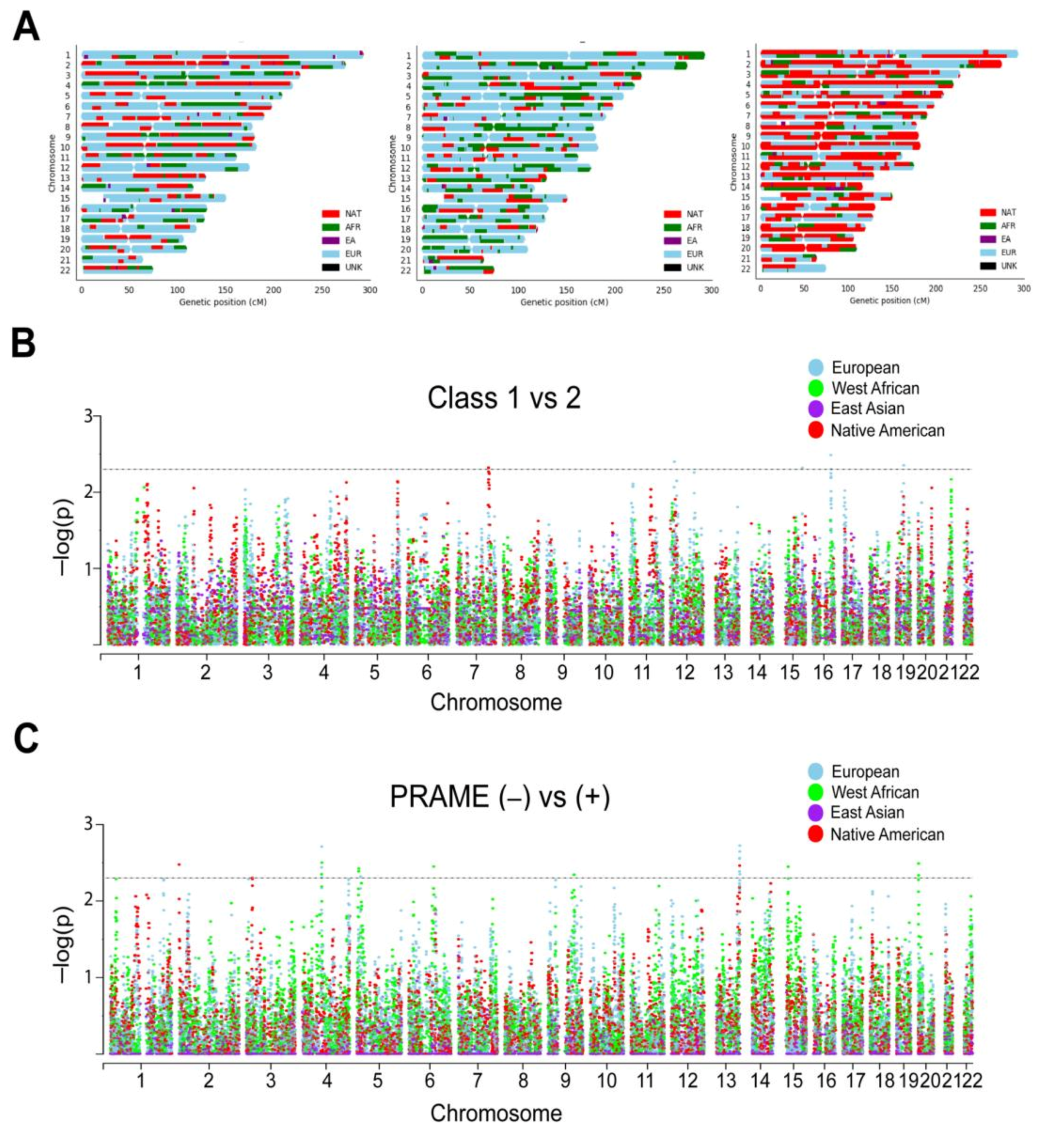
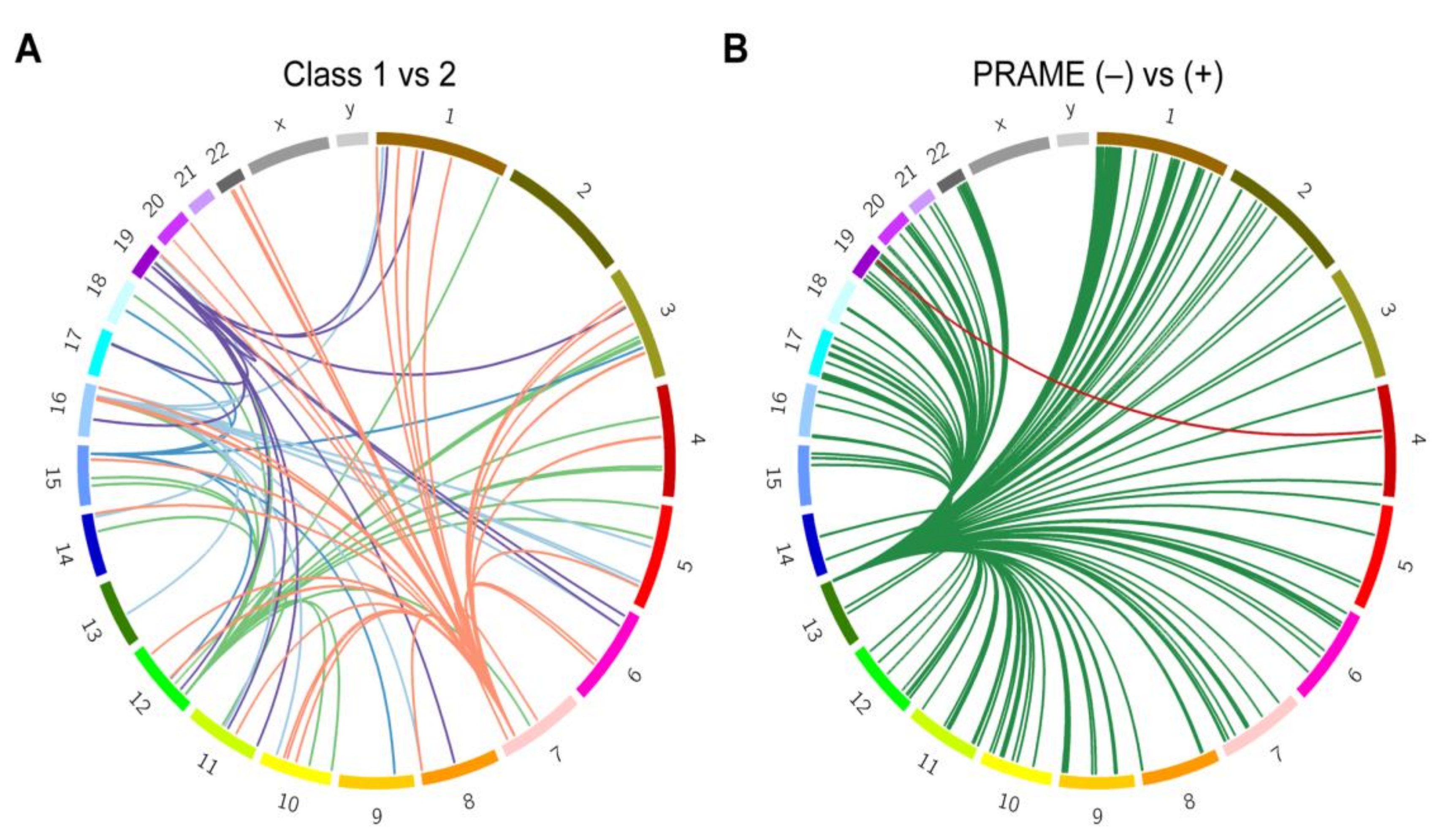
2.3. Local Genetic Ancestry and Association with Prognosis
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Genotyping
4.2. Reference Populations and Ancestry Estimation
4.3. Quantitative Trait Loci
4.4. Global Associations and Admixture Mapping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Bustamante, C.D.; De La Vega, F.M.; Burchard, E.G. Genomics for the world. Nat. Cell Biol. 2011, 475, 163–165. [Google Scholar] [CrossRef]
- Petrovski, S.; Goldstein, D.B. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 2016, 17, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Hu, Z.; Mahal, B.A.; Zhao, S.D.; Kensler, K.H.; Pi, J.; Hu, X.; Zhang, Y.; Wang, Y.; Jiang, J.; et al. Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer Cell 2018, 34, 549–560.e9. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.H.; Qian, H.; Hou, Z.; Rosen, J.D.; Tapia, A.L.; Shan, Y.; Jain, D.; Argos, M.; Arnett, D.K.; Avery, C.; et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019, 15, e1008500. [Google Scholar] [CrossRef] [PubMed]
- Aaberg, T.M.; Cook, R.W.; Oelschlager, K.; Maetzold, D.; Rao, P.K.; Mason, J.O. Current clinical practice: Differential management of uveal melanoma in the era of molecular tumor analyses. Clin. Ophthalmol. 