Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives

 ,
,
Simple Summary
Abstract
1. Introduction
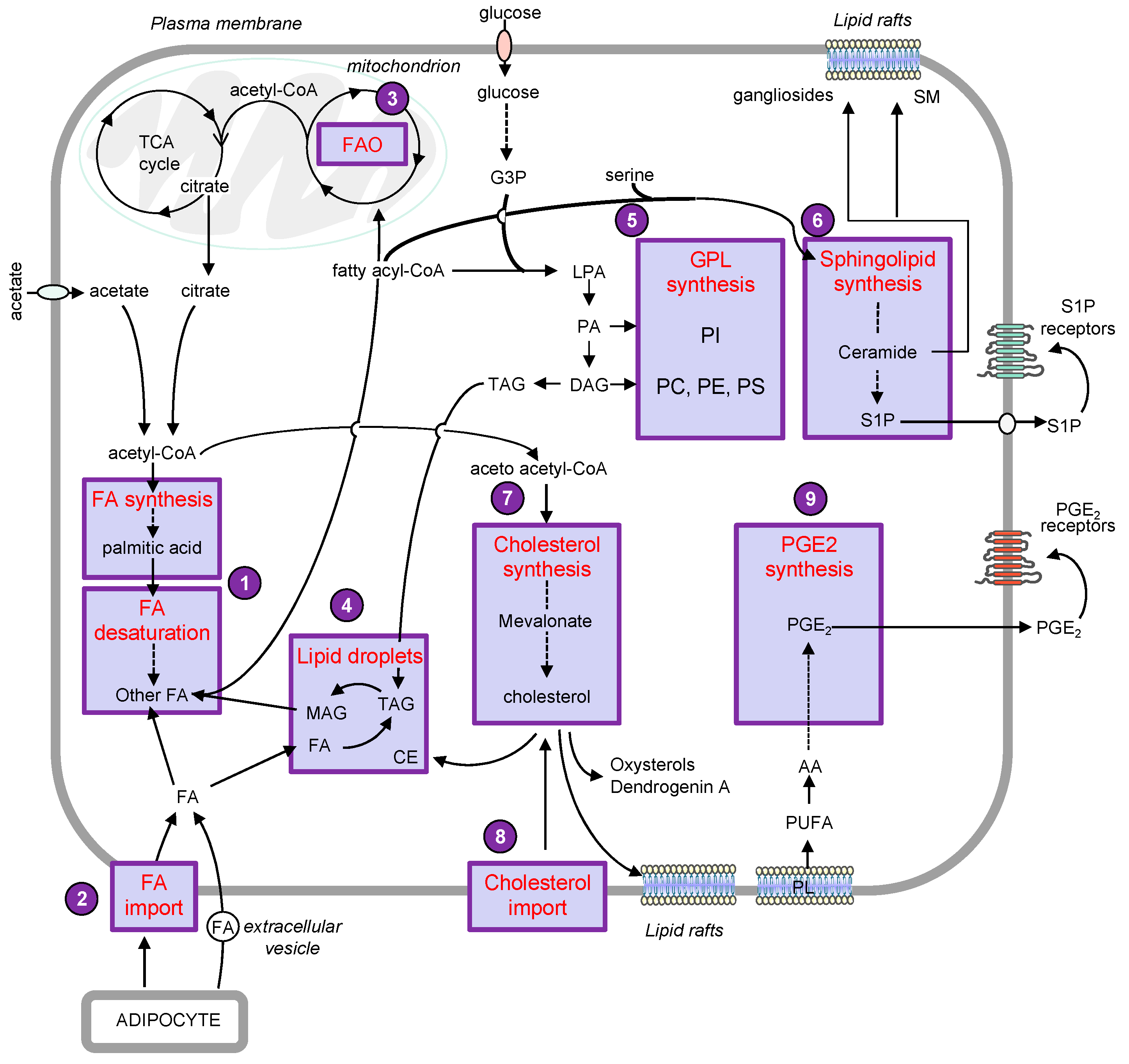
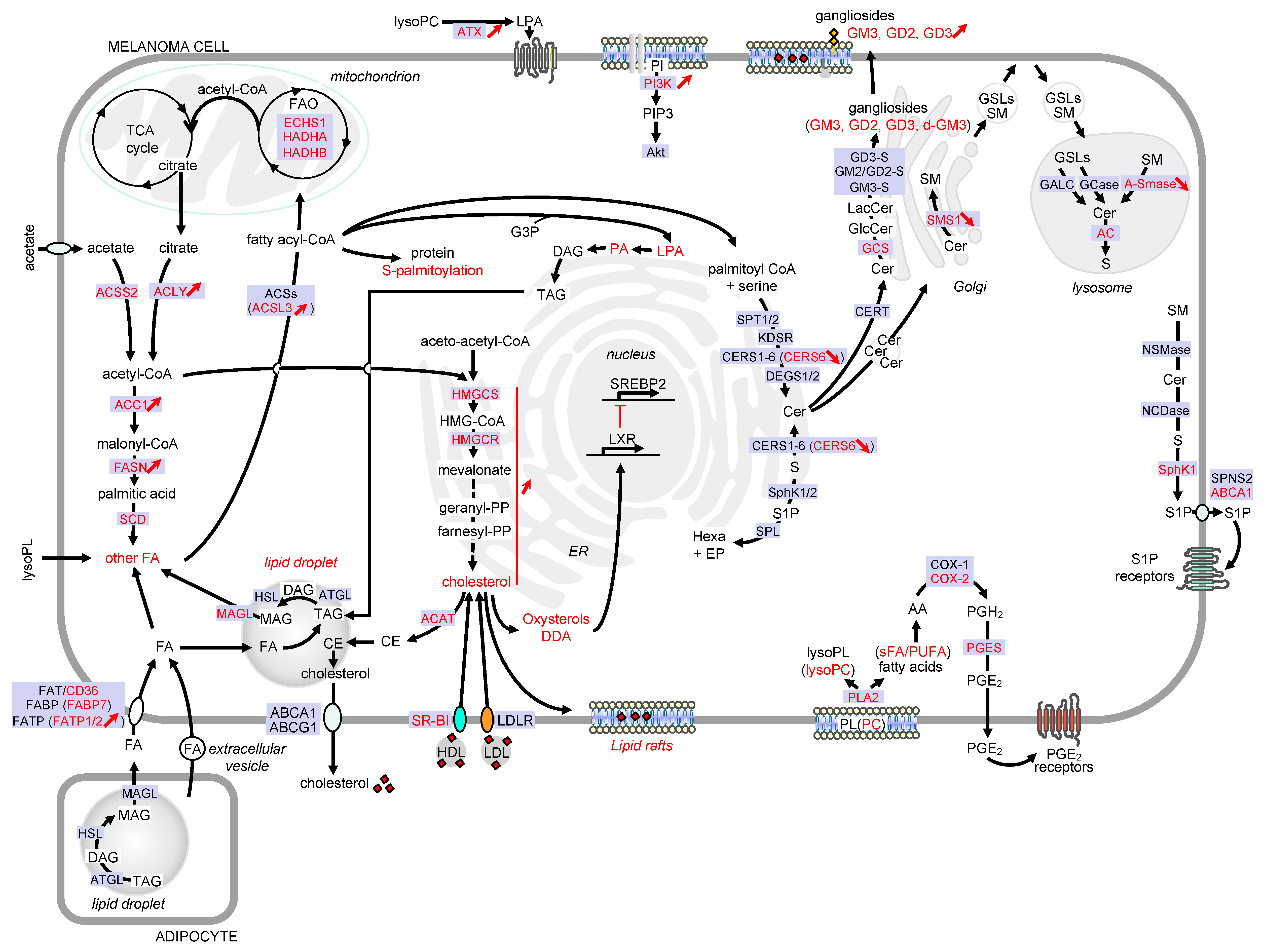
2. Lipid Droplets
3. Phospholipids
3.1. Glycerophospholipids: Potential Roles in Melanoma Progression and Therapeutic Approaches
3.2. Sphingolipids
3.2.1. Potential Roles of Sphingolipids in Melanoma Progression
Role of Ceramide and S1P in Melanoma Progression
Role of Gangliosides in Melanoma Progression
3.2.2. Therapeutic Approaches Targeting Sphingolipids in Melanoma
4. Sterols
4.1. Potential Roles of Sterols in Melanoma Progression
4.2. Therapeutic Approaches Targeting Sterols in Melanoma
5. Eicosanoids
5.1. Potential Roles of Prostanoids in Melanoma Progression
5.2. Therapeutic Approaches Targeting COX-2 in Melanoma
6. Obesity and Melanoma: Role of Adipose Tissue in Tumor Progression
7. Conclusions
Funding
Conflicts of Interest
References
- Baenke, F.; Dhomen, N.; Gottlieb, E.; Marais, R. Melanoma metabolism. In Melanoma; Fisher, D.E., Bastian, B.C., Eds.; Springer: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanita, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic plasticity of melanoma cells and their crosstalk with tumor microenvironment. Front. Oncol. 2020, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell Melanoma. Res. 2018, 31, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Naar, A.M. SREBP1-dependent de novo fatty acid synthesis gene expression is elevated in malignant melanoma and represents a cellular survival trait. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Ma, J.; Yang, Y.; Guo, S.; Zhang, W.; Zhao, T.; Yi, X.; Wang, H.; Wang, S.; Liu, Y.; et al. ATP-citrate lyase epigenetically potentiates oxidative phosphorylation to promote melanoma growth and adaptive resistance to MAPK inhibition. Clin. Cancer Res. 2020, 26, 2725–2739. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 1–17. [Google Scholar] [CrossRef]
- Yoshii, Y.; Furukawa, T.; Saga, T.; Fujibayashi, Y. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: Overview and application. Cancer Lett. 2015, 356, 211–216. [Google Scholar] [CrossRef]
- Lakhter, A.J.; Hamilton, J.; Konger, R.L.; Brustovetsky, N.; Broxmeyer, H.E.; Naidu, S.R. Glucose-independent acetate metabolism promotes melanoma cell survival and tumor growth. J. Biol. Chem. 2016, 291, 21869–21879. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Du, H.; Huang, V.; Sun, B.; Harris, J.P.; Richardson, Q.; Shen, X.; Jin, R.; Li, G.; et al. Withaferin A suppresses the up-regulation of acetyl-coA carboxylase 1 and skin tumor formation in a skin carcinogenesis mouse model. Mol. Carcinog. 2016, 55, 1739–1746. [Google Scholar] [CrossRef]
- Kapur, P.; Rakheja, D.; Roy, L.C.; Hoang, M.P. Fatty acid synthase expression in cutaneous melanocytic neoplasms. Mod. Pathol. 2005, 18, 1107–1112. [Google Scholar] [CrossRef]
