Plasma Predictive Features in Treating EGFR-Mutated Non-Small Cell Lung Cancer


 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics
2.2. Presence of Concomitant Mutations (besides EGFR Primary Mutation and p.T790M)
2.3. Presence of TP53 Mutations
2.4. Resistance Mechanisms
2.4.1. First Line Cohort (1st Generation EGFR-TKI)
2.4.2. Second Line Cohort (Osimertinib)
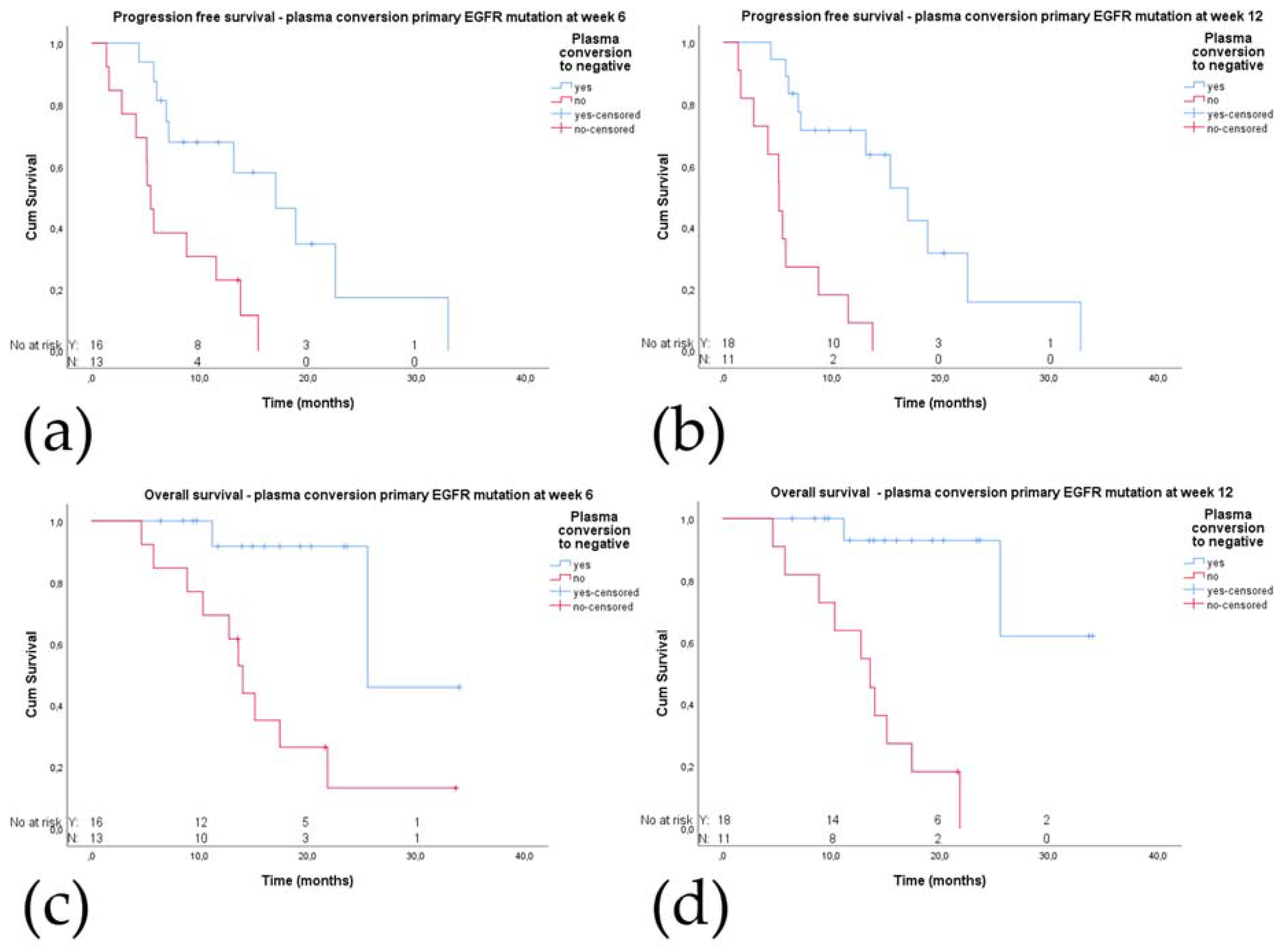
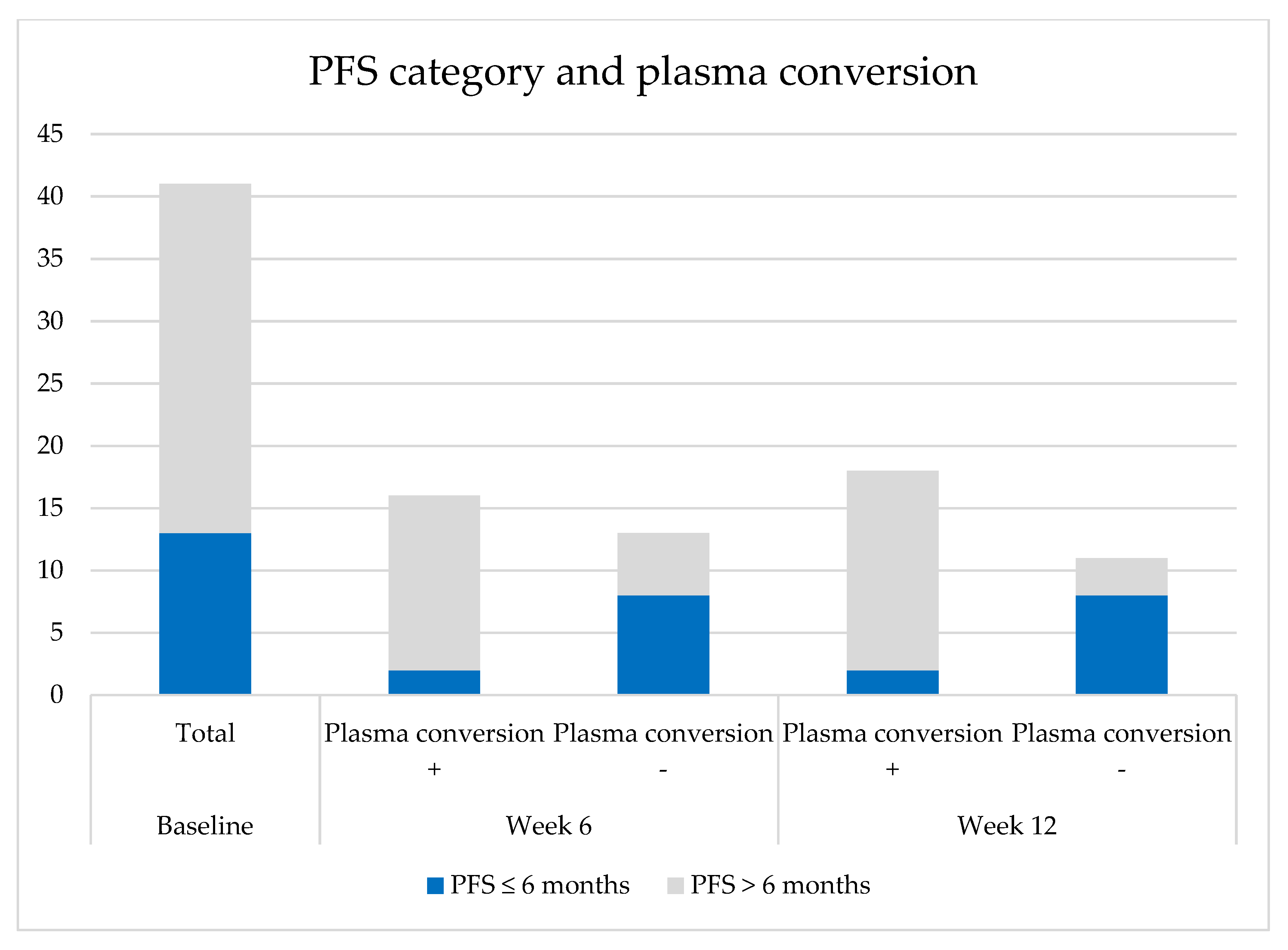
2.5. Plasma Conversion
2.5.1. Plasma Conversion to cfDNA Negative for the Primary EGFR Mutation
2.5.2. Plasma Conversion to cfDNA Negative for the EGFR p.T790M Mutation
2.5.3. Plasma Mutation Levels during Treatment
2.6. Short Responders
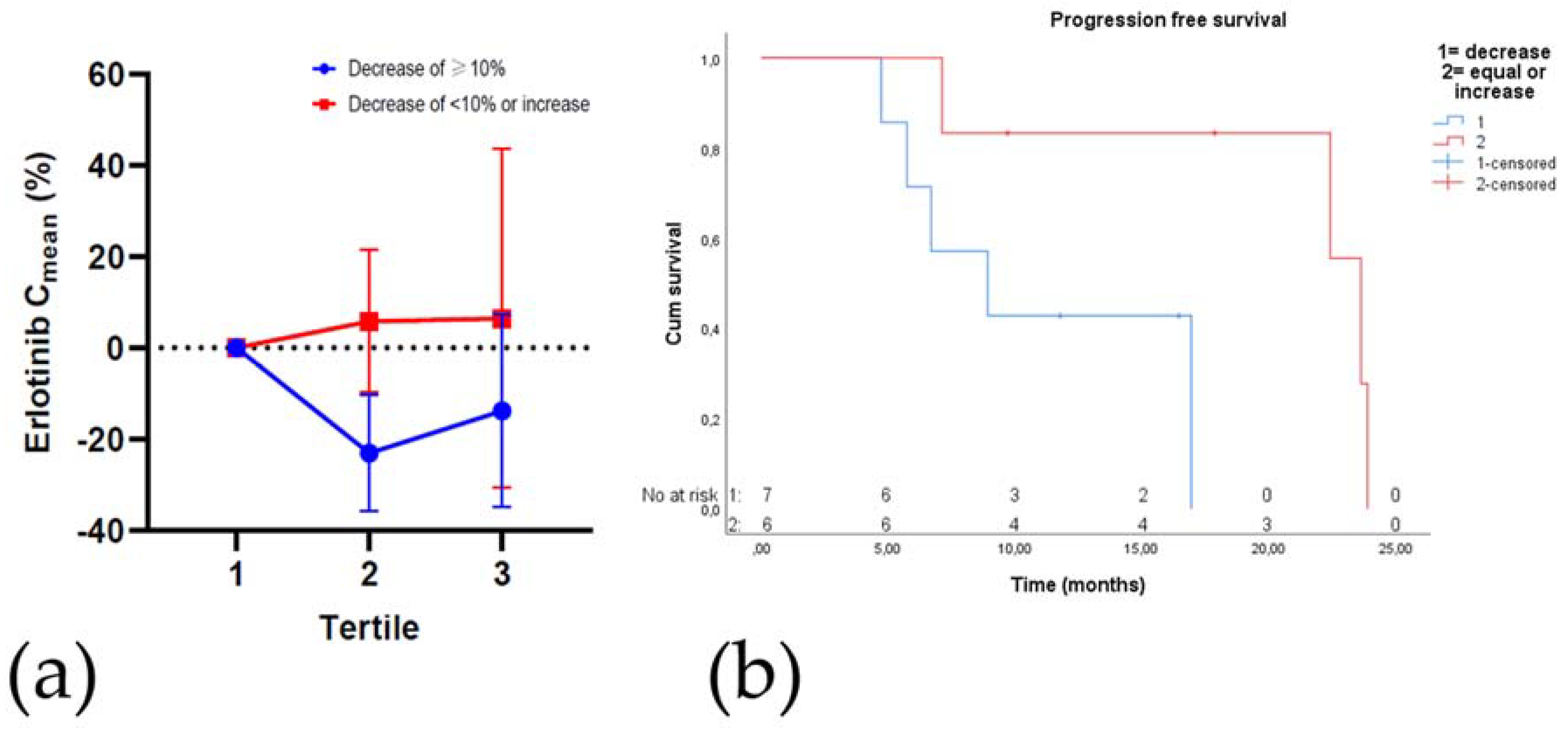
2.7. Plasma Drug Concentrations
Toxicity
2.8. Brain Metastasis
2.9. Multivariate Analysis
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Blood Collection and Processing
4.2.1. Next Generation Sequencing (NGS)
4.2.2. Plasma Drug Concentrations
4.3. Toxicity
4.4. Brain Metastasis
4.5. Objectives
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Hsu, W.H.; Yang, J.C.; Mok, T.S.; Loong, H.H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29, i3–i9. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, B.; Shim, J.H.; Lee, S.H.; Park, W.Y.; Choi, Y.L.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K. Concurrent genetic alterations predict the progression to target therapy in EGFR-mutated advanced NSCLC. J. Thorac. Oncol. 2019, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Lai, Y.C.; Chang, C.Y.; Huang, L.K.; Chen, S.J.; Tan, K.T.; Yu, P.N.; Lai, J.I. Concomitant genetic alterations are associated with worse clinical outcome in EGFR mutant NSCLC patients treated with tyrosine kinase inhibitors. Transl. Oncol. 2019, 12, 1425–1431. [Google Scholar] [CrossRef]
- Jiao, X.D.; Qin, B.D.; You, P.; Cai, J.; Zang, Y.S. The prognostic value of tp53 and its correlation with EGFR mutation in advanced non-small cell lung cancer, an analysis based on cbioportal data base. Lung Cancer 2018, 123, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 mutations on outcome in EGFR-mutated patients treated with first-line tyrosine kinase inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef]
