RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status

 , , ,
, , ,  and
and 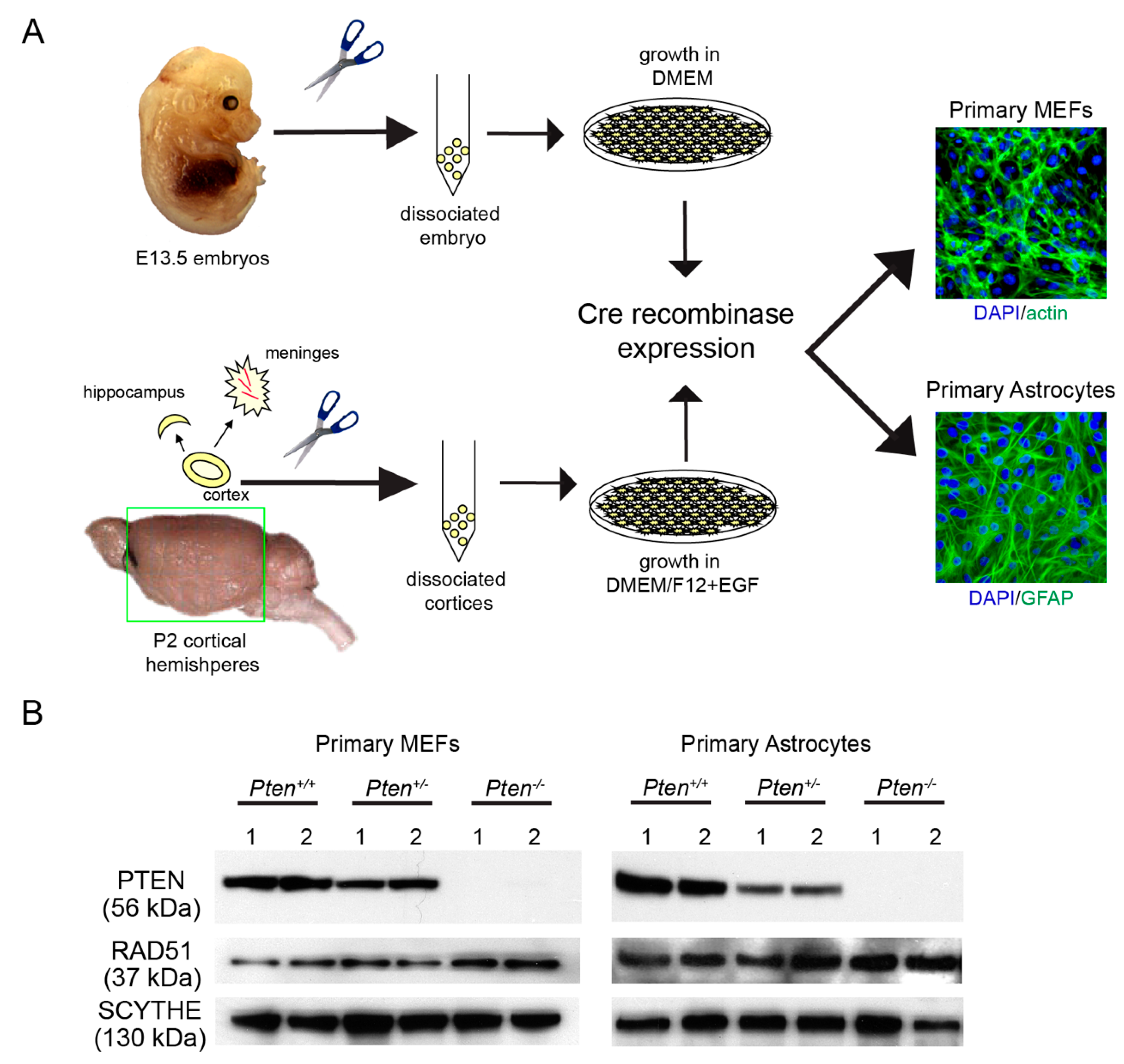{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Primary Cells from Pten-Deficient Mice
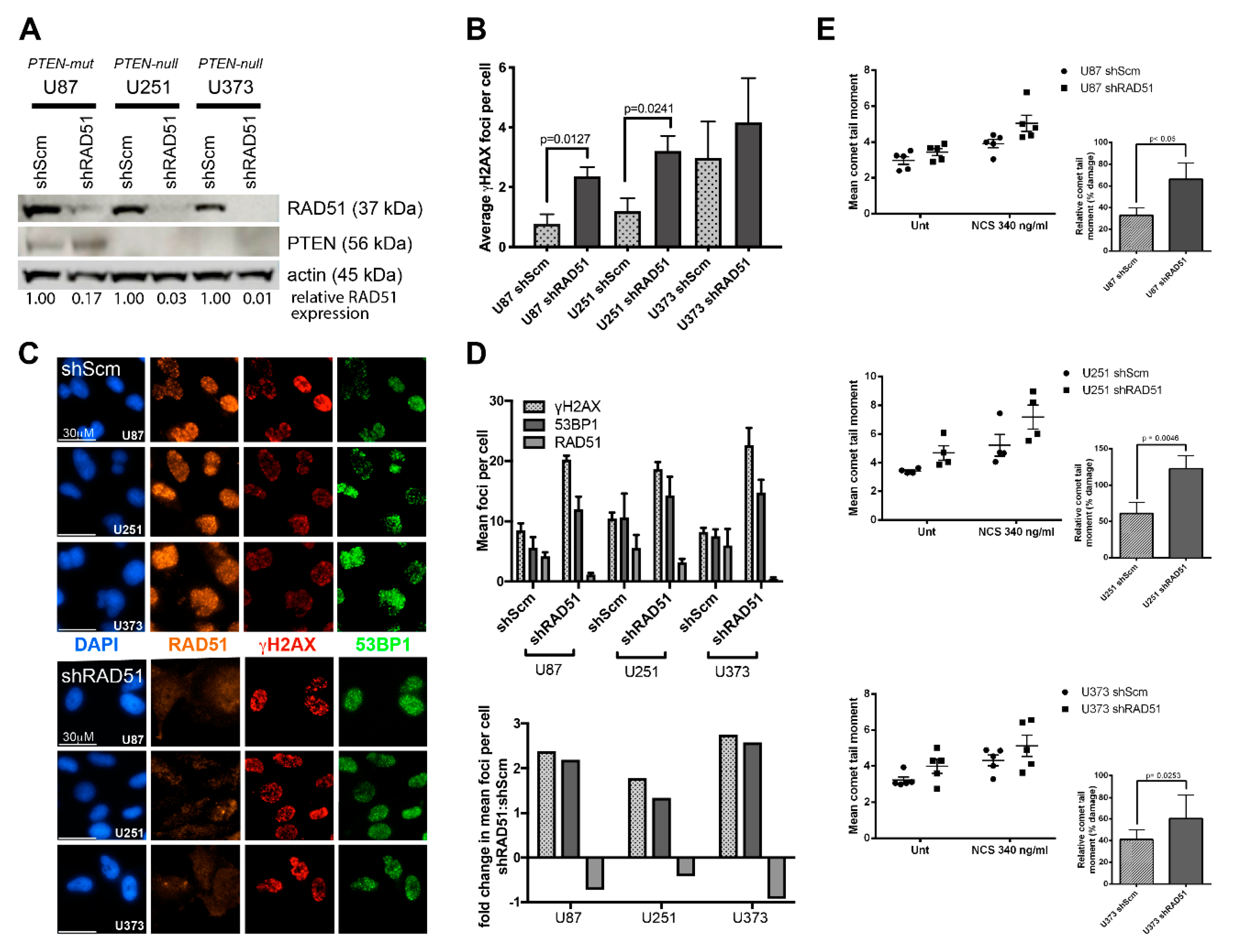
2.2. PTEN-Deficient Cancer Cell Lines and RNA Interference
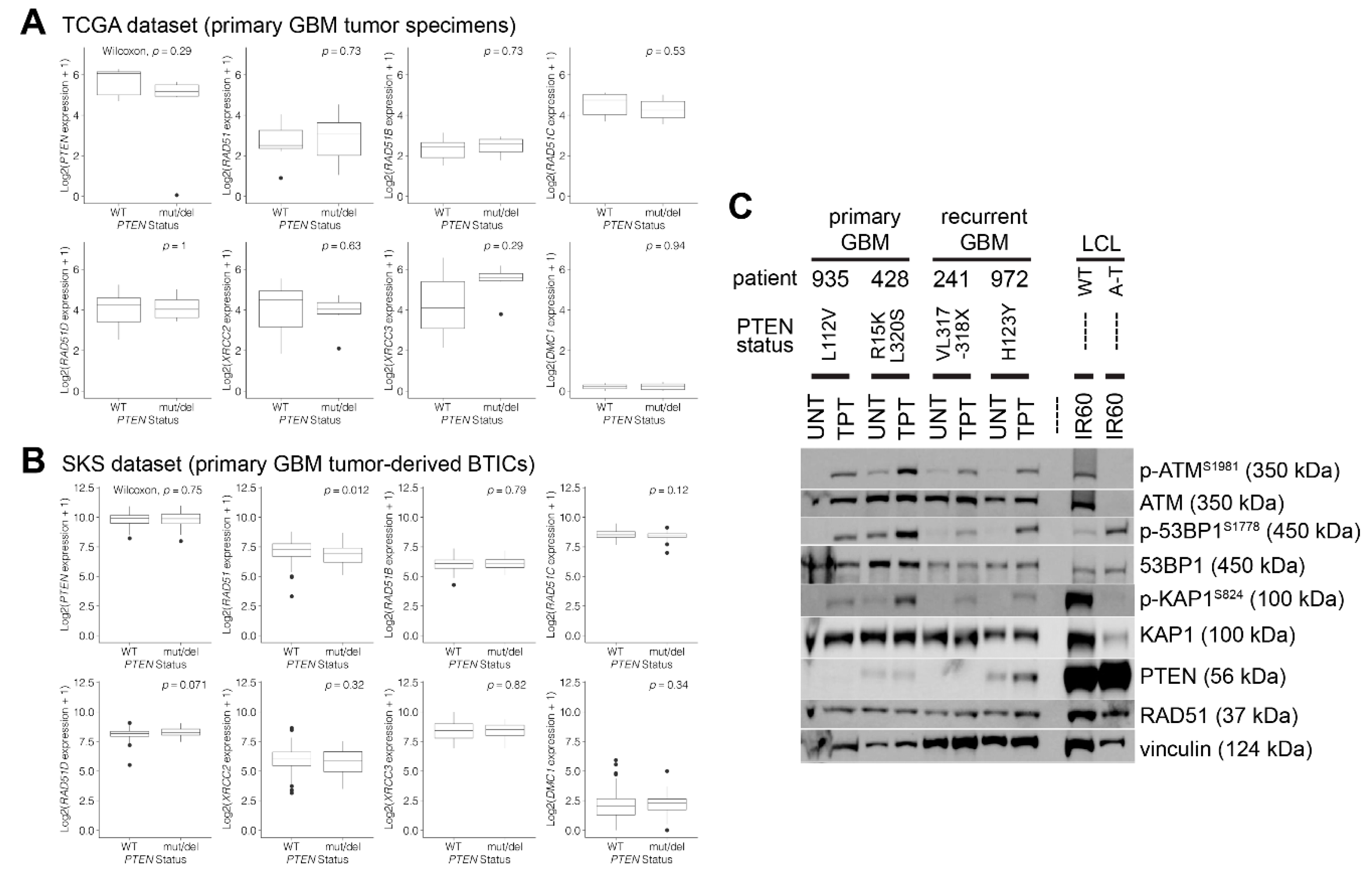
2.3. Isolation and Propagation of GBM Patient Brain Tumor Initiating Cells (BTICs)
2.4. Western Analysis
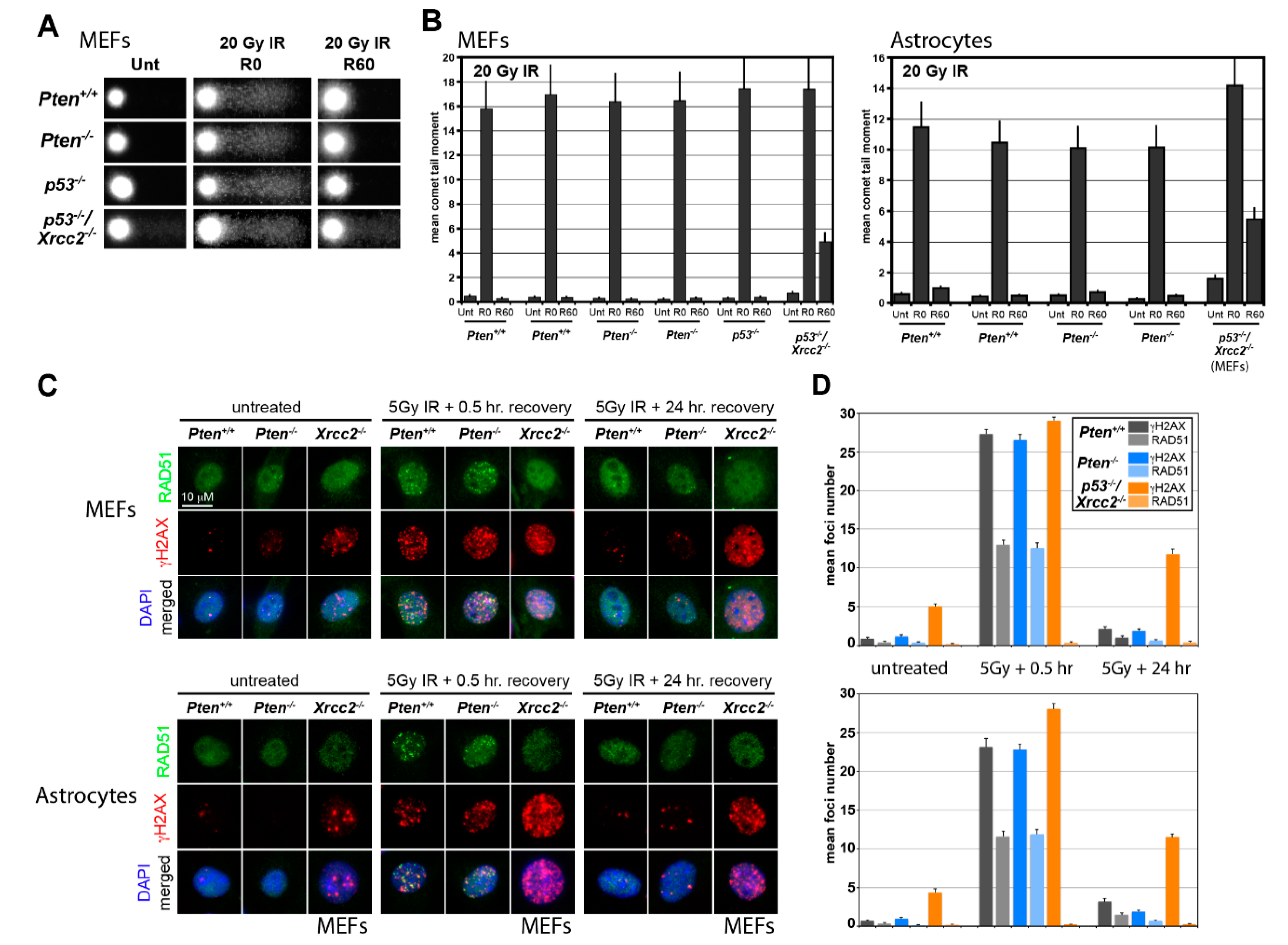
2.5. Alkali Comet Assay
2.6. Quantification of DNA Damage-Induced Nuclear Foci
2.7. Cell Viability Assays
2.8. Gene Expression Analyses According to the PTEN Status (TCGA and SKS Datasets)
3. Results
3.1. RAD51 Expression and HR Activity Are Not Impaired in PTEN-Deficient Primary Cells
3.2. PTEN-Deficient Cancer Lines Express RAD51 and Exhibit Robust DNA Repair Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a Candidate Tumour Suppressor Gene, MMAC1, At Chromosome 10q23.3 That Is Mutated in Multiple Advanced Cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Endersby, R.; Baker, S.J. PTEN Signaling in Brain: Neuropathology and Tumorigenesis. Oncogene 2008, 27, 5416–5430. [Google Scholar] [CrossRef]
- Cantley, L.C.; Neel, B.G. New Insights Into Tumor Suppression: PTEN Suppresses Tumor Formation by Restraining the Phosphoinositide 3-Kinase/AKT Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef]
- Simpson, L.; Parsons, R. PTEN: Life as a Tumor Suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef]
- Hou, S.Q.; Ouyang, M.; Brandmaier, A.; Hao, H.; Shen, W.H. PTEN in the Maintenance of Genome Integrity: From DNA Replication to Chromosome Segregation. BioEssays 2017, 39. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; McKinnon, P.J. DNA Strand Breaks, Neurodegeneration and Aging in the Brain. Mech. Ageing Dev. 2008, 129, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; McKinnon, P.J. Disconnecting XRCC1 and DNA Ligase III. Cell Cycle 2011, 10, 2269–2275. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural Basis of Homologous Recombination. Cell. Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A Mutational Signature Reveals Alterations Underlying Deficient Homologous Recombination Repair in Breast Cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C. Signatures of DNA-Repair Deficiencies in Breast Cancer. N. Engl. J. Med. 2017, 377, 2490–2492. [Google Scholar] [CrossRef]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reuterswärd, C.; Häkkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-Genome Sequencing of Triple-Negative Breast Cancers in a Population-Based Clinical Study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Morganella, S. Mutational Signatures in Breast Cancer: The Problem at the DNA Level. Clin. Cancer Res. 2017, 23, 2617–2629. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.D.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef]
- Jeggo, P.A. DNA Repair: PARP—Another Guardian Angel? Curr. Biol. 1998, 8, R49–R51. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R. Homologous Recombination Deficiency and Ovarian Cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Garje, R.; Vaddepally, R.K.; Zakharia, Y. PARP Inhibitors in Prostate and Urothelial Cancers. Front. Oncol. 2020, 10, 114. [Google Scholar] [CrossRef]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP Inhibitors in Pancreatic Cancer: Molecular Mechanisms and Clinical Applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V.; Walia, J. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef]
- O’Sullivan, C.C.; Moon, D.H.; Kohn, E.C.; Lee, J.M. Beyond Breast and Ovarian Cancers: PARP Inhibitors for BRCA Mutation-Associated and BRCA-Like Solid Tumors. Front. Oncol. 2014, 4, 42. [Google Scholar] [CrossRef]