- Innocenzi, D.; Alo, P.L.; Balzani, A.; Sebastiani, V.; Silipo, V.; La Torre, G.; Ricciardi, G.; Bosman, C.; Calvieri, S. Fatty acid synthase expression in melanoma. J. Cutan. Pathol. 2003, 30, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.A.; Zecchin, K.G.; Seguin, F.; Bastos, D.C.; Agostini, M.; Rangel, A.L.; Veiga, S.S.; Raposo, H.F.; Oliveira, H.C.; Loda, M.; et al. Fatty acid synthase inhibition with orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int. J. Cancer 2008, 123, 2557–2565. [Google Scholar] [CrossRef]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic analysis of gene expression alterations and clinical outcomes for long-chain Acyl-Coenzyme a synthetase family in cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sun, Y.; Niu, J.; Jarugumilli, G.K.; Wu, X. Protein lipidation in cell signaling and diseases: Function, regulation, and therapeutic opportunities. Cell Chem. Biol. 2018, 25, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, B.; Yin, C.; Liu, W.; Han, C.; Chen, B.; Liu, T.; Li, X.; Chen, X.; Li, C.; et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017, 549, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef]
- Thompson, B.J. YAP/TAZ: Drivers of tumor growth, metastasis, and resistance to therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The role of cholesterol in cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed]
- Sumantran, V.N.; Mishra, P.; Sudhakar, N. Microarray analysis of differentially expressed genes regulating lipid metabolism during melanoma progression. Indian J. Biochem. Biophys. 2015, 52, 125–131. [Google Scholar] [PubMed]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.F.; Obre, E.; de Melo, F.H.; Santos, G.C.J.; Galina, A.; Jasiulionis, M.G.; Rossignol, R.; Rumjanek, F.D.; Amoedo, N.D. Enhanced OXPHOS, glutaminolysis and beta-oxidation constitute the metastatic phenotype of melanoma cells. Biochem. J. 2016, 473, 703–715. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 2018, 8, 1006–1025. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: A Novel mechanism linking obesity and cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Fu, X.; Liu, B.; Chao, X.; Chan, C.L.; Cao, H.; Su, T.; Tse, A.K.; Fong, W.F.; Yu, Z.L. Subcutaneous adipocytes promote melanoma cell growth by activating the Akt signaling pathway: Role of palmitic acid. J. Biol. Chem. 2014, 289, 30525–30537. [Google Scholar] [CrossRef]
- Alicea, G.M.; Rebecca, V.W.; Goldman, A.R.; Fane, M.E.; Douglass, S.M.; Behera, R.; Webster, M.R.; Kugel, C.H., III; Ecker, B.L.; Caino, M.C.; et al. Changes in aged fibroblast lipid metabolism induce age-dependent melanoma cell resistance to targeted therapy via the fatty acid transporter FATP2. Cancer Discov. 2020, 10, 1282–1295. [Google Scholar] [CrossRef]