- Labbe, C.; Cabanero, M.; Korpanty, G.J.; Tomasini, P.; Doherty, M.K.; Mascaux, C.; Jao, K.; Pitcher, B.; Wang, R.; Pintilie, M.; et al. Prognostic and predictive effects of tp53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 2017, 111, 23–29. [Google Scholar] [CrossRef]
- Hou, H.; Qin, K.; Liang, Y.; Zhang, C.; Liu, D.; Jiang, H.; Liu, K.; Zhu, J.; Lv, H.; Li, T.; et al. Concurrent tp53 mutations predict poor outcomes of EGFR-tki treatments in chinese patients with advanced NSCLC. Cancer Manag. Res. 2019, 11, 5665–5675. [Google Scholar] [CrossRef]
- Verheijen, R.B.; Yu, H.; Schellens, J.H.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin. Pharmacol. Ther. 2017, 102, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Veerman, G.D.M.; Hussaarts, K.; Jansman, F.G.A.; Koolen, S.W.L.; van Leeuwen, R.W.F.; Mathijssen, R.H.J. Clinical implications of food-drug interactions with small-molecule kinase inhibitors. Lancet Oncol. 2020, 21, e265–e279. [Google Scholar] [CrossRef]
- Hussaarts, K.; Veerman, G.D.M.; Jansman, F.G.A.; van Gelder, T.; Mathijssen, R.H.J.; van Leeuwen, R.W.F. Clinically relevant drug interactions with multikinase inhibitors: A review. Ther. Adv. Med. Oncol. 2019, 11, 1758835918818347. [Google Scholar] [CrossRef]
- Brown, K.; Comisar, C.; Witjes, H.; Maringwa, J.; de Greef, R.; Vishwanathan, K.; Cantarini, M.; Cox, E. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br. J. Clin. Pharmacol. 2017, 83, 1216–1226. [Google Scholar] [CrossRef]
- Hidalgo, M.; Siu, L.L.; Nemunaitis, J.; Rizzo, J.; Hammond, L.A.; Takimoto, C.; Eckhardt, S.G.; Tolcher, A.; Britten, C.D.; Denis, L.; et al. Phase i and pharmacologic study of osi-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J. Clin. Oncol. 2001, 19, 3267–3279. [Google Scholar] [CrossRef] [PubMed]