- McEllin, B.; Camacho, C.V.; Mukherjee, B.; Hahm, B.; Tomimatsu, N.; Bachoo, R.M.; Burma, S. PTEN Loss Compromises Homologous Recombination Repair in Astrocytes: Implications for Glioblastoma Therapy with Temozolomide or Poly(ADP-Ribose) Polymerase Inhibitors. Cancer Res. 2010, 70, 5457–5464. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- Mukherjee, A.; Karmakar, P. Attenuation of PTEN Perturbs Genomic Stability via Activation of Akt and Down-Regulation of Rad51 in Human Embryonic Kidney Cells. Mol. Carcinog. 2013, 52, 611–618. [Google Scholar] [CrossRef]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic Lethal Targeting of PTEN Mutant Cells with PARP Inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN Deficiency in Endometrioid Endometrial Adenocarcinomas Predicts Sensitivity to PARP Inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.R.; Gupta, A.; Horikoshi, N.; Pandita, T.K. Does PTEN Loss Impair DNA Double-Strand Break Repair by Homologous Recombination? Clin. Cancer Res. 2011, 18, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Zhao, H.; Luoto, K.R.; Lundin, C.; Coackley, C.; Chan, N.; Joshua, A.M.; Bismar, T.A.; Evans, A.; Helleday, T.; et al. PTEN Deletion in Prostate Cancer Cells Does Not Associate with Loss of Rad51 Function: Implications for Radiotherapy and Chemotherapy. Clin. Cancer Res. 2012, 18, 1015–1027. [Google Scholar] [CrossRef]
- Gupta, A.; Yang, Q.; Pandita, R.K.; Hunt, C.R.; Xiang, T.; Misri, S.; Zeng, S.; Pagan, J.; Jeffery, J.; Puc, J.; et al. Cell Cycle Checkpoint Defects Contribute to Genomic Instability in PTEN Deficient Cells Independent of DNA DSB Repair. Cell Cycle 2009, 8, 2198–2210. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN Deficiency Sensitizes Endometrioid Endometrial Cancer to Compound PARP-PI3K Inhibition but not PARP Inhibition as Monotherapy. Oncogene 2018, 37, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; El-Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 Facilitates Chromosomal Single-Strand Break Repair in Neurons and is Neuroprotective in Vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yamaguchi, M.T.; Ohteki, T.; Sasaki, T.; Kaisho, T.; Kimura, Y.; Yoshida, R.; Wakeham, A.; Higuchi, T.; Fukumoto, M.; et al. T Cell-Specific Loss of PTEN Leads to Defects in Central and Peripheral Tolerance. Immunity 2001, 14, 523–534. [Google Scholar] [CrossRef]
