Innate Lymphoid Cells in the Malignant Melanoma Microenvironment

 , , and
, , and
Simple Summary
Abstract
1. Introduction
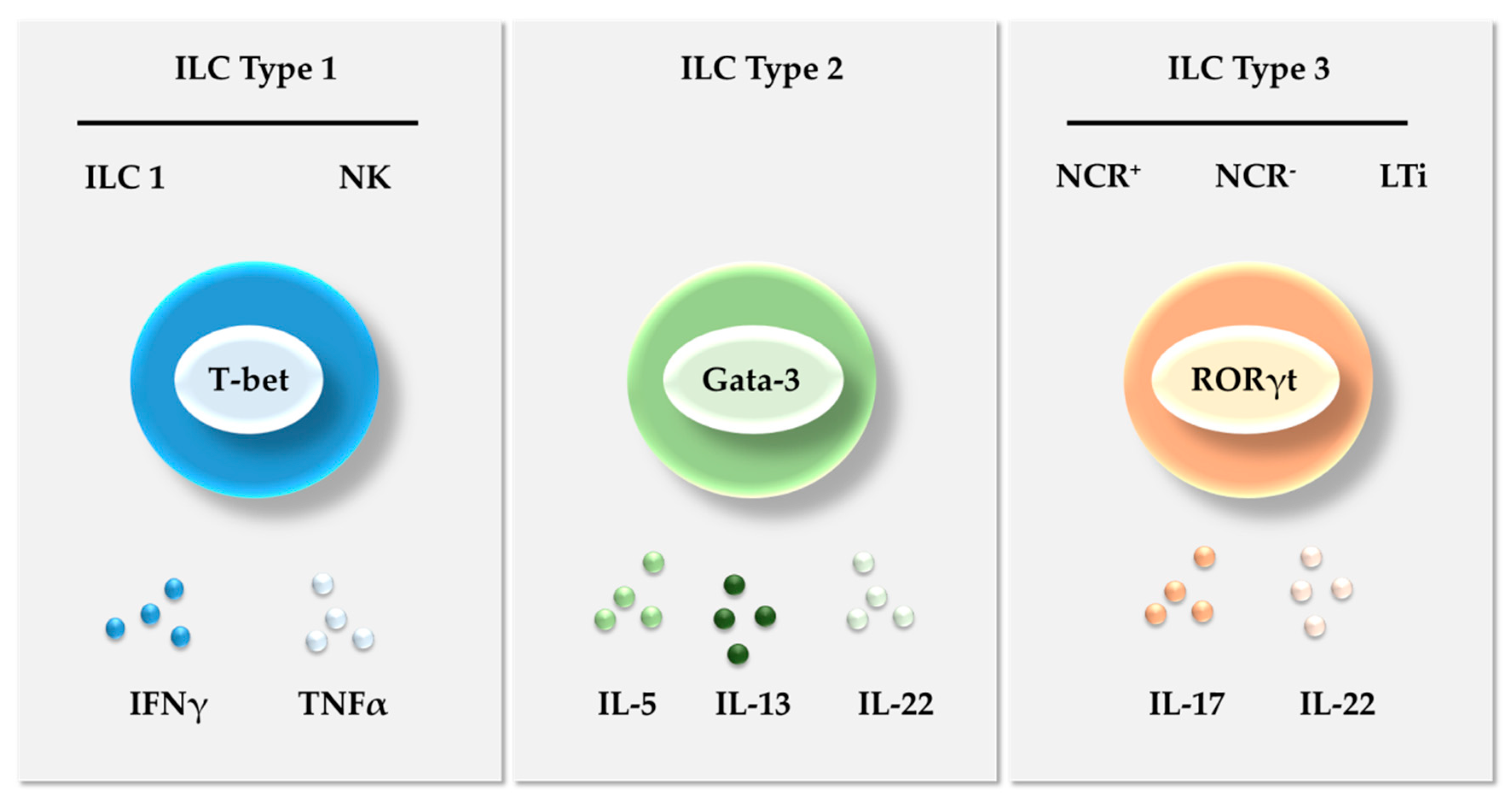
2. Innate Lymphoid Cells
3. ILCs in the Skin, Liver, and Lung
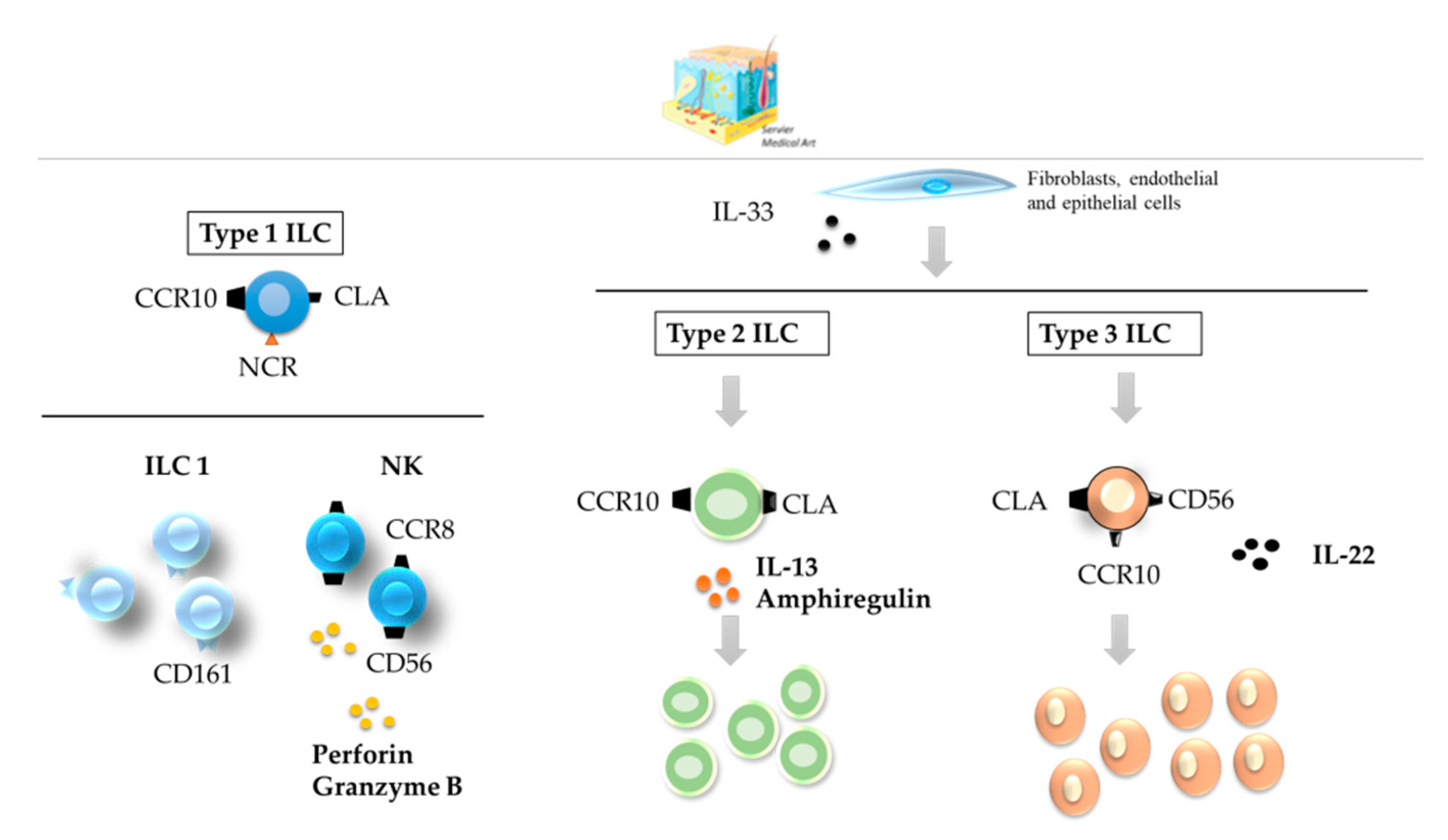
3.1. Skin
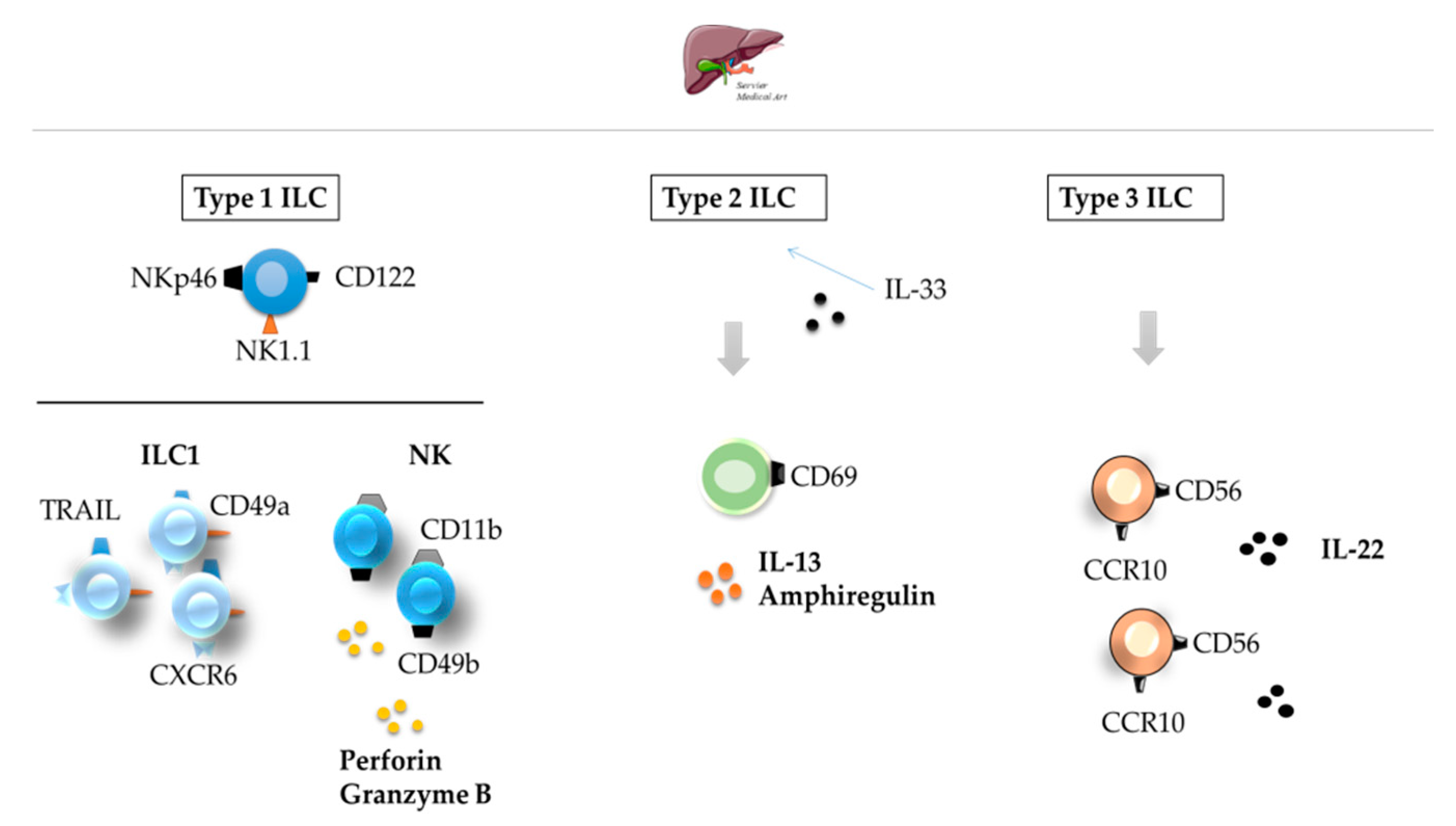
3.2. Liver
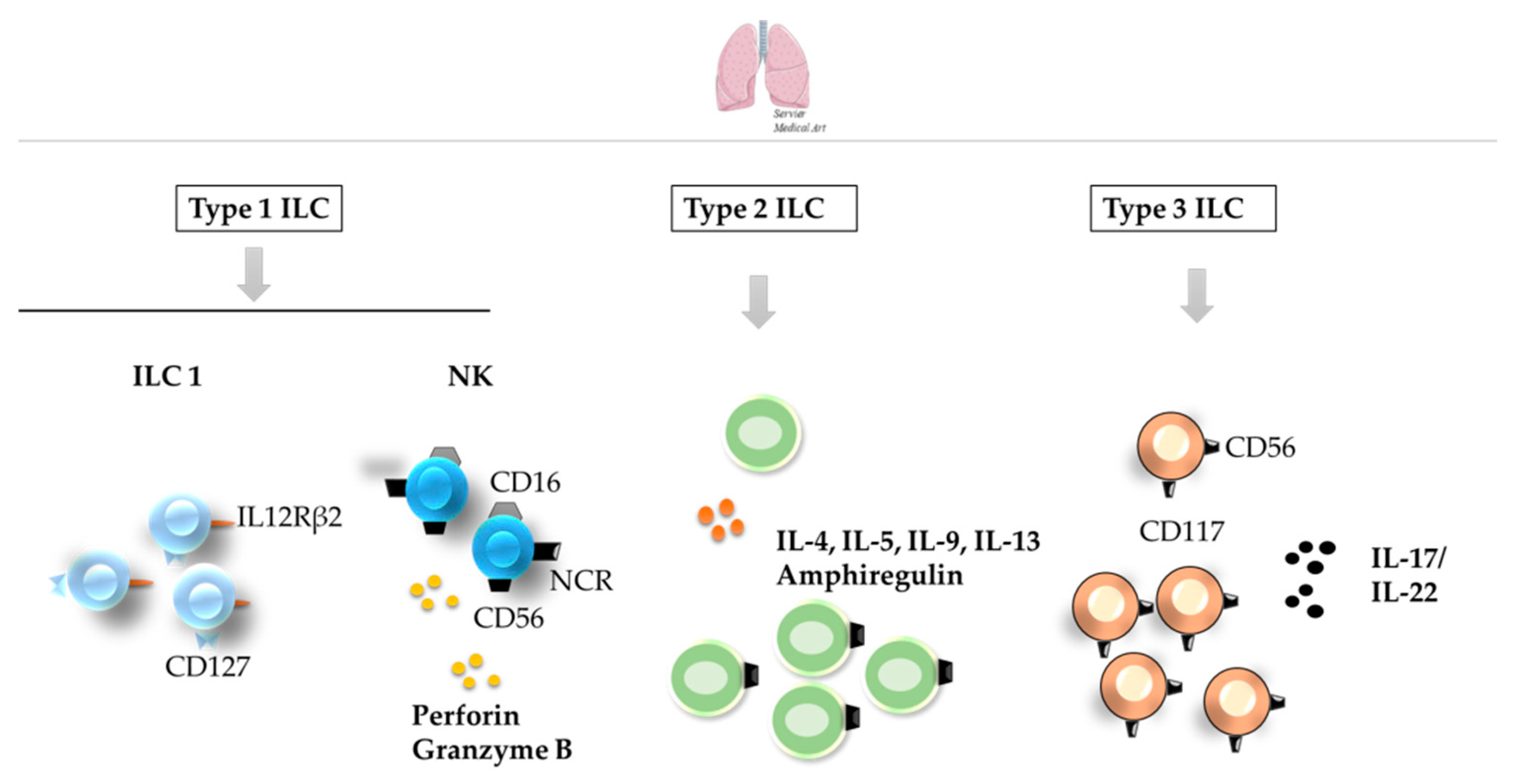
3.3. Lungs
4. The Function of ILCs in Melanoma
4.1. ILCs in Primary Cutaneous Melanoma
ILCs and the Stromal Compartment in the Primary Melanoma
4.2. ILCs in the Metastatic Progression of Cutaneous Melanoma
4.2.1. ILCs and Inflammation during Metastatic Progression in the Liver
4.2.2. ILCs and Inflammation during Metastatic Progression to Lungs
4.2.3. ILCs and the Stromal Compartment in the Metastatic Microenvironment
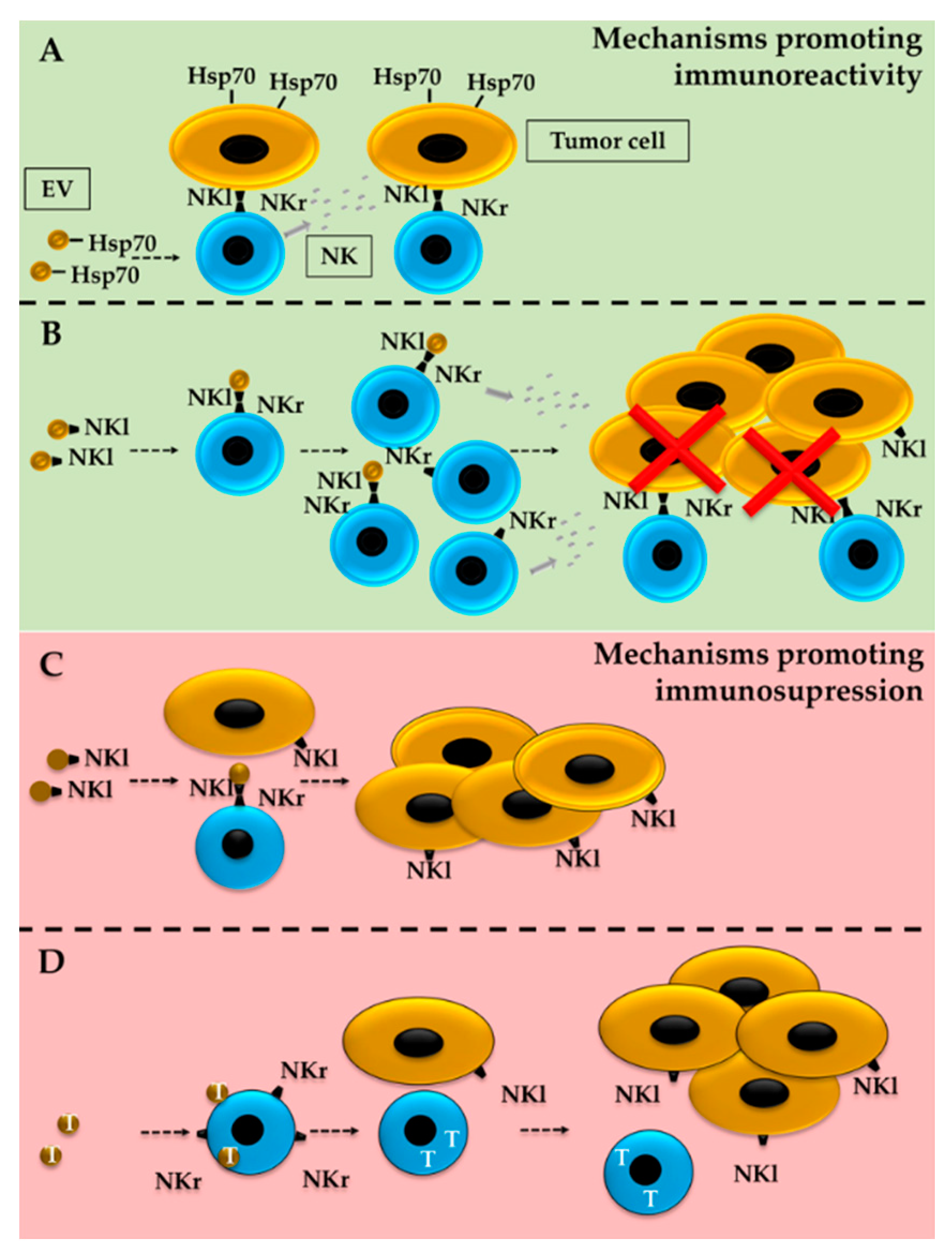
5. ILCs and EVs in the TME
5.1. Effect of Tumor-Derived EVs on ILCs
5.2. Tumor Modulation by ILC-Derived EVs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ultraviolet (UV) Radiation and Skin Cancer. Available online: https://www.who.int/news-room/q-a-detail/ultraviolet-(uv)-radiation-and-skin-cancer (accessed on 15 October 2020).
- Matthews, N.H.; Li, W.-Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of melanoma. Cutan. Melanoma Etiol. 2017, 65, 3–22. [Google Scholar] [CrossRef]
- Tas, F. Metastatic behavior in melanoma: Timing, pattern, survival and influencing factors. J. Oncol. 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of melanoma: Facts and hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current advances in the treatment of BRAF-mutant melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharm. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Kakavand, H.; Rawson, R.V.; Pupo, G.M.; Yang, J.Y.H.; Menzies, A.M.; Carlino, M.S.; Kefford, R.F.; Howle, J.R.; Saw, R.P.M.; Thompson, J.F.; et al. PD-L1 expression and immune escape in melanoma resistance to MAPK inhibitors. Clin. Cancer Res. 2017, 23, 6054–6061. [Google Scholar] [CrossRef]
- Tallerico, R.; Cristiani, C.M.; Staaf, E.; Garofalo, C.; Sottile, R.; Capone, M.; Pico de Coaña, Y.; Madonna, G.; Palella, E.; Wolodarski, M.; et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Gurzu, S.; Beleaua, M.A.; Jung, I. The role of tumor microenvironment in development and progression of malignant melanomas—A systematic review. Rom. J. Morphol. Embryol. 2018, 59, 23–28. [Google Scholar]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Briseño, C.G.; Murphy, T.L.; Murphy, K.M. Complementary diversification of dendritic cells and innate lymphoid cells. Curr. Opin. Immunol. 2014, 29, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Crome, S.Q.; Nguyen, L.T.; Lopez-Verges, S.; Yang, S.Y.C.; Martin, B.; Yam, J.Y.; Johnson, D.J.; Nie, J.; Pniak, M.; Yen, P.H.; et al. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat. Med. 2017, 23, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 Receptor-mediated interferon-γ production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity 2018, 48, 107–119.e4. [Google Scholar] [CrossRef]
- Richards, C.D. Innate immune cytokines, fibroblast phenotypes, and regulation of extracellular matrix in lung. J. Interf. Cytokine Res. 2017, 37, 52–61. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Knolle, P.A.; Wohlleber, D. Immunological functions of liver sinusoidal endothelial cells. Cell. Mol. Immunol. 2016, 13, 347–353. [Google Scholar] [CrossRef]
- Constantinides, M.G.; McDonald, B.D.; Verhoef, P.A.; Bendelac, A. A committed precursor to innate lymphoid cells. Nature 2014, 508, 397–401. [Google Scholar] [CrossRef]
- Yu, Y.; Tsang, J.C.H.; Wang, C.; Clare, S.; Wang, J.; Chen, X.; Brandt, C.; Kane, L.; Campos, L.S.; Lu, L.; et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature 2016, 539, 102–106. [Google Scholar] [CrossRef]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells-a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Sungur, C.M.; Murphy, W.J. Positive and negative regulation by NK cells in cancer. Crit. Rev. Oncog. 2014, 19, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N. Role of natural killer cells in control of cancer metastasis. Cancer Metastasis Rev. 1982, 1, 45–64. [Google Scholar] [CrossRef]
- Pikovskaya, O.; Chaix, J.; Rothman, N.J.; Collins, A.; Chen, Y.-H.; Scipioni, A.M.; Vivier, E.; Reiner, S.L. Cutting edge: Eomesodermin is sufficient to direct type 1 innate lymphocyte development into the conventional NK lineage. J. Immunol. 2016, 196, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Marotel, M.; Fauteux-Daniel, S.; Mathieu, A.-L.L.; Viel, S.; Marçais, A.; Walzer, T. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur. J. Immunol. 2018, 48, 738–750. [Google Scholar] [CrossRef]
- Wang, X.; Peng, H.; Cong, J.; Wang, X.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. Memory formation and long-term maintenance of IL-7Rα+ ILC1s via a lymph node-liver axis. Nat. Commun. 2018, 9, 4854. [Google Scholar] [CrossRef]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORα is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef]