- Goto, Y.; Matsuzaki, Y.; Kurihara, S.; Shimizu, A.; Okada, T.; Yamamoto, K.; Murata, H.; Takata, M.; Aburatani, H.; Hoon, D.S.; et al. A new melanoma antigen fatty acid-binding protein 7, involved in proliferation and invasion, is a potential target for immunotherapy and molecular target therapy. Cancer Res. 2006, 66, 4443–4449. [Google Scholar] [CrossRef] [PubMed]
- Slipicevic, A.; Jorgensen, K.; Skrede, M.; Rosnes, A.K.; Troen, G.; Davidson, B.; Florenes, V.A. The fatty acid binding protein 7 (FABP7) is involved in proliferation and invasion of melanoma cells. BMC Cancer 2008, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Koyanagi, K.; Narita, N.; Kawakami, Y.; Takata, M.; Uchiyama, A.; Nguyen, L.; Nguyen, T.; Ye, X.; Morton, D.L.; et al. Aberrant fatty acid-binding protein-7 gene expression in cutaneous malignant melanoma. J. Invest. Dermatol. 2010, 130, 221–229. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Arozarena, I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015, 28, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Feige, E.; Yokoyama, S.; Levy, C.; Khaled, M.; Igras, V.; Lin, R.J.; Lee, S.; Widlund, H.R.; Granter, S.R.; Kung, A.L.; et al. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc. Natl. Acad. Sci. USA 2011, 108, E924–E933. [Google Scholar] [CrossRef] [PubMed]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Louphrasitthiphol, P.; Ledaki, I.; Chauhan, J.; Falletta, P.; Siddaway, R.; Buffa, F.M.; Mole, D.R.; Soga, T.; Goding, C.R. MITF controls the TCA cycle to modulate the melanoma hypoxia response. Pigment Cell Melanoma Res. 2019, 32, 792–808. [Google Scholar] [CrossRef]
- Ferguson, J.; Smith, M.; Zudaire, I.; Wellbrock, C.; Arozarena, I. Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 2017, 8, 32946–32959. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 1–16. [Google Scholar] [CrossRef]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018, 33, 890.e5–904.e5. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward minimal residual disease-directed therapy in melanoma. Cell 2018, 174, 843.e19–855.e19. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Vivas-Garcia, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-restricted regulation of SCD and fatty acid saturation by MITF controls melanoma phenotypic plasticity. Mol. Cell 2020, 77, 120–137. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-kappaB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Su, Y.; Qian, C.; Yuan, D.; Miao, K.; Lee, D.; Ng, A.H.C.; Wijker, R.S.; Ribas, A.; Levine, R.D.; et al. Raman-guided subcellular pharmaco-metabolomics for metastatic melanoma cells. Nat. Commun. 2020, 11, 4830. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar] [CrossRef]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Le, T.T.; Johlfs, M.G.; Fiscus, R.R.; Wilsch-Brauninger, M.; Corbeil, D.; Lorico, A. Wnt interaction and extracellular release of prominin-1/CD133 in human malignant melanoma cells. Exp. Cell. Res. 2013, 319, 810–819. [Google Scholar] [CrossRef]