- Lankheet, N.A.; Knapen, L.M.; Schellens, J.H.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib, and sunitinib in routine clinical outpatient cancer care. Ther. Drug Monit. 2014, 36, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Eppler, S.M.; Wolf, J.; Hamilton, M.; Rakhit, A.; Bruno, R.; Lum, B.L. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin. Pharmacol. Ther. 2006, 80, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; Becker, K.; Levy, B.P. Epidermal growth factor receptor tyrosine kinase inhibitors for central nervous system metastases from non-small cell lung cancer. Oncologist 2018, 23, 1199–1209. [Google Scholar] [CrossRef]
- Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Han, J.Y.; Katakami, N.; Kim, H.R.; Hodge, R.; Kaur, P.; Brown, A.P.; Ghiorghiu, D.; et al. Cns efficacy of osimertinib in patients with t790m-positive advanced non-small-cell lung cancer: Data from a randomized phase iii trial (aura3). J. Clin. Oncol. 2018, 36, 2702–2709. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): A statement paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.; Barrett, J.C.; Jänne, P.A. Association between plasma genotyping and outcomes of treatment with osimertinib (azd9291) in advanced non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef]
- Akamatsu, H.; Koh, Y.; Okamoto, I.; Fujimoto, D.; Bessho, A.; Azuma, K.; Morita, S.; Yamamoto, N.; Nakagawa, K. Clinical significance of monitoring EGFR mutation in plasma using multiplexed digital pcr in EGFR mutated patients treated with afatinib (west japan oncology group 8114LTR study). Lung Cancer 2019, 131, 128–133. [Google Scholar] [CrossRef]
- Ebert, E.B.F.; McCulloch, T.; Hansen, K.H.; Linnet, H.; Sorensen, B.; Meldgaard, P. Clearing of circulating tumour DNA predicts clinical response to first line tyrosine kinase inhibitors in advanced epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2020, 141, 37–43. [Google Scholar] [CrossRef]
- Buder, A.; Hochmair, M.J.; Setinek, U.; Pirker, R.; Filipits, M. EGFR mutation tracking predicts survival in advanced EGFR-mutated non-small cell lung cancer patients treated with osimertinib. Transl. Lung Cancer Res. 2020, 9, 239–245. [Google Scholar] [CrossRef]