2014, 8, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Char, D.H.; Augsburger, J.J.; Correa, Z.M.; Nudleman, E.; Aaberg, T.M.; Altaweel, M.M.; Bardenstein, D.S.; Finger, P.T.; et al. Collaborative Ocular Oncology Group Report Number 1: Prospective Validation of a Multi-Gene Prognostic Assay in Uveal Melanoma. Ophthalmology 2012, 119, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Roberson, E.D.O.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Decatur, C.L.; Tarlan, B.; Oelschlager, K.M.; Stone, J.F.; Kuznetsov, J.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Epigenetic reprogramming and aberrant expression of PRAME are associated with increased metastatic risk in Class 1 and Class 2 uveal melanomas. Oncotarget 2016, 7, 59209–59219. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; Van Der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. HaploReg v4: Systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2015, 44, D877–D881. [Google Scholar] [CrossRef]
- Onken, M.D.; Ehlers, J.P.; Worley, L.A.; Makita, J.; Yokota, Y.; Harbour, J.W. Functional Gene Expression Analysis Uncovers Phenotypic Switch in Aggressive Uveal Melanomas. Cancer Res. 2006, 66, 4602–4609. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-H.; Worley, L.A.; Onken, M.D.; Harbour, J.W. Prognostic biomarkers in uveal melanoma: Evidence for a stem cell-like phenotype associated with metastasis. Melanoma Res. 2008, 18, 191–200. [Google Scholar] [CrossRef]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Van Essen, T.; Van Pelt, S.I.; Bronkhorst, I.H.G.; Versluis, M.; Némati, F.; Laurent, C.; Luyten, G.P.M.; Hall, T.E.; Elsen, P.J.V.D.; Van Der Velden, P.A.; et al. Upregulation of HLA Expression in Primary Uveal Melanoma by Infiltrating Leukocytes. PLoS ONE 2016, 11, e0164292. [Google Scholar] [CrossRef]