- Scott, C.C.; Vossio, S.; Vacca, F.; Snijder, B.; Larios, J.; Schaad, O.; Guex, N.; Kuznetsov, D.; Martin, O.; Chambon, M.; et al. Wnt directs the endosomal flux of LDL-derived cholesterol and lipid droplet homeostasis. EMBO Rep. 2015, 16, 741–752. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Olkhanud, P.B.; O’Connell, M.P.; Carter, A.; French, A.D.; Camilli, T.C.; Emeche, C.D.; Hewitt, K.J.; Rosenthal, D.T.; Leotlela, P.D.; et al. Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008, 68, 10205–10214. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.G.; Chammas, R.; Monteiro, R.Q.; Moreira, M.E.; Barcinski, M.A. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009, 283, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Henderson, F.; Johnston, H.R.; Badrock, A.P.; Jones, E.A.; Forster, D.; Nagaraju, R.T.; Evangelou, C.; Kamarashev, J.; Green, M.; Fairclough, M.; et al. Enhanced fatty acid scavenging and glycerophospholipid metabolism accompany melanocyte neoplasia progression in zebrafish. Cancer Res. 2019, 79, 2136–2151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Du, M.; Wu, M.; Zhu, Y.; Zhao, X.; Cao, X.; Li, X.; Long, P.; Li, W.; Hu, B. Phosphatidic acid improves reprogramming to pluripotency by reducing apoptosis. Stem. Cells Dev. 2016, 25, 43–54. [Google Scholar] [CrossRef]
- Muinonen-Martin, A.J.; Susanto, O.; Zhang, Q.; Smethurst, E.; Faller, W.J.; Veltman, D.M.; Kalna, G.; Lindsay, C.; Bennett, D.C.; Sansom, O.J.; et al. Melanoma cells break down LPA to establish local gradients that drive chemotactic dispersal. PLoS Biol. 2014, 12, e1001966. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Park, S.H.; Kwon, S.B.; Youn, S.W.; Park, K.C. Effects of lysophosphatidic acid on melanogenesis. Chem. Phys. Lipids 2004, 127, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Raynor, A.; Jantscheff, P.; Ross, T.; Schlesinger, M.; Wilde, M.; Haasis, S.; Dreckmann, T.; Bendas, G.; Massing, U. Saturated and mono-unsaturated lysophosphatidylcholine metabolism in tumour cells: A potential therapeutic target for preventing metastases. Lipids Health Dis. 2015, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.; Jakubzig, B.; Grundmann, M.; Massing, U.; Kostenis, E.; Schlesinger, M.; Bendas, G. The molecular mechanism by which saturated lysophosphatidylcholine attenuates the metastatic capacity of melanoma cells. FEBS Open Bio 2016, 6, 1297–1309. [Google Scholar] [CrossRef]
- Jankowski, M. Autotaxin: Its role in biology of melanoma cells and as a pharmacological target. Enzym. Res. 2011, 2011, 1–5. [Google Scholar] [CrossRef]