- Hoyler, T.; Klose, C.S.N.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef]
- Kotas, M.E.; Locksley, R.M. Why innate lymphoid cells? Immunity 2018, 48, 1081–1090. [Google Scholar] [CrossRef]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 2018, 48, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Gronke, K.; Kofoed-Nielsen, M.; Diefenbach, A. Innate lymphoid cells, precursors and plasticity. Immunol. Lett. 2016, 179, 9–18. [Google Scholar] [CrossRef]
- Vonarbourg, C.; Mortha, A.; Bui, V.L.; Hernandez, P.P.; Kiss, E.A.; Hoyler, T.; Flach, M.; Bengsch, B.; Thimme, R.; Hölscher, C.; et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt+ innate lymphocytes. Immunity 2010, 33, 736–751. [Google Scholar] [CrossRef]
- Ohne, Y.; Silver, J.S.; Thompson-Snipes, L.; Collet, M.A.; Blanck, J.P.; Cantarel, B.L.; Copenhaver, A.M.; Humbles, A.A.; Liu, Y.-J. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat. Immunol. 2016, 17, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Newell, E.W. Dissecting human ILC heterogeneity: More than just three subsets. Immunology 2018, 153, 297–303. [Google Scholar] [CrossRef]
- Williamson, T.; Sultanpuram, N.; Sendi, H. The role of liver microenvironment in hepatic metastasis. Clin. Transl. Med. 2019, 8, 21. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Yang, J.; Hu, S.; Zhao, L.; Kaplan, D.H.; Perdew, G.H.; Xiong, N. Selective programming of CCR10+ innate lymphoid cells in skin-draining lymph nodes for cutaneous homeostatic regulation. Nat. Immunol. 2016, 17, 48–56. [Google Scholar] [CrossRef]
- Kim, B.S. Innate Lymphoid Cells in the Skin. J. Invest. Derm. 2015, 135, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Ebert, L.M.; Meuter, S.; Moser, B. Homing and function of human skin γδ T cells and NK Cells: Relevance for tumor surveillance. J. Immunol. 2006, 176, 4331–4336. [Google Scholar] [CrossRef]
- Tsuchiyama, J.; Yoshino, T.; Toba, K.; Harada, N.; Nishiuchi, R.; Akagi, T.; Furukawa, T.; Takahashi, M.; Fuse, I.; Aizawa, Y.; et al. Induction and characterization of cutaneous lymphocyte antigen on natural killer cells. Br. J. Haematol. 2002, 118, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Ogg, G. Innate lymphoid cells and the skin. BMC Derm. 2014, 14, 18. [Google Scholar] [CrossRef]
- Teunissen, M.B.M.; Munneke, J.M.; Bernink, J.H.; Spuls, P.I.; Res, P.C.M.; Te Velde, A.; Cheuk, S.; Brouwer, M.W.D.; Menting, S.P.; Eidsmo, L.; et al. Composition of innate lymphoid cell subsets in the human skin: Enrichment of NCR + ILC3 in lesional skin and blood of psoriasis patients. J. Investig. Derm. 2014, 134, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Villanova, F.; Flutter, B.; Tosi, I.; Grys, K.; Sreeneebus, H.; Perera, G.K.; Chapman, A.; Smith, C.H.; Di Meglio, P.; Nestle, F.O. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J. Investig. Derm. 2014, 134, 984–991. [Google Scholar] [CrossRef]
- Dyring-Andersen, B.; Geisler, C.; Agerbeck, C.; Lauritsen, J.P.H.P.H.; Gúdjonsdottir, S.D.D.; Skov, L.; Bonefeld, C.M.M. Increased number and frequency of group 3 innate lymphoid cells in nonlesional psoriatic skin. Br. J. Derm. 2014, 170, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Karakawa, M.; Komine, M.; Hanakawa, Y.; Tsuda, H.; Sayama, K.; Tamaki, K.; Ohtsuki, M. CCL27 is downregulated by interferon gamma via epidermal growth factor receptor in normal human epidermal keratinocytes. J. Cell. Physiol. 2014, 229, 1935–1945. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, C. The role of innate lymphoid cells in immune-mediated liver diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Seillet, C.; Belz, G.T.; Huntington, N.D. Development, homeostasis and heterogeneity of NK cells and ILC1. In Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2015; Volume 395, pp. 37–61. [Google Scholar]
- Peng, H.; Tian, Z. Re-examining the origin and function of liver-resident NK cells. Trends Immunol. 2015, 36, 293–299. [Google Scholar] [CrossRef]
- Shen, Y.; Li, J.; Wang, S.-Q.; Jiang, W. Ambiguous roles of innate lymphoid cells in chronic development of liver diseases. World J. Gastroenterol. 2018, 24, 1962–1977. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Jiang, X.; Chen, Y.; Sojka, D.K.; Wei, H.; Gao, X.; Sun, R.; Yokoyama, W.M.; Tian, Z. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J. Clin. Investig. 2013, 123, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cretney, E.; Hayakawa, Y.; Ota, T.; Akiba, H.; Ogasawara, K.; Yagita, H.; Kinoshita, K.; Okumura, K.; Smyth, M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 2005, 105, 2082–2089. [Google Scholar] [CrossRef]
- Krueger, P.D.; Narayanan, S.; Surette, F.A.; Brown, M.G.; Sung, S.-S.J.; Hahn, Y.S. Murine liver-resident group 1 innate lymphoid cells regulate optimal priming of anti-viral CD8 + T cells. J. Leukoc. Biol. 2017, 101, 329–338. [Google Scholar] [CrossRef]