- Endersby, R.; Zhu, X.; Hay, N.; Ellison, D.W.; Baker, S.J. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res. 2011, 71, 4106–4116. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; Lee, Y.; Nitiss, K.C.; Downing, S.M.; Li, Y.; Shimada, M.; Zhao, J.; Russell, H.R.; Petrini, J.H.; Nitiss, J.L.; et al. Aberrant Topoisomerase-1 DNA Lesions are Pathogenic in Neurodegenerative Genome Instability Syndromes. Nat. Neurosci. 2014, 17, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Desmots, F.; Russell, H.R.; Lee, Y.; Boyd, K.; McKinnon, P.J. The Reaper-Figure Binding Protein Scythe Modulates Apoptosis and Proliferation during Mammalian Development. Mol. Cell. Biol. 2005, 25, 10329–10337. [Google Scholar] [CrossRef] [PubMed]
- Orii, K.E.; Lee, Y.; Kondo, N.; McKinnon, P.J. Selective Utilization of Nonhomologous End-Joining and Homologous Recombination DNA Repair Pathways During Nervous System Development. Proc. Natl. Acad. Sci. USA 2006, 103, 10017–10022. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Manoranjan, B.; Chokshi, C.; Venugopal, C.; Subapanditha, M.; Savage, N.; Tatari, N.; Provias, J.P.; Murty, N.K.; Moffat, J.; Doble, B.W.; et al. A CD133-AKT-Wnt Signaling Axis Drives Glioblastoma Brain Tumor-Initiating Cells. Oncogene 2020, 39, 1590–1599. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol. Cell. Biol. 2012, 33, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Heinen, C.D.; Schmutte, C.; Fishel, R. DNA Repair and Tumorigenesis: Lessons from Hereditary Cancer Syndromes. Cancer Biol. Ther. 2002, 1, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Stark, J.M.; Araujo, F.D.; Moynahan, M.E.; Berwick, M.; Jasin, M. Double-Strand Breaks and Tumorigenesis. Trends Cell Biol. 2001, 11, S52–S59. [Google Scholar] [CrossRef]