- Lligona Trulla, L.; Magistrelli, A.; Salmona, M.; Tacconi, M.T. Phospholipid composition, phosphoinositide metabolism and metastatic capacity in murine melanoma B16 variants at different stages of growth. Melanoma Res. 1992, 2, 235–240. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.; Kim, S.H.; Jin, H.; Bae, J.; Choi, H.K. Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Schroeder, F.; Gardiner, J.M. Membrane lipids and enzymes of cultured high- and low-metastatic B16 melanoma variants. Cancer Res. 1984, 44, 3262–3269. [Google Scholar]
- Pulido, R. PTEN inhibition in human disease therapy. Molecules 2018, 23, 285. [Google Scholar] [CrossRef]
- Aziz, S.A.; Davies, M.; Pick, E.; Zito, C.; Jilaveanu, L.; Camp, R.L.; Rimm, D.L.; Kluger, Y.; Kluger, H.M. Phosphatidylinositol-3-kinase as a therapeutic target in melanoma. Clin. Cancer Res. 2009, 15, 3029–3036. [Google Scholar] [CrossRef]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Kim, K.H.; Sung, H.; Paraiso, K.H.; Dannenberg, J.H.; Bosenberg, M.; Chin, L.; Kim, M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010, 29, 6222–6232. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic significance of activated Akt expression in melanoma: A clinicopathologic study of 292 cases. J. Clin. Oncol. 2005, 23, 1473–1482. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, L.; Su, Q.; Fan, X.; Wang, Y.; Gao, S.; Wang, H.; Chen, H.; Chan, C.B.; Liu, Z. Phosphorylation of MITF by AKT affects its downstream targets and causes TP53-dependent cell senescence. Int. J. Biochem. Cell Biol. 2016, 80, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Garandeau, D.; Mrad, M.; Levade, T.; Perrotta, C.; Diab-Assaf, M.; Andrieu-abadie, N. Dysregulation of sphingolipid metabolism in melanoma: Roles in pigmentation, cell survival and tumor progression. In Bioactive Sphingolipids in Cancer Biology and Therapy; Hannun, Y.A., Luberto, C., Mao, C., Obeid, L.M., Eds.; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar] [CrossRef]
- Carrie, L.; Virazels, M.; Dufau, C.; Montfort, A.; Levade, T.; Segui, B.; Andrieu-Abadie, N. New insights into the role of sphingolipid metabolism in melanoma. Cells 2020, 9, 1967. [Google Scholar] [CrossRef]
- Tang, Y.; Cao, K.; Wang, Q.; Chen, J.; Liu, R.; Wang, S.; Zhou, J.; Xie, H. Silencing of CerS6 increases the invasion and glycolysis of melanoma WM35, WM451 and SK28 cell lines via increased GLUT1-induced downregulation of WNT5A. Oncol. Rep. 2016, 35, 2907–2915. [Google Scholar] [CrossRef]
- Bizzozero, L.; Cazzato, D.; Cervia, D.; Assi, E.; Simbari, F.; Pagni, F.; De Palma, C.; Monno, A.; Verdelli, C.; Querini, P.R.; et al. Acid sphingomyelinase determines melanoma progression and metastatic behaviour via the microphtalmia-associated transcription factor signalling pathway. Cell Death Differ. 2014, 21, 507–520. [Google Scholar] [CrossRef]
- Liu, R.; Cao, K.; Tang, Y.; Liu, J.; Li, J.; Chen, J.; Wang, S.; Chen, Z.; Zhou, J. C16:0 ceramide effect on melanoma malignant behavior and glycolysis depends on its intracellular or exogenous location. Am. J. Transl. Res. 2020, 12, 1123–1135. [Google Scholar]