- Matsumoto, A.; Kanai, T.; Mikami, Y.; Chu, P.-S.; Nakamoto, N.; Ebinuma, H.; Saito, H.; Sato, T.; Yagita, H.; Hibi, T. IL-22-producing RORγt-dependent innate lymphoid cells play a novel protective role in murine acute hepatitis. PLoS ONE 2013, 8, e62853. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.-M.; Shu, Q.; Fan, J. The origin and role of innate lymphoid cells in the lung. Mil. Med. Res. 2016, 3, 25. [Google Scholar] [CrossRef]
- Barlow, J.L.; McKenzie, A.N.J. Innate Lymphoid Cells of the Lung. Annu. Rev. Physiol. 2019, 81, 429–452. [Google Scholar] [CrossRef]
- De Grove, K.C.; Provoost, S.; Verhamme, F.M.; Bracke, K.R.; Joos, G.F.; Maes, T.; Brusselle, G.G. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS ONE 2016, 11, e0145961. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Marquardt, N.; Kekäläinen, E.; Chen, P.; Kvedaraite, E.; Wilson, J.N.; Ivarsson, M.A.; Mjösberg, J.; Berglin, L.; Säfholm, J.; Manson, M.L.; et al. Human lung natural killer cells are predominantly comprised of highly differentiated hypofunctional CD69−CD56dim cells. J. Allergy Clin. Immunol. 2017, 139, 1321–1330.e4. [Google Scholar] [CrossRef]
- Dutton, E.E.; Camelo, A.; Sleeman, M.; Herbst, R.; Carlesso, G.; Belz, G.T.; Withers, D.R. Characterisation of innate lymphoid cell populations at different sites in mice with defective T cell immunity. Wellcome Open Res. 2017, 2, 117. [Google Scholar] [CrossRef]
- Weizman, O.-E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E.; et al. ILC1 confer early host protection at initial sites of viral infection. Cell 2017, 171, 795–808.e12. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.Y.; Kita, H. Group 2 innate lymphoid cells in the lung. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2014; pp. 1–16. [Google Scholar]
- Mohapatra, A.; Van Dyken, S.J.; Schneider, C.; Nussbaum, J.C.; Liang, H.E.; Locksley, R.M. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol. 2016, 9, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Borger, J.G.; Lau, M.; Hibbs, M.L. The influence of innate lymphoid cells and unconventional T Cells in chronic inflammatory lung disease. Front. Immunol. 2019, 10, 1597. [Google Scholar] [CrossRef] [PubMed]
- Ardain, A.; Porterfield, J.Z.; Kløverpris, H.N.; Leslie, A. Type 3 ILCs in lung disease. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Ardain, A.; Domingo-Gonzalez, R.; Das, S.; Kazer, S.W.; Howard, N.C.; Singh, A.; Ahmed, M.; Nhamoyebonde, S.; Rangel-Moreno, J.; Ogongo, P.; et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 2019, 570, 528–532. [Google Scholar] [CrossRef]
- Almeida, F.F.; Tognarelli, S.; Marçais, A.; Kueh, A.J.; Friede, M.E.; Liao, Y.; Willis, S.N.; Luong, K.; Faure, F.; Mercier, F.E.; et al. A point mutation in the Ncr1 signal peptide impairs the development of innate lymphoid cell subsets. Oncoimmunology 2018, 7, e1475875. [Google Scholar] [CrossRef]
- Tarazona, R.; Duran, E.; Solana, R. Natural killer cell recognition of melanoma: New clues for a more effective immunotherapy. Front. Immunol. 2016, 6. [Google Scholar] [CrossRef]
- Pietra, G.; Manzini, C.; Vitale, M.; Balsamo, M.; Ognio, E.; Boitano, M.; Queirolo, P.; Moretta, L.; Mingari, M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009, 21, 793–801. [Google Scholar] [CrossRef]
- Cristiani, C.M.; Turdo, A.; Ventura, V.; Apuzzo, T.; Capone, M.; Madonna, G.; Mallardo, D.; Garofalo, C.; Giovannone, E.D.; Grimaldi, A.M.; et al. Accumulation of circulating CCR7þ natural killer cells marks melanoma evolution and reveals a CCL19-dependent metastatic pathway. Cancer Immunol. Res. 2019, 7, 841–852. [Google Scholar] [CrossRef]
- Rosinsky, C.; Antony, P.A. A role for pre-mNK cells in tumor progression. J. Immunother. Cancer 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Takeda, K.; Kawano, M.; Takai, T.; Ishii, N.; Ogasawara, K. Natural killer (NK)-dendritic cell interactions generate MHC class II-dressed NK cells that regulate CD4+ T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 18360–18365. [Google Scholar] [CrossRef] [PubMed]
- Terme, M.; Mignot, G.; Ullrich, E.; Bonmort, M.; Minard-Colin, V.; Jacquet, A.; Schultze, J.L.; Kroemer, G.; Leclerc, C.; Chaput, N.; et al. The dendritic cell-like functions of IFN-producing killer dendritic cells reside in the CD11b+ subset and are licensed by tumor cells. Cancer Res. 2009, 69, 6590–6597. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.A.; Goding, S.R.; Neely, H.R.; Harris, K.M.; Antony, P.A. Depletion of B220 + NK1.1 + cells enhances the rejection of established melanoma by tumor-specific CD4 + T cells. Oncoimmunology 2015, 4, e1019196. [Google Scholar] [CrossRef] [PubMed]
- Tietze, J.K.; Angelova, D.; Heppt, M.V.; Ruzicka, T.; Berking, C. Low baseline levels of NK cells may predict a positive response to ipilimumab in melanoma therapy. Exp. Derm. 2017, 26, 622–629. [Google Scholar] [CrossRef]