- Buttitta, F.; Felicioni, L.; Lorito, A.D.; Cortellini, A.; Irtelli, L.; Brocco, D.; Marino, P.D.; Traisci, D.; D’Ostilio, N.; Paolo, A.D.; et al. Early prediction of resistance to tyrosine kinase inhibitors by plasma monitoring of EGFR mutations in NSCLC: A new algorithm for patient selection and personalized treatment. Oncotarget 2020, 11, 982–991. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Liu, G.; Stockley, T.L.; Shabir, M.; Zhang, T.; Law, J.H.; Le, L.W.; Sacher, A.; Shepherd, F.A.; Bradbury, P.A.; et al. The presence and variant allele fraction of EGFR mutations in ctdna and development of resistance. Lung Cancer 2019, 131, 86–89. [Google Scholar] [CrossRef]
- Romero, A.; Serna-Blasco, R.; Alfaro, C.; Sánchez-Herrero, E.; Barquín, M.; Turpin, M.C.; Chico, S.; Sanz-Moreno, S.; Rodrigez-Festa, A.; Laza-Briviesca, R.; et al. Ctdna analysis reveals different molecular patterns upon disease progression in patients treated with osimertinib. Transl. Lung Cancer Res. 2020, 9, 532–540. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR t790m-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, X.; Zhao, C.; Jiang, T.; Jia, Y.; Shi, J.; He, Y.; Li, J.; Zhou, F.; Gao, G.; et al. Loss of t790m mutation is associated with early progression to osimertinib in chinese patients with advanced NSCLC who are harboring EGFR t790m. Lung Cancer 2019, 128, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Aggarwal, C.; Davis, C.W.; Mick, R.; Thompson, J.C.; Ahmed, S.; Jeffries, S.; Bagley, S.; Gabriel, P.; Evans, T.L.; Bauml, J.M.; et al. Influence of tp53 mutation on survival in patients with advanced EGFR-mutant non-small-cell lung cancer. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef]
- Lu, H.Y.; Qin, J.; Han, N.; Lei, L.; Xie, F.; Li, C. EGFR, KRAS, BRAF, PTEN, and PIK3CA mutation in plasma of small cell lung cancer patients. OncoTargets Ther. 2018, 11, 2217–2226. [Google Scholar] [CrossRef]