- Furnari, F.B.; Lin, H.; Huang, H.S.; Cavenee, W.K. Growth Suppression of Glioma Cells by PTEN Requires a Functional Phosphatase Catalytic Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Motaln, H.; Koren, A.; Gruden, K.; Ramšak, Ž.; Schichor, C.; Lah, T.T. Heterogeneous Glioblastoma Cell Cross-Talk Promotes Phenotype Alterations and Enhanced Drug Resistance. Oncotarget 2015, 6, 40998–41017. [Google Scholar] [CrossRef]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the Human PTEN Gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Nijman, S.M. Synthetic Lethality: General Principles, Utility and Detection Using Genetic Screens in Human Cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.J.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Stordal, B.; Timms, K.; Farrelly, A.; Gallagher, D.; Busschots, S.; Renaud, M.; Thery, J.; Williams, D.; Potter, J.; Tran, T.; et al. Brca1/2 Mutation Analysis In 41 Ovarian Cell Lines Reveals Only One Functionally Deleterious Brca1 Mutation. Mol. Oncol. 2013, 7, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Yap, T.A.; De Bono, J.S. The Emerging Role of Poly(ADP-Ribose) Polymerase Inhibitors in Cancer Treatment. Curr. Drug Targets 2011, 12, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Rabenau, K.; Hofstatter, E. DNA Damage Repair and the Emerging Role of Poly(ADP-ribose) Polymerase Inhibition in Cancer Therapeutics. Clin. Ther. 2016, 38, 1577–1588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Min, A.; Im, S.-A.; Yoon, Y.-K.; Song, S.-H.; Nam, H.-J.; Hur, H.-S.; Kim, H.-P.; Lee, K.-H.; Han, S.-W.; Oh, D.-Y.; et al. Rad51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib. Mol. Cancer Ther. 2013, 12, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. New Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.I.; Butterworth, K.T.; Chen, C.; Harkin, D.P.; et al. Mechanistic Rationale to Target PTEN-Deficient Tumor Cells with Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res. 2015, 75, 2159–2165. [Google Scholar] [CrossRef]