- De Jonge, K.; Ebering, A.; Nassiri, S.; Maby-El Hajjami, H.; Ouertatani-Sakouhi, H.; Baumgaertner, P.; Speiser, D.E. Circulating CD56 bright NK cells inversely correlate with survival of melanoma patients. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Huarte, E.; Rynda-Apple, A.; Riccardi, C.; Skyberg, J.A.; Golden, S.; Rollins, M.C.F.; Ramstead, A.G.; Jackiw, L.O.; Maddaloni, M.; Pascual, D.W. Tolerogen-induced interferon-producing killer dendritic cells (IKDCs) protect against EAE. J. Autoimmun. 2011, 37, 328–341. [Google Scholar] [CrossRef]
- Lee, H.; Quek, C.; Silva, I.; Tasker, A.; Batten, M.; Rizos, H.; Lim, S.Y.; Nur Gide, T.; Shang, P.; Attrill, G.H.; et al. Integrated molecular and immunophenotypic analysis of NK cells in anti-PD-1 treated metastatic melanoma patients. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef]
- Schuster, I.S.; Wikstrom, M.E.; Brizard, G.; Coudert, J.D.; Estcourt, M.J.; Manzur, M.; O’Reilly, L.A.; Smyth, M.J.; Trapani, J.A.; Hill, G.R.; et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity 2014, 41, 646–656. [Google Scholar] [CrossRef]
- Lu, L.; Ikizawa, K.; Hu, D.; Werneck, M.B.F.; Wucherpfennig, K.W.; Cantor, H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity 2007, 26, 593–604. [Google Scholar] [CrossRef]
- Ercolano, G.; Garcia-Garijo, A.; Salomé, B.; Gomez-Cadena, A.; Vanoni, G.; Mastelic-Gavillet, B.; Ianaro, A.; Speiser, D.E.; Romero, P.; Trabanelli, S.; et al. Immunosuppressive mediators impair proinflammatory innate lymphoid cell function in human malignant melanoma. Cancer Immunol. Res. 2020, 8, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Dominguez, D.; Qin, L.; Chen, S.; Fan, J.; Zhang, M.; Fang, D.; Zhang, Y.; Kuzel, T.M.; Zhang, B. Type 2 innate lymphoid cells impede IL-33–mediated tumor suppression. J. Immunol. 2018, 201, 3456–3464. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Ealey, K.N.; Tetsu, H.; Kiniwa, T.; Motomura, Y.; Moro, K.; Koyasu, S. Tumor-derived lactic acid contributes to the paucity of intratumoral ILC2s. Cell Rep. 2020, 30, 2743–2757.e5. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, W.; Moon, U.J.; Kim, H.J.; Choi, H.-J.; Sin, J.-I.; Park, N.H.; Cho, H.R.; Kwon, B. Intratumorally establishing type 2 innate lymphoid cells blocks tumor growth. J. Immunol. 2016, 196, 2410–2423. [Google Scholar] [CrossRef]
- Mjösberg, J.; Bernink, J.; Peters, C.; Spits, H. Transcriptional control of innate lymphoid cells. Eur. J. Immunol. 2012, 42, 1916–1923. [Google Scholar] [CrossRef]
- Li, Z.; Hodgkinson, T.; Gothard, E.J.; Boroumand, S.; Lamb, R.; Cummins, I.; Narang, P.; Sawtell, A.; Coles, J.; Leonov, G.; et al. Epidermal Notch1 recruits RORγ(+) group 3 innate lymphoid cells to orchestrate normal skin repair. Nat. Commun. 2016, 7, 11394. [Google Scholar] [CrossRef]
- Tang, Q.; Li, J.; Zhu, H.; Li, P.; Zou, Z.; Xiao, Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 Axis promotes tumor growth in murine models of melanoma—PubMed. Mediat. Inflamm. 2013, 713859. [Google Scholar]
- Chen, Y.S.; Huang, T.H.; Liu, C.L.; Chen, H.S.; Lee, M.H.; Chen, H.W.; Shen, C.R. Locally targeting the IL-17/IL-17RA axis reduced tumor growth in a Murine B16F10 melanoma model. Hum. Gene Ther. 2019, 30, 273–285. [Google Scholar] [CrossRef]
- Leijten, E.F.A.; van Kempen, T.S.; Boes, M.; Michels-van Amelsfort, J.M.R.; Hijnen, D.; Hartgring, S.A.Y.; van Roon, J.A.G.; Wenink, M.H.; Radstake, T.R.D.J. Brief report: Enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol. 2015, 67, 2673–2678. [Google Scholar] [CrossRef]
- Irshad, S.; Flores-Borja, F.; Lawler, K.; Monypenny, J.; Evans, R.; Male, V.; Gordon, P.; Cheung, A.; Gazinska, P.; Noor, F.; et al. RORγt+ innate lymphoid cells promote lymph node metastasis of breast cancers. Cancer Res. 2017, 77, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.; Kim, H.Y.; Lee, Y.; Park, I.K.; Kang, C.H.; Kim, Y.T.; Kim, J.E.; Choi, M.; Lee, W.W.; Jeon, Y.K.; et al. IL23-producing human lung cancer cells promote tumor growth via conversion of innate lymphoid cell 1 (ILC1) into ILC3. Clin. Cancer Res. 2019, 25, 4026–4037. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Kanitakis, J. Anatomy, histology and immunohistochemistry of normal human skin. Eur. J. Dermatol. 2002, 12, 390–401. [Google Scholar] [PubMed]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef]
- Byun, J.S.; Gardner, K. Wounds that will not heal. Am. J. Pathol. 2013, 182, 1055–1064. [Google Scholar] [CrossRef]
- An, Z.; Flores-Borja, F.; Irshad, S.; Deng, J.; Ng, T. Pleiotropic role and bidirectional immunomodulation of innate lymphoid cells in cancer. Front. Immunol. 2020, 10, 3111. [Google Scholar] [CrossRef]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef]
- Roy, M.; Marchetti, D. Cell surface heparan sulfate released by Heparanase promotes melanoma cell migration and angiogenesis. J. Cell. Biochem. 2009, 106, 200–209. [Google Scholar] [CrossRef]
- Vornicova, O.; Boyango, I.; Feld, S.; Naroditsky, I.; Kazarin, O.; Zohar, Y.; Tiram, Y.; Ilan, N.; Ben-Izhak, O.; Vlodavsky, I.; et al. The prognostic significance of heparanase expression in metastatic melanoma. Oncotarget 2016, 7, 74678–74685. [Google Scholar] [CrossRef]