- Seki, Y.; Fujiwara, Y.; Kohno, T.; Yoshida, K.; Goto, Y.; Horinouchi, H.; Kanda, S.; Nokihara, H.; Yamamoto, N.; Kuwano, K.; et al. Circulating cell-free plasma tumour DNA shows a higher incidence of EGFR mutations in patients with extrathoracic disease progression. ESMO Open 2018, 3, e000292. [Google Scholar] [CrossRef]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Jänne, P.A.; et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef]
- Rolfo, C.; Cardona, A.F.; Cristofanilli, M.; Paz-Ares, L.; Diaz Mochon, J.J.; Duran, I.; Raez, L.E.; Russo, A.; Lorente, J.A.; Malapelle, U.; et al. Challenges and opportunities of cfdna analysis implementation in clinical practice: Perspective of the international society of liquid biopsy (ISLB). Crit. Rev. Oncol. Hematol. 2020, 151, 102978. [Google Scholar] [CrossRef]
- Steendam, C.M.J.; Atmodimedjo, P.; De Jonge, E.; Paats, M.S.; Van der Leest, C.; Oomen-de Hoop, E.; Jansen, M.P.H.M.; Del Re, M.; Von der Thüsen, J.H.; Dinjens, W.N.M.; et al. Plasma cell-free DNA testing of patients with EGFR mutant non–small-cell lung cancer: Droplet digital PCR versus next-generation sequencing compared with tissue-based results. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Pruis, M.A.; Geurts-Giele, W.R.R.; von der, T.J.H.; Meijssen, I.C.; Dinjens, W.N.M.; Aerts, J.; Dingemans, A.M.C.; Lolkema, M.P.; Paats, M.S.; Dubbink, H.J. Highly accurate DNA-based detection and treatment results of met exon 14 skipping mutations in lung cancer. Lung Cancer 2020, 140, 46–54. [Google Scholar] [CrossRef]
- Veerman, G.D.M.; Lam, M.H.; Mathijssen, R.H.J.; Koolen, S.L.W.; de Bruijn, P. Quantification of afatinib, alectinib, crizotinib and osimertinib in human plasma by liquid chromatography/triple-quadrupole mass spectrometry; focusing on the stability of osimertinib. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1113, 37–44. [Google Scholar] [CrossRef]