- Cerrato, A.; Morra, F.; Celetti, A. Use of Poly ADP-Ribose Polymerase [PARP] Inhibitors in Cancer Cells Bearing DDR Defects: The Rationale for their Inclusion in the Clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Guan, J.; Zhang, Z.; Lv, J.; Wang, Y.; Liu, L.; Zhou, Q.; Mao, W. Inhibition of Rad51 Sensitizes Breast Cancer Cells with Wild-Type PTEN to Olaparib. Biomed. Pharmacother. 2017, 94, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss Of PTEN-Assisted G2/M Checkpoint Impedes Homologous Recombination Repair and Enhances Radio-Curability and Parp Inhibitor Treatment Response in Prostate Cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Liang, F.; Jia, Z.L.; Song, S.T.; Jiang, Z.F. PTEN Mutation, Methylation and Expression in Breast Cancer Patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef]
- Russell, J.S.; Brady, K.; Burgan, W.E.; Cerra, M.A.; Oswald, K.A.; Camphausen, K.; Tofilon, P.J. Gleevec-Mediated Inhibition of Rad51 Expression and Enhancement of Tumor Cell Radiosensitivity. Cancer Res. 2003, 63, 7377–7383. [Google Scholar]
- Miyasaka, A.; Oda, K.; Ikeda, Y.; Wada-Hiraike, O.; Kashiyama, T.; Enomoto, A.; Hosoya, N.; Koso, T.; Fukuda, T.; Inaba, K.; et al. Anti-Tumor Activity of Olaparib, a Poly(ADP-ribose) Polymerase (PARP) Inhibitor, in Cultured Endometrial Carcinoma Cells. BMC Cancer 2014, 14, 179. [Google Scholar] [CrossRef]
- Turchick, A.; Liu, Y.; Zhao, W.; Cohen, I.; Glazer, P.M. Synthetic Lethality of a Cell-Penetrating Anti-Rad51 Antibody in PTEN-Deficient Melanoma and Glioma Cells. Oncotarget 2019, 10, 1272–1283. [Google Scholar] [CrossRef]
- Sun, H.; Lesche, R.; Li, D.-M.; Liliental, J.; Zhang, H.; Gao, J.; Gavrilova, N.; Mueller, B.; Liu, X.; Wu, H. PTEN Modulates Cell Cycle Progression and Cell Survival by Regulating Phosphatidylinositol 3,4,5,-Trisphosphate and Akt/Protein Kinase B Signaling Pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 6199–6204. [Google Scholar] [CrossRef]