- Mathieu, V.; De Lassalle, E.M.; Toelen, J.; Mohr, T.; Bellahcène, A.; Van Goietsenoven, G.; Verschuere, T.; Bouzin, C.; Debyser, Z.; De Vleeschouwer, S.; et al. Galectin-1 in melanoma biology and related neo-angiogenesis processes. J. Investig. Derm. 2012, 132, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Radosavljevic, G.; Jovanovic, I.; Majstorovic, I.; Mitrovic, M.; Lisnic, V.J.; Arsenijevic, N.; Jonjic, S.; Lukic, M.L. Deletion of galectin-3 in the host attenuates metastasis of murine melanoma by modulating tumor adhesion and NK cell activity. Clin. Exp. Metastasis 2011, 28, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Kageshita, T.; Kashio, Y.; Yamauchi, A.; Seki, M.; Abedin, M.J.; Nishi, N.; Shoji, H.; Nakamura, T.; Ono, T.; Hirashima, M. Possible role of galectin-9 in cell aggregation and apoptosis of human melanoma cell lines and its clinical significance. Int. J. Cancer 2002, 99, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.W.; Kim, A.B.; Li, P.J.; Wang, J.; Jackson, B.T.; Huang, K.T.H.; Zhang, L.; Raulet, D.H. Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife 2017, 6. [Google Scholar] [CrossRef]
- Levi, I.; Amsalem, H.; Nissan, A.; Darash-Yahana, M.; Peretz, T.; Mandelboim, O.; Rachmilewitz, J. Characterization of tumor infiltrating Natural Killer cell subset. Oncotarget 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural killer cells as key players of tumor progression and angiogenesis: Old and novel tools to divert their pro-tumor activities into potent anti-tumor effects. Cancers 2019, 11, 461. [Google Scholar] [CrossRef]
- Ziani, L.; Safta-Saadoun, T.B.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef]
- Goda, S.; Inoue, H.; Umehara, H.; Miyaji, M.; Nagano, Y.; Harakawa, N.; Imai, H.; Lee, P.; Macarthy, J.B.; Ikeo, T.; et al. Matrix metalloproteinase-1 produced by human CXCL12-stimulated natural killer cells. Am. J. Pathol. 2006, 169, 445–458. [Google Scholar] [CrossRef]
- Kim, M.H.; Jin, S.P.; Jang, S.; Choi, J.Y.; Chung, D.H.; Lee, D.H.; Kim, K.H.; Kim, H.Y. IL-17A–Producing innate lymphoid cells promote skin inflammation by inducing IL-33–driven type 2 immune responses. J. Investig. Derm. 2020, 140, 827–837.e9. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.S.; Wong, C.K. IL33: Roles in allergic inflammation and therapeutic perspectives. Front. Immunol. 2019, 10, 364. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Irshad, S.; Gordon, P.; Wong, F.; Sheriff, I.; Tutt, A.; Ng, T. Crosstalk between innate lymphoid cells and other immune cells in the tumor microenvironment. J. Immunol. Res. 2016, 2016. [Google Scholar] [CrossRef]
- Eisenring, M.; vom Berg, J.; Kristiansen, G.; Saller, E.; Becher, B. IL-12 initiates tumor rejection via lymphoid tissue–inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 2010, 11, 1030–1038. [Google Scholar] [CrossRef]
- Murugaiyan, G.; Saha, B. Protumor vs. antitumor functions of IL-17. J. Immunol. 2009, 183, 4169–4175. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, L.; Zhang, Q.; Liu, S.; Ge, D.; You, Z. Interleukin-17 indirectly promotes M2 macrophage differentiation through stimulation of COX-2/PGE2 pathway in the cancer cells. Cancer Res. Treat. 2014, 46, 297–306. [Google Scholar] [CrossRef]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef]
- Kondo, T.; Okabayashi, K.; Hasegawa, H.; Tsuruta, M.; Shigeta, K.; Kitagawa, Y. The impact of hepatic fibrosis on the incidence of liver metastasis from colorectal cancer. Br. J. Cancer 2016, 115, 34–39. [Google Scholar] [CrossRef]
- Brodt, P. Role of the microenvironment in liver metastasis: From pre- to prometastatic niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef]
- Ducimetière, L.; Vermeer, M.; Tugues, S. The interplay between innate lymphoid cells and the tumor microenvironment. Front. Immunol. 2019, 10, 2895. [Google Scholar] [CrossRef]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 family members in cancer; two sides to every story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Yang, C.; Cao, H.; Liu, N.; Xu, K.; Ding, M.; Mao, L.J. Oncolytic adenovirus expressing interleukin-18 improves antitumor activity of dacarbazine for malignant melanoma. Drug Des. Dev. Ther. 2016, 10, 3755–3761. [Google Scholar] [CrossRef]
- Park, S.; Cheon, S.; Cho, D. The dual effects of interleukin-18 in tumor progression. Cell. Mol. Immunol. 2007, 4, 329–335. [Google Scholar]
- Gil, M.; Kim, K.E. Interleukin-18 is a prognostic biomarker correlated with CD8+ T cell and natural killer cell infiltration in skin cutaneous melanoma. J. Clin. Med. 2019, 8, 1993. [Google Scholar] [CrossRef]
- Salado, C.; Olaso, E.; Gallot, N.; Valcarcel, M.; Egilegor, E.; Mendoza, L.; Vidal-Vanaclocha, F. Resveratrol prevents inflammation-dependent hepatic melanoma metastasis by inhibiting the secretion and effects of interleukin-18. J. Transl. Med. 2011, 9. [Google Scholar] [CrossRef]