- Han, W.S.; Yoo, J.Y.; Youn, S.W.; Kim, D.S.; Park, K.C.; Kim, S.Y.; Kim, K.H. Effects of C2-ceramide on the Malme-3M melanoma cell line. J. Dermatol. Sci. 2002, 30, 10–19. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, S.Y.; Chung, J.H.; Kim, K.H.; Eun, H.C.; Park, K.C. Delayed ERK activation by ceramide reduces melanin synthesis in human melanocytes. Cell. Signal. 2002, 14, 779–785. [Google Scholar] [CrossRef]
- Bilal, F.; Montfort, A.; Gilhodes, J.; Garcia, V.; Riond, J.; Carpentier, S.; Filleron, T.; Colacios, C.; Levade, T.; Daher, A.; et al. Sphingomyelin synthase 1 (SMS1) downregulation is associated with sphingolipid reprogramming and a worse prognosis in melanoma. Front. Pharmacol. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Realini, N.; Palese, F.; Pizzirani, D.; Pontis, S.; Basit, A.; Bach, A.; Ganesan, A.; Piomelli, D. Acid ceramidase in melanoma: Expression, localization, and effects of pharmacological inhibition. J. Biol. Chem. 2016, 291, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Garandeau, D.; Pandiani, C.; Gaudel, C.; Bille, K.; Nottet, N.; Garcia, V.; Colosetti, P.; Pagnotta, S.; Bahadoran, P.; et al. Lysosomal acid ceramidase ASAH1 controls the transition between invasive and proliferative phenotype in melanoma cells. Oncogene 2019, 38, 1282–1295. [Google Scholar] [CrossRef]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete acid ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Madhunapantula, S.V.; Hengst, J.; Gowda, R.; Fox, T.E.; Yun, J.K.; Robertson, G.P. Targeting sphingosine kinase-1 to inhibit melanoma. Pigment. Cell Melanoma. Res. 2012, 25, 259–274. [Google Scholar] [CrossRef]
- Albinet, V.; Bats, M.L.; Huwiler, A.; Rochaix, P.; Chevreau, C.; Segui, B.; Levade, T.; Andrieu-Abadie, N. Dual role of sphingosine kinase-1 in promoting the differentiation of dermal fibroblasts and the dissemination of melanoma cells. Oncogene 2014, 33, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Mrad, M.; Imbert, C.; Garcia, V.; Rambow, F.; Therville, N.; Carpentier, S.; Segui, B.; Levade, T.; Azar, R.; Marine, J.C.; et al. Downregulation of sphingosine kinase-1 induces protective tumor immunity by promoting M1 macrophage response in melanoma. Oncotarget 2016, 7, 71873–71886. [Google Scholar] [CrossRef]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kitayama, J.; Takuwa, N.; Arikawa, K.; Inoki, I.; Takehara, K.; Nagawa, H.; Takuwa, Y. Sphingosine-1-phosphate receptor subtype-specific positive and negative regulation of Rac and haematogenous metastasis of melanoma cells. Biochem. J. 2003, 374, 715–722. [Google Scholar] [CrossRef]
- Pierrat, M.J.; Marsaud, V.; Mauviel, A.; Javelaud, D. Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-beta. J. Biol. Chem. 2012, 287, 17996–18004. [Google Scholar] [CrossRef]