- Heppt, M.V.; Steeb, T.; Schlager, J.G.; Rosumeck, S.; Dressler, C.; Ruzicka, T.; Nast, A.; Berking, C. Immune checkpoint blockade for unresectable or metastatic uveal melanoma: A systematic review. Cancer Treat. Rev. 2017, 60, 44–52. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Jack, J.; Havener, T.M.; McLeod, H.L.; Motsinger-Reif, A.A.; Foster, M. Abstract LB-246: Evaluating the role of admixture in cancer therapy via in vitro drug response and multivariate genome-wide associations. Exp. Mol. Ther. 2015, 75, 1451–1463. [Google Scholar] [CrossRef]
- Gezgin, G.; Dogrusöz, M.; Van Essen, T.H.; Kroes, W.G.M.; Luyten, G.P.M.; Van Der Velden, P.A.; Walter, V.; Verdijk, R.M.; Hall, T.E.; Van Der Burg, S.H.; et al. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol. Immunother. 2017, 66, 903–912. [Google Scholar] [CrossRef]
- Sharma, A.; Liu, H.; Tobar-Tosse, F.; Noll, A.; Dakal, T.C.; Li, H.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Genome organization in proximity to the BAP1 locus appears to play a pivotal role in a variety of cancers. Cancer Sci. 2020, 111, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Castle Biosciences: Molecular Diagnostics for Cancer. Available online: https://castlebiosciences.com (accessed on 10 January 2020).