- Ikeda, A.; Aoki, N.; Kido, M.; Iwamoto, S.; Nishiura, H.; Maruoka, R.; Chiba, T.; Watanabe, N. Progression of autoimmune hepatitis is mediated by IL-18-producing dendritic cells and hepatic CXCL9 expression in mice. Hepatology 2014, 60, 224–236. [Google Scholar] [CrossRef]
- Souza-Fonseca-Guimaraes, F.; Huntington, N.D. A new checkpoint for natural killer cell activation. Immunol. Cell Biol. 2018, 96, 5–7. [Google Scholar] [CrossRef]
- Timmers, M.; Vekemans, K.; Vermijlen, D.; Asosingh, K.; Kuppen, P.; Bouwens, L.; Wisse, E.; Braet, F. Interactions between rat colon carcinoma cells and Kupffer cells during the onset of hepatic metastasis. Int. J. Cancer 2004, 112, 793–802. [Google Scholar] [CrossRef]
- Liu, J.; Lin, P.; Zhou, B. Inflammation fuels tumor progress and metastasis. Curr. Pharm. Des. 2015, 21, 3032–3040. [Google Scholar] [CrossRef]
- Chiba, S.; Ikushima, H.; Ueki, H.; Yanai, H.; Kimura, Y.; Hangai, S.; Nishio, J.; Negishi, H.; Tamura, T.; Saijo, S.; et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife 2014, 3, 1–20. [Google Scholar] [CrossRef]
- Reid, D.M.; Montoya, M.; Taylor, P.R.; Borrow, P.; Gordon, S.; Brown, G.D.; Wong, S.Y.C. Expression of the β-glucan receptor, Dectin-1, on murine leukocytes in situ correlates with its function in pathogen recognition and reveals potential roles in leukocyte interactions. J. Leukoc. Biol. 2004, 76, 86–94. [Google Scholar] [CrossRef]
- Aydin, E.; Johansson, J.; Nazir, F.H.; Hellstrand, K.; Martner, A. Role of NOX2-derived reactive oxygen species in NK cell–mediated control of murine melanoma metastasis. Cancer Immunol. Res. 2017, 5, 804–811. [Google Scholar] [CrossRef]
- Eckelhart, E.; Warsch, W.; Zebedin, E.; Simma, O.; Stoiber, D.; Kolbe, T.; Rülicke, T.; Mueller, M.; Casanova, E.; Sexl, V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011, 117, 1565–1573. [Google Scholar] [CrossRef]
- Leong, J.W.; Schneider, S.E.; Sullivan, R.P.; Parikh, B.A.; Anthony, B.A.; Singh, A.; Jewell, B.A.; Schappe, T.; Wagner, J.A.; Link, D.C.; et al. PTEN regulates natural killer cell trafficking in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, E700–E709. [Google Scholar] [CrossRef]
- Briercheck, E.L.; Trotta, R.; Chen, L.; Hartlage, A.S.; Cole, J.P.; Cole, T.D.; Mao, C.; Banerjee, P.P.; Hsu, H.-T.; Mace, E.M.; et al. PTEN is a negative regulator of NK cell cytolytic function. J. Immunol. 2015, 194, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Kato, A. Group 2 innate lymphoid cells in airway diseases. Chest 2019, 156, 141–149. [Google Scholar] [CrossRef]
- Hong, J.; Kim, S.; Lin, P.C. Interleukin-33 and ST2 signaling in tumor microenvironment. J. Interf. Cytokine Res. 2019, 39, 61–71. [Google Scholar] [CrossRef]
- Mills, K.H.G.; Dungan, L.S.; Jones, S.A.; Harris, J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J. Leukoc. Biol. 2013, 93, 489–497. [Google Scholar] [CrossRef]
- Melo-Gonzalez, F.; Hepworth, M.R. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology 2017, 150, 265–275. [Google Scholar] [CrossRef]
- Scherbarth, S.; Orr, F.W. Intravital videomicroscopic evidence for regulation of metastasis by the hepatic microvasculature: Effects of interleukin-1α on metastasis and the location of B16F1 melanoma cell arrest. Cancer Res. 1997, 57, 4105–4110. [Google Scholar]
- Langley, R.R.; Carlisle, R.; Specian, R.D.; Gerritsen, M.E.; Granger, D.N. Endothelial expression of vascular cell adhesion molecule-1 correlates with metastatic pattern in spontaneous melanoma. Microcirculation 2001, 8, 335–345. [Google Scholar] [CrossRef]
- Zhang, P.; Goodrich, C.; Fu, C.; Dong, C. Melanoma upregulates ICAM-1 expression on endothelial cells through engagement of tumor CD44 with endothelial E-selectin and activation of a PKCα–p38-SP-1 pathway. FASEB J. 2014, 28, 4591–4609. [Google Scholar] [CrossRef]
- Renkonen, R.; Mattila, P.; Majuri, M.L.; Paavonen, T.; Silvennoinen, O. IL-4 decreases IFN-gamma-induced endothelial ICAM-1 expression by a transcriptional mechanism. Scand. J. Immunol. 1992, 35, 525–530. [Google Scholar] [CrossRef]
- Benedicto, A.; Herrero, A.; Romayor, I.; Marquez, J.; Smedsrød, B.; Olaso, E.; Arteta, B. Liver sinusoidal endothelial cell ICAM-1 mediated tumor/endothelial crosstalk drives the development of liver metastasis by initiating inflammatory and angiogenic responses. Sci. Rep. 2019, 9, 13111. [Google Scholar] [CrossRef]
- Karta, M.R.; Rosenthal, P.S.; Beppu, A.; Vuong, C.Y.; Miller, M.; Das, S.; Kurten, R.C.; Doherty, T.A.; Broide, D.H. β2 integrins rather than β1 integrins mediate Alternaria-induced group 2 innate lymphoid cell trafficking to the lung. J. Allergy Clin. Immunol. 2018, 141, 329–338.e12. [Google Scholar] [CrossRef] [PubMed]