- Lesche, R.; Groszer, M.; Gao, J.; Wang, Y.; Messing, A.; Sun, H.; Liu, X.; Wu, H. Cre/loxP-Mediated Inactivation of the Murine PTEN Tumor Suppressor Gene. Genesis 2002, 32, 148–149. [Google Scholar] [CrossRef]
- Camacho, C.V.; Todorova, P.K.; Hardebeck, M.C.; Tomimatsu, N.; Gil Del Alcazar, C.R.; Ilcheva, M.; Mukherjee, B.; McEllin, B.; Vemireddy, V.; Hatanpaa, K.; et al. DNA Double-Strand Breaks Cooperate with Loss of Ink4 and Arf Tumor Suppressors to Generate Glioblastomas with Frequent Met Amplification. Oncogene 2015, 34, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Ho, S.-C.; Tan, E.; Ng, A.W.T.; McPherson, J.R.; Goh, G.Y.L.; Teh, B.T.; Bard, F.; Rozen, S.G. Functional Genomics Identifies Specific Vulnerabilities in PTEN-Deficient Breast Cancer. Breast Cancer Res. 2018, 20, 22. [Google Scholar] [CrossRef]
- Jividen, K.; Kedzierska, K.Z.; Yang, C.-S.; Szlachta, K.; Ratan, A.; Paschal, B.M. Genomic Analysis of DNA Repair Genes and Androgen Signaling in Prostate Cancer. BMC Cancer 2018, 18, 960. [Google Scholar] [CrossRef] [PubMed]
- Gaymes, T.J.; Mohamedali, A.M.; Patterson, M.; Matto, N.; Smith, A.; Kulasekararaj, A.; Chelliah, R.; Curtin, N.J.; Farzaneh, F.; Shall, S.; et al. Microsatellite Instability Induced Mutations in DNA Repair Genes CtIP and MRE11 Confer Hypersensitivity to Poly(ADP-ribose) Polymerase Inhibitors in Myeloid Malignancies. Haematologica 2013, 98, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Aguado, M.; Fallik, D.; Praz, F. The Role of the DNA Mismatch Repair System in the Cytotoxicity of the Topoisomerase Inhibitors Camptothecin and Etoposide to Human Colorectal Cancer Cells. Cancer Res. 2001, 61, 6555–6562. [Google Scholar] [PubMed]