- Braal, C.L.; Veerman, G.D.M.; Peric, R.; Aerts, J.; Mathijssen, R.H.J.; Koolen, S.L.W.; de Bruijn, P. Quantification of the tyrosine kinase inhibitor erlotinib in human scalp hair by liquid chromatography-tandem mass spectrometry: Pitfalls for clinical application. J. Pharm. Biomed. Anal. 2019, 172, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Veerman, G.D.M.; Hussaarts, K.; Peric, R.; Oomen-de Hoop, E.; Landa, K.D.; van der Leest, C.H.; Broerse, S.D.; Rutten, H.B.; Belderbos, H.N.A.; Steendam, C.M.J.; et al. Influence of cow’s milk and esomeprazole on the absorption of erlotinib: A randomized, crossover pharmacokinetic study in lung cancer patients. Clin. Pharmacokinet. 2020. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Health. Cancer Therapy Evaluation Program, Common Terminology Criteria for Adverse Events (CTCAE). Available online: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf (accessed on 1 December 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Patient Characteristics | n = 41 |
|---|---|
| Gender | |
| Male | 18 (44%) |
| Female | 23 (56%) |
| Age (median, range) | 62 (42–83) |
| Ethnicity | |
| Caucasian | 36 (88%) |
| Asian | 4 (10%) |
| Unknown | 1 (2%) |
| Smoking status | |
| Current | 2 (5%) |
| Former | 24 (58%) |
| Never | 15 (37%) |
| Former pack years (median, range) | 6 (0–40) |
| 0 | 15 (37%) |
| 1–15 | 11 (27%) |
| 15–30 | 9 (22%) |
| ≥30 | 1 (2%) |
| Unknown | 5 (12%) |
| Histology | |
| NSCLC; adenocarcinoma | 39 (95%) |
| NSCLC NOS | 1 (2.5%) |
| Unknown | 1 (2.5%) |
| Type of primary EGFR mutation | |
| Exon 19 | 28 (68%) |
| Deletion | 23 (56%) |
| Deletion-insertion | 4 (9.5%) |
| Other (VUS) | 1 (2.5%) |
| Exon 21 | 13 (32%) |
| p.L858R | 12 (29.5%) |
| Other | 1 (2.5%) |
| Exon 20 concomitant mutation | 28 (68%) |
| p.T790M | 27 (66%) |
| Exon 19 + exon 20 | 1 (2.5%) |
| Plasma available at baseline | 38 (93%) |
| Tissue available at baseline | 32 (78%) |
| Available Treatment Lines during Study | Total Cohort * (n = 41) | 1st Line Cohort (n = 19) | 2nd Line Cohort (n = 27) |
|---|---|---|---|
| Best response on treatment | |||
| PR | 25 (61%) | 13 (68%) | 15 (56%) |
| SD | 12 (29%) | 4 (21%) | 10 (37%) |
| PD | 3 (7%) | 2 (11%) | 1 (3.5%) |
| Unknown | 1 (3%) | 0 (0%) | 1 (3.5%) |
| Progression or death event | 28 (68%) | 15 (79%) | 18 (67%) |
| Radiological progression | 24 (59%) | 13 (68%) | 16 (59%) |