- Olaso, E.; Salado, C.; Egilegor, E.; Gutierrez, V.; Santisteban, A.; Sancho-Bru, P.; Friedman, S.L.; Vidal-Vanaclocha, F. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 2003, 37, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Goding, S.; Hagenaars, M.; Carlos, T.; Albertsson, P.; Kuppen, P.; Nannmark, U.; Hokland, M.E.; Basse, P.H. Morphological appearance, content of extracellular matrix and vascular density of lung metastases predicts permissiveness to infiltration by adoptively transferred natural killer and T cells. Cancer Immunol. Immunother. 2006, 55, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Loh, Z.; Fitzsimmons, R.L.; Reid, R.C.; Ramnath, D.; Clouston, A.; Gupta, P.K.; Irvine, K.M.; Powell, E.E.; Schroder, K.; Stow, J.L.; et al. Inhibitors of class I histone deacetylases attenuate thioacetamide-induced liver fibrosis in mice by suppressing hepatic type 2 inflammation. Br. J. Pharm. 2019, 176, 3775–3790. [Google Scholar] [CrossRef]
- Yamada, M.; Ichikawa, Y.; Yamagishi, S.; Momiyama, N.; Ota, M.; Fujii, S.; Tanaka, K.; Togo, S.; Ohki, S.; Shimada, H. Amphiregulin is a promising prognostic marker for liver metastases of colorectal cancer. Clin. Cancer Res. 2008, 14, 2351–2356. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Cao, W.; Mielke, L.A.; Seillet, C.; Belz, G.T.; Jacquelot, N. Innate Lymphoid Cells in Colorectal Cancers: A Double-Edged Sword. Front. Immunol. 2020, 10, 3080. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, H.C.; McDowell, P.; Lutz, P.; Wawman, R.E.; Roberts, S.; Bagnall, C.; Birtwistle, J.; Adams, D.H.; Oo, Y.H. Human intrahepatic ILC2 are IL-13positive amphiregulinpositive and their frequency correlates with model of end stage liver disease score. PLoS ONE 2017, 12, e0188649. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Loiacono, F.; Di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR + ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280. [Google Scholar] [CrossRef]
- Zhou, L.; Sonnenberg, G.F. In Situ Support of ILC Precursors. Immunity 2020, 52, 207–209. [Google Scholar] [CrossRef]
- Le Coz, V.; Zhu, C.; Devocelle, A.; Vazquez, A.; Boucheix, C.; Azzi, S.; Gallerne, C.; Eid, P.; Lecourt, S.; Giron-Michel, J. IGF-1 contributes to the expansion of melanoma-initiating cells through an epithelial-mesenchymal transition process. Oncotarget 2016, 7, 82511–82527. [Google Scholar] [CrossRef]
- Han, L.; Lam, E.W.F.; Sun, Y. Extracellular vesicles in the tumor microenvironment: Old stories, but new tales. Mol. Cancer 2019, 18, 59. [Google Scholar] [CrossRef]
- Anderson, H.C. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 1969, 41, 59–72. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Leijendekker, R.; Harding, C.V.; Melief, C.J.M.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.L.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef]
- Gyukity-Sebestyén, E.; Harmati, M.; Dobra, G.; Németh, I.B.; Mihály, J.; Zvara, Á.; Hunyadi-Gulyás, É.; Katona, R.; Nagy, I.; Horváth, P.; et al. Melanoma-derived exosomes induce PD-1 overexpression and tumor progression via mesenchymal stem cell oncogenic reprogramming. Front. Immunol. 2019, 10, 2459. [Google Scholar] [CrossRef]
- Tracey, E.H.; Vij, A. Updates in melanoma. Derm. Clin. 2019, 37, 73–82. [Google Scholar] [CrossRef]
- Sheehan, C.; D’Souza-Schorey, C. Tumor-derived extracellular vesicles: Molecular parcels that enable regulation of the immune response in cancer. J. Cell Sci. 2019, 132, jcs235085. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Dai, S. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-Specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin. Cancer Res. 2005, 11, 7554–7563. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring anticancer immune response by targeting tumor-derived exosomes with a HSP70 peptide aptamer. J. Natl. Cancer Inst. 2016, 108, djv330. [Google Scholar] [CrossRef]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano, J.; Malzahn, D.; Bickeböller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Zinn, K.; Wang, J.; Zhang, L.; Jia, Y.; Kappes, J.C.; Barnes, S.; Kimberly, R.P.; Grizzle, W.E.; et al. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J. Immunol. 2006, 176, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Pogge von Strandmann, E.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Böll, B.; Simhadri, V.L.; Borchmann, P.; et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef]
- Simhadri, V.R.; Reiners, K.S.; Hansen, H.P.; Topolar, D.; Simhadri, V.L.; Nohroudi, K.; Kufer, T.A.; Engert, A.; Pogge von Strandmann, E. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS ONE 2008, 3, e3377. [Google Scholar] [CrossRef]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; André, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Rα. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Israelsson, P.; Ottander, U.; Lundin, E.; Nagaev, I.; Nagaeva, O.; Dehlin, E.; Baranov, V.; Mincheva-Nilsson, L. Differential expression of ligands for NKG2D and DNAM-1 receptors by epithelial ovarian cancer-derived exosomes and its influence on NK cell cytotoxicity. Tumor Biol. 2016, 37, 5455–5466. [Google Scholar] [CrossRef]
- López-Cobo, S.; Campos-Silva, C.; Moyano, A.; Oliveira-Rodríguez, M.; Paschen, A.; Yáñez-Mó, M.; Blanco-López, M.C.; Valés-Gómez, M. Immunoassays for scarce tumour-antigens in exosomes: Detection of the human NKG2D-Ligand, MICA, in tetraspanin-containing nanovesicles from melanoma. J. Nanobiotechnol. 2018, 16, 47. [Google Scholar] [CrossRef]