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed]
- Beagle Reference. Available online: http://bochet.gcc.biostat.washington.edu/beagle/1000_Genomes_phase3_v5a/READ_ME_beagle_ref (accessed on 1 March 2018).
- Reich, D.; Patterson, N.; Campbell, D.; Tandon, A.; Mazieres, S.; Ray, N.; Parra, M.V.; Rojas, W.; Duque, C.; Mesa, N.; et al. Reconstructing Native American population history. Nat. Cell Biol. 2012, 488, 370–374. [Google Scholar] [CrossRef]
- Rosenberg, N.A.; Pritchard, J.K.; Weber, J.L.; Cann, H.M.; Kidd, K.K.; Zhivotovsky, L.A.; Feldman, M.W. Genetic Structure of Human Populations. Science 2002, 298, 2381–2385. [Google Scholar] [CrossRef]
- Rosenberg, N.A.; Mahajan, S.; Ramachandran, S.; Zhao, C.; Pritchard, J.K.; Feldman, M.W. Clines, Clusters, and the Effect of Study Design on the Inference of Human Population Structure. PLoS Genet. 2005, 1, e70. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Wang, C.; The FUSION Study; Zhan, X.; Bragggresham, J.L.; Kang, H.M.; Stambolian, D.; Chew, E.Y.; Branham, K.E.; Heckenlively, J.R.; Fulton, R.; et al. Ancestry estimation and control of population stratification for sequence-based association studies. Nat. Genet. 2014, 46, 409–415. [Google Scholar] [CrossRef]
- Wang, C.; Zhan, X.; Liang, L.; Abecasis, G.R.; Lin, X. Improved Ancestry Estimation for both Genotyping and Sequencing Data using Projection Procrustes Analysis and Genotype Imputation. Am. J. Hum. Genet. 2015, 96, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Population Architecture using Genomics and Epidemiology (PAGE). Available online: http://www.pagestudy.org (accessed on 1 March 2018).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: https://broadinstitute.github.io/picard (accessed on 1 February 2018).
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Maples, B.K.; Gravel, S.; Kenny, E.E.; Bustamante, C.D. RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef]
- Martin, E.R.; Tunc, I.; Liu, Z.; Slifer, S.H.; Beecham, A.H.; Beecham, G.W. Properties of global- and local-ancestry adjustments in genetic association tests in admixed populations. Genet. Epidemiol. 2017, 42, 214–229. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA). Available online: https://www.cancer.gov/tcga (accessed on 1 January 2019).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Delaneau, O.; Ongen, H.; Brown, A.A.; Fort, A.; Panousis, N.I.; Dermitzakis, E.T. A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 2017, 8, 15452. [Google Scholar] [CrossRef]
- QTLtools. Available online: https://qtltools.github.io/qtltools (accessed on 1 January 2018).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ambrosini, G.; Bucher, P. SNP2TFBS—A database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017, 45, D139–D144. [Google Scholar] [CrossRef] [PubMed]
- sCMplot: A High-Quality Drawing Tool Designed for Manhattan Plot of Genomic Analysis. Available online: https://github.com/YinLiLin/R-CMplot (accessed on 1 January 2018).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Status | White Non-Latino | Other | ALL | q Value a |