| Death without radiological progression | 4 (10%) | 2 (11%) | 2 (28%) |
| Progression free survival category | |||
| ≤6 months | 13 (32%) | 4 (21%) | 9 (33%) |
| >6 months | 28 (68%) | 15 (79%) | 18 (67%) |
| Baseline TP53 mutation present (n = 41) | 23 (56%) |
| Present in plasma (n = 23) | 6 (26%) |
| Present in tissue (n = 23) | 22 (96%) |
| No plasma sample baseline | 2 (9%) |
| No ctDNA baseline | 4 (17%) |
| Not covered by cfDNA panel | 6 (26%) |
| Covered by cfDNA panel | 4 (17%) |
| Location of mutation | |
| Exon 5 | 6 (26%) |
| Exon 6 | 3 (13%) |
| Exon 7 | 8 (35%) |
| Exon 8 | 2 (9%) |
| Other | 4 (17%) |
| Type of mutation | |
| Missense | 15 (65%) |
| Nonsense | 1 (5%) |
| Other | 7 (30%) |
| Variable | Event | Erlotinib | Osimertinib | ||
|---|---|---|---|---|---|
| n (Events) | Log-Rank p-Value | n (Events) | Log-Rank p-Value | ||
| Cmean of total treatment † | PFS | 15 (11) | 0.517 | 22 (13) | 0.631 |
| Cmean first six weeks † | PFS | 12 (8) | 0.197 | 20 (11) | 0.972 |
| Cmean two months before PD † | PFS | 7 (7) | 0.779 | 15 (9) | 0.221 |
| Cmean two months before PD/Cmean first six weeks ‡ | PFS | 5 (5) | 0.039* | 7 (7) | 0.561 |
| Cmean third tertile/Cmean first tertile ‡ | PFS | 8 (8) | 0.855 | 17 (10) | 0.821 |
| Cmean second tertile/Cmean first tertile ‡ | PFS | 13 (9) | 0.037* | 17 (10) | 0.169 |
| Cmean third tertile/Cmean second tertile ‡ | PFS | 9 (9) | 0.415 | 15 (8) | 0.517 |
| Cmean until severe toxicity or end of study † | Tox | 15 (10) | 0.430 | 22 (3) | 0.134 |
| Cmean third tertile/Cmean first tertile ‡ | Tox | 8 (4) | 0.433 | 17 (2) | 0.705 |
| Cmean second tertile/Cmean first tertile ‡ | Tox | 13 (8) | 0.031 * | 17 (3) | 0.460 |
| Cmean third tertile/Cmean second tertile ‡ | Tox | 9 (5) | 0.786 | 15 (2) | 0.564 |
| Erlotinib (n = 18) | |
|---|---|
| Severe toxicity | 10 (56%) |
| CTCAE grade ≥ 3 | 5 † (28%) |
| Hospital admission | 2 (11%) |
| Dose reduction | 8 (44%) |
| TKI interruption/discontinuation | 2 (11%) |
| Erlotinib specific toxicity (all grade) | |
| Rash | 12 (72%) |
| Alopecia | 8 (44%) |