- Sharma, P.; Ludwig, S.; Muller, L.; Hong, C.S.; Kirkwood, J.M.; Ferrone, S.; Whiteside, T.L. Immunoaffinity-based isolation of melanoma cell-derived exosomes from plasma of patients with melanoma. J. Extracell. Vesicles 2018, 7, 1435138. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schlößer, H.A.; Wang, Z.; Qin, J.; Li, J.; Popp, F.; Popp, M.C.; Alakus, H.; Chon, S.-H.H.; Hansen, H.P.; et al. Tumor-derived extracellular vesicles inhibit natural killer cell function in pancreatic cancer. Cancers 2019, 11, 874. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Krasagakis, K.; Thölke, D.; Farthmann, B.; Eberle, J.; Mansmann, U.; Orfanos, C. Elevated plasma levels of transforming growth factor (TGF)-β1 and TGF-β2 in patients with disseminated malignant melanoma. Br. J. Cancer 1998, 77, 1492–1494. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Karabulut, S.; Yasasever, C.T.; Duranyildiz, D. Serum transforming growth factor-beta 1 (TGF-β1) levels have diagnostic, predictive, and possible prognostic roles in patients with melanoma. Tumor Biol. 2014, 35, 7233–7237. [Google Scholar] [CrossRef] [PubMed]
- Düchler, M.; Czernek, L.; Peczek, L.; Cypryk, W.; Sztiller-Sikorska, M.; Czyz, M. Melanoma-derived extracellular vesicles bear the potential for the induction of antigen-specific tolerance. Cells 2019, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef]
- Jong, A.Y.; Wu, C.-H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef] [PubMed]
- Shoae-Hassani, A.; Behfar, M.; Mortazavi-Tabatabaei, S.A.; Ai, J.; Mohseni, R.; Hamidieh, A.A. Natural killer cells from the subcutaneous adipose tissue underexpress the NKp30 and NKp44 in obese persons and are less active against major histocompatibility complex class I non-expressing neoplastic cells. Front. Immunol. 2017, 8, 1486. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Skin | Liver | Lung | |||||
|---|---|---|---|---|---|---|---|
| Healthy | Melanoma | Healthy | Melanoma | Healthy | Melanoma | ||
| Type 1 ILC | ILC1 | + | ++ | ++ | + | ++ | NA * |
| NK | ++ | ++++ | +++ | ++++ | ++ | ++++ | |
| Type 2 ILC | ++++ | +++ | ++ | ++ | +++ | ++++ | |
| Type 3 ILC | NCR(+) | + | +++ | + | ++ | ++ | +++ |
| NCR(−) | +++ | + | + | ++ | +++ | NA * | |
| Skin | Liver | Lung | |||||
|---|---|---|---|---|---|---|---|
| Healthy | Melanoma | Healthy | Melanoma | Healthy | Melanoma | ||
| Type 1 ILC | ILC1 | Immune cell recruitment. | Antitumoral (antigen recognition), protumoral (PD-L1 expression). | Normal hepatic function, local immune tolerance. | Inflammatory tumor microenvironment, tumor growth. | Homeostasis, host protection, immune surveillance. | Antitumoral (IFNγ-production) |
| NK | Cytotoxicity. | Antitumoral (cytotoxic), protumoral (T cell exhaustion, ILC2 modulation). | Local immune response. | Antitumoral (cytotoxicity). | Cytotoxicity. | Antitumoral (cytotoxicity). | |
| Type 2 ILC | Wound healing. | Antitumoral (IL-5), protumoral (NK cell-impairment, fibroblast transdifferentiation, increase NK PD-1 expression). | Response to chronic stress, local immune-suppression, counteract inflammatory injury. | Antitumoral (NA *), protumoral (favour a protumoral desmoplastic reaction) | Host protection, immune surveillance. | Antitumoral (tumor immunesurveillance, induction of IFNγ producing ILC1), protumoral (NK cell imapirment). | |
| Type 3 ILC | NCR(+) | Immune cell recruitment. | Proangiogenic, immunesuppressive cell recruitment, IL-6 and Stat-3 activation | Response to chronic stress, counteract inflammatory injury. | Upregulation of adhesion molecules | Regulation of inflammation. | Antitumor immune response |
| NCR(−) | Wound healing. | Proangiogenic, immunesuppressive cell recruitment, IL-6 and Stat-3 activation | Response to chronic stress, counteract inflammatory injury. | NA * | Regulation of inflammation. | NA * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apraiz, A.; Benedicto, A.; Marquez, J.; Agüera-Lorente, A.; Asumendi, A.; Olaso, E.; Arteta, B. Innate Lymphoid Cells in the Malignant Melanoma Microenvironment. Cancers 2020, 12, 3177. https://doi.org/10.3390/cancers12113177
Apraiz A, Benedicto A, Marquez J, Agüera-Lorente A, Asumendi A, Olaso E, Arteta B. Innate Lymphoid Cells in the Malignant Melanoma Microenvironment. Cancers. 2020; 12(11):3177. https://doi.org/10.3390/cancers12113177
Chicago/Turabian StyleApraiz, Aintzane, Aitor Benedicto, Joana Marquez, Andrea Agüera-Lorente, Aintzane Asumendi, Elvira Olaso, and Beatriz Arteta. 2020. "Innate Lymphoid Cells in the Malignant Melanoma Microenvironment" Cancers 12, no. 11: 3177. https://doi.org/10.3390/cancers12113177
APA StyleApraiz, A., Benedicto, A., Marquez, J., Agüera-Lorente, A., Asumendi, A., Olaso, E., & Arteta, B. (2020). Innate Lymphoid Cells in the Malignant Melanoma Microenvironment. Cancers, 12(11), 3177. https://doi.org/10.3390/cancers12113177