- Nishimura, E.K.; Suzuki, M.; Igras, V.; Du, J.; Lonning, S.; Miyachi, Y.; Roes, J.; Beermann, F.; Fisher, D.E. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem. Cell 2010, 6, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Ren, S.; Kleuser, B.; Shabahang, S.; Eberhardt, W.; Radeke, H.; Schafer-Korting, M.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J. Biol. Chem. 2004, 279, 35255–35262. [Google Scholar] [CrossRef] [PubMed]
- Sauer, B.; Vogler, R.; von Wenckstern, H.; Fujii, M.; Anzano, M.B.; Glick, A.B.; Schafer-Korting, M.; Roberts, A.B.; Kleuser, B. Involvement of Smad signaling in sphingosine 1-phosphate-mediated biological responses of keratinocytes. J. Biol. Chem. 2004, 279, 38471–38479. [Google Scholar] [CrossRef]
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J. Biol. Chem. 2004, 279, 53994–54001. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, S.Y.; Jeong, Y.M.; Yun, H.Y.; Baek, K.J.; Kwon, N.S.; Park, K.C.; Kim, D.S. The regulatory mechanism of melanogenesis by FTY720, a sphingolipid analogue. Exp. Dermatol. 2011, 20, 237–241. [Google Scholar] [CrossRef]
- Yasumoto, K.; Takeda, K.; Saito, H.; Watanabe, K.; Takahashi, K.; Shibahara, S. Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J. 2002, 21, 2703–2714. [Google Scholar] [CrossRef]
- Tsuchida, T.; Saxton, R.E.; Morton, D.L.; Irie, R.F. Gangliosides of human melanoma. J. Natl. Cancer Inst. 1987, 78, 45–54. [Google Scholar] [CrossRef]
- Hamamura, K.; Furukawa, K.; Hayashi, T.; Hattori, T.; Nakano, J.; Nakashima, H.; Okuda, T.; Mizutani, H.; Hattori, H.; Ueda, M.; et al. Ganglioside GD3 promotes cell growth and invasion through p130Cas and paxillin in malignant melanoma cells. Proc. Natl. Acad. Sci. USA 2005, 102, 11041–11046. [Google Scholar] [CrossRef]
- Nakano, J.; Raj, B.K.; Asagami, C.; Lloyd, K.O. Human melanoma cell lines deficient in GD3 ganglioside expression exhibit altered growth and tumorigenic characteristics. J. Invest. Dermatol. 1996, 107, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Kambe, M.; Miyata, M.; Ohkawa, Y.; Tajima, O.; Furukawa, K. Ganglioside GD3 induces convergence and synergism of adhesion and hepatocyte growth factor/Met signals in melanomas. Cancer Sci. 2014, 105, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, Y.; Miyazaki, S.; Hamamura, K.; Kambe, M.; Miyata, M.; Tajima, O.; Ohmi, Y.; Yamauchi, Y.; Furukawa, K.; Furukawa, K. Ganglioside GD3 enhances adhesion signals and augments malignant properties of melanoma cells by recruiting integrins to glycolipid-enriched microdomains. J. Biol. Chem. 2010, 285, 27213–27223. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Kambe, M.; Ohkawa, Y.; Hamamura, K.; Tajima, O.; Takeuchi, R.; Furukawa, K.; Furukawa, K. Differential roles of gangliosides in malignant properties of melanomas. PLoS ONE 2018, 13, e0206881. [Google Scholar] [CrossRef]