| Diarrhea | 6 (33%) |
| Sicca (dry eyes, mouth and/or skin) | 6 (33%) |
| Paronychia | 5 (28%) |
| Hand-Foot Syndrome | 1 (6%) |
| Hypertrichosis | 1 (6%) |
| Osimertinib (n = 27) | |
| Severe toxicity | 4 (15%) |
| CTCAE grade ≥ 3 | 4 * (15%) |
| Hospital admission | 2 (7%) |
| Reduction | 2 (7%) |
| TKI interruption/discontinuation | 3 (11%) |
| Osimertinib specific toxicity (all grade) | |
| CK elevation | 6 (22%) |
| Paronychia | 5 (19%) |
| Diarrhea | 4 (15%) |
| Dry skin | 4 (15%) |
| QTc prolongation | 4 ‡ (17%) |
| Pneumonitis | 3 (11%) |
| Rash | 2 (7%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steendam, C.M.J.; Veerman, G.D.M.; Pruis, M.A.; Atmodimedjo, P.; Paats, M.S.; van der Leest, C.; von der Thüsen, J.H.; Yick, D.C.Y.; Oomen-de Hoop, E.; Koolen, S.L.W.; et al. Plasma Predictive Features in Treating EGFR-Mutated Non-Small Cell Lung Cancer. Cancers 2020, 12, 3179. https://doi.org/10.3390/cancers12113179
Steendam CMJ, Veerman GDM, Pruis MA, Atmodimedjo P, Paats MS, van der Leest C, von der Thüsen JH, Yick DCY, Oomen-de Hoop E, Koolen SLW, et al. Plasma Predictive Features in Treating EGFR-Mutated Non-Small Cell Lung Cancer. Cancers. 2020; 12(11):3179. https://doi.org/10.3390/cancers12113179
Chicago/Turabian StyleSteendam, Christi M. J., G. D. Marijn Veerman, Melinda A. Pruis, Peggy Atmodimedjo, Marthe S. Paats, Cor van der Leest, Jan H. von der Thüsen, David C. Y. Yick, Esther Oomen-de Hoop, Stijn L. W. Koolen, and et al. 2020. "Plasma Predictive Features in Treating EGFR-Mutated Non-Small Cell Lung Cancer" Cancers 12, no. 11: 3179. https://doi.org/10.3390/cancers12113179
APA StyleSteendam, C. M. J., Veerman, G. D. M., Pruis, M. A., Atmodimedjo, P., Paats, M. S., van der Leest, C., von der Thüsen, J. H., Yick, D. C. Y., Oomen-de Hoop, E., Koolen, S. L. W., Dinjens, W. N. M., van Schaik, R. H. N., Mathijssen, R. H. J., Aerts, J. G. J. V., Dubbink, H. J., & Dingemans, A.-M. C. (2020). Plasma Predictive Features in Treating EGFR-Mutated Non-Small Cell Lung Cancer. Cancers, 12(11), 3179. https://doi.org/10.3390/cancers12113179