- Wu, M.; Wang, X.; McGregor, N.; Pienta, K.J.; Zhang, J. Dynamic Regulation of Rad51 by E2F1 and P53 in Prostate Cancer Cells upon Drug-Induced DNA Damage under Hypoxia. Mol. Pharmacol. 2014, 85, 866–876. [Google Scholar] [CrossRef]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; De Albuquerque, C.P.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 816. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The Poly(ADP-ribose) Polymerase Inhibitor Niraparib (MK4827) in BRCA Mutation Carriers and Patients with Sporadic Cancer: A Phase 1 Dose-Escalation Trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Wang, J. Complete Pathological Remission after Treatment with Olaparib in a Patient with PTEN-Deficient Sarcomatoid Prostate Cancer. J. Mol. Cancer. 2018, 1, 17–19. [Google Scholar]
- Gupta, M.; Iyer, R.; Fountzilas, C. Poly(ADP-Ribose) Polymerase Inhibitors in Pancreatic Cancer: A New Treatment Paradigms and Future Implications. Cancers 2019, 11, 1980. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, A.; Saleh, A.; Endersby, R.; Yuan, S.H.; Chokshi, C.R.; Brown, K.R.; Kuzio, B.; Kauppinen, T.; Singh, S.K.; Baker, S.J.; et al. RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status. Cancers 2020, 12, 3178. https://doi.org/10.3390/cancers12113178
Sinha A, Saleh A, Endersby R, Yuan SH, Chokshi CR, Brown KR, Kuzio B, Kauppinen T, Singh SK, Baker SJ, et al. RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status. Cancers. 2020; 12(11):3178. https://doi.org/10.3390/cancers12113178
Chicago/Turabian StyleSinha, Asha, Ali Saleh, Raelene Endersby, Shek H. Yuan, Chirayu R. Chokshi, Kevin R. Brown, Bozena Kuzio, Tiina Kauppinen, Sheila K. Singh, Suzanne J. Baker, and et al. 2020. "RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status" Cancers 12, no. 11: 3178. https://doi.org/10.3390/cancers12113178
APA StyleSinha, A., Saleh, A., Endersby, R., Yuan, S. H., Chokshi, C. R., Brown, K. R., Kuzio, B., Kauppinen, T., Singh, S. K., Baker, S. J., McKinnon, P. J., & Katyal, S. (2020). RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status. Cancers, 12(11), 3178. https://doi.org/10.3390/cancers12113178