|---|---|---|---|---|---|
| Patient age (years) | Mean | 63.04 | 60.24 | 61.71 | 0.163 |
| Median | 65 | 62 | 65 | ||
| Minimum-Maximum | 22–87 | 18–87 | 18–87 | ||
| Sex | Male | 33 | 39 | 72 | 0.116 |
| Female | 41 | 28 | 69 | ||
| Largest tumor diameter (mm) | Mean | 14.75 | 14.32 | 14.55 | 0.291 |
| Median | 14.65 | 14 | 14.5 | ||
| Minimum-Maximum | 7.5–24 | 6.9–23 | 6.9–24 | ||
| Tumor thickness (mm) | Mean | 5.97 | 6.81 | 6.36 | 0.115 |
| Median | 4.6 | 5.95 | 5 | ||
| Minimum-Maximum | 1.5–14.9 | 1.7–15.3 | 1.5–15.3 | ||
| Ciliary Body Involvement | Yes | 21 | 16 | 37 | 0.291 |
| No | 51 | 49 | 100 | ||
| Melanocytosis | Yes | 1 | 6 | 7 | 0.117 |
| No | 43 | 48 | 91 | ||
| GEP class | Class 1 | 43 | 37 | 80 | 0.293 |
| Class 2 | 31 | 30 | 61 | ||
| PRAME status | Positive | 31 | 20 | 51 | 0.116 |
| Negative | 37 | 46 | 83 | ||
| Primary treatment | Enucleation | 26 | 21 | 47 | 0.290 |
| Brachytherapy | 48 | 46 | 94 | ||
| Metastasis | Yes | 16 | 7 | 23 | 0.110 |
| No | 58 | 60 | 118 |
| Ancestral Group | Subpopulation | Number of Reference Samples | Number of Reference Samples Postprocessing |
|---|---|---|---|
| European | British, Iberian, Toscani, Finnish, Utah residents | 659 | 358 |
| West African | Gambian Mandinka, Mende, Esan, Yoruba, Luhya | 604 | 398 |
| East Asian | Dai Chinese, Han Chinese, Japanese, Southern Han Chinese, Kinh Vietnamese | 601 | 384 |
| Native American | Aleutian, Algonquin, Arara, Arhuaco, Aymara, Bribri, Cabecar, Chane, Chilote, Chipewyan, Chono, Chorotega, Cree, Diaguita, EGInuit, Eguahibo, Guarani, Guaymi, Huetar, Hulliche, Inga, Jamamadi, Kaingang, Kaqchikel, Karitiana, Kogi, Maleku, Maya, Mixe, Mixtec, Ojibwa, Palikur, Parakana, Piapoco, Pima, Purepecha, Quechua, Surui, Tepehuano, Teribe, Ticuna, Toba, Waunana, Wayuu, WGInuit, Wichi, Yaghan, Yaqui, Zapotec | 493 | 241 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, D.A.; Sanchez, M.I.; Decatur, C.L.; Correa, Z.M.; Martin, E.R.; Harbour, J.W. Impact of Genetic Ancestry on Prognostic Biomarkers in Uveal Melanoma. Cancers 2020, 12, 3208. https://doi.org/10.3390/cancers12113208
Rodriguez DA, Sanchez MI, Decatur CL, Correa ZM, Martin ER, Harbour JW. Impact of Genetic Ancestry on Prognostic Biomarkers in Uveal Melanoma. Cancers. 2020; 12(11):3208. https://doi.org/10.3390/cancers12113208
Chicago/Turabian StyleRodriguez, Daniel A., Margaret I. Sanchez, Christina L. Decatur, Zelia M. Correa, Eden R. Martin, and J. William Harbour. 2020. "Impact of Genetic Ancestry on Prognostic Biomarkers in Uveal Melanoma" Cancers 12, no. 11: 3208. https://doi.org/10.3390/cancers12113208
APA StyleRodriguez, D. A., Sanchez, M. I., Decatur, C. L., Correa, Z. M., Martin, E. R., & Harbour, J. W. (2020). Impact of Genetic Ancestry on Prognostic Biomarkers in Uveal Melanoma. Cancers, 12(11), 3208. https://doi.org/10.3390/cancers12113208