- Saha, S.; Mohanty, K.C. Enhancement of metastatic potential of mouse B16-melanoma cells to lung after treatment with gangliosides of B-16-melanoma cells of higher metastatic potential to lung. Indian J. Exp. Biol. 2003, 41, 1253–1258. [Google Scholar] [PubMed]
- Liu, J.W.; Sun, P.; Yan, Q.; Paller, A.S.; Gerami, P.; Ho, N.; Vashi, N.; Le Poole, I.C.; Wang, X.Q. De-N-acetyl GM3 promotes melanoma cell migration and invasion through urokinase plasminogen activator receptor signaling-dependent MMP-2 activation. Cancer Res. 2009, 69, 8662–8669. [Google Scholar] [CrossRef]
- Levade, T.; Andrieu-Abadie, N.; Micheau, O.; Legembre, P.; Segui, B. Sphingolipids modulate the epithelial-mesenchymal transition in cancer. Cell Death Discov. 2015, 1, 1–2. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef]
- Hosain, S.B.; Khiste, S.K.; Uddin, M.B.; Vorubindi, V.; Ingram, C.; Zhang, S.; Hill, R.A.; Gu, X.; Liu, Y.Y. Inhibition of glucosylceramide synthase eliminates the oncogenic function of p53 R273H mutant in the epithelial-mesenchymal transition and induced pluripotency of colon cancer cells. Oncotarget 2016, 7, 60575–60592. [Google Scholar] [CrossRef]
- Bedia, C.; Casas, J.; Andrieu-Abadie, N.; Fabrias, G.; Levade, T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. J. Biol. Chem. 2011, 286, 28200–28209. [Google Scholar] [CrossRef]
- Ishitsuka, A.; Fujine, E.; Mizutani, Y.; Tawada, C.; Kanoh, H.; Banno, Y.; Seishima, M. FTY720 and cisplatin synergistically induce the death of cisplatin-resistant melanoma cells through the downregulation of the PI3K pathway and the decrease in epidermal growth factor receptor expression. Int. J. Mol. Med. 2014, 34, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Abe, N.; Kanoh, H.; Banno, Y.; Seishima, M. Synergistic effects of vemurafenib and fingolimod (FTY720) in vemurafenibresistant melanoma cell lines. Mol. Med. Rep. 2018, 18, 5151–5158. [Google Scholar] [CrossRef] [PubMed]
- Garandeau, D.; Noujarede, J.; Leclerc, J.; Imbert, C.; Garcia, V.; Bats, M.L.; Rambow, F.; Gilhodes, J.; Filleron, T.; Meyer, N.; et al. Targeting the Sphingosine 1-Phosphate axis exerts potent antitumor activity in BRAFi-resistant melanomas. Mol. Cancer Ther. 2019, 18, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-survival lipid sphingosine-1-phosphate metabolically programs T cells to limit anti-tumor activity. Cell Rep. 2019, 28, 1879–1893. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Hettmer, S.; Smith, P.; Ladisch, S. Inhibition of melanoma tumor growth by a novel inhibitor of glucosylceramide synthase. Cancer Res. 2003, 63, 3654–3658. [Google Scholar] [PubMed]
- Guthmann, M.D.; Bitton, R.J.; Carnero, A.J.; Gabri, M.R.; Cinat, G.; Koliren, L.; Lewi, D.; Fernandez, L.E.; Alonso, D.F.; Gomez, D.E.; et al. Active specific immunotherapy of melanoma with a GM3 ganglioside-based vaccine: A report on safety and immunogenicity. J. Immunother. 2004, 27, 442–451. [Google Scholar] [CrossRef]
- Irie, R.F.; Ollila, D.W.; O’Day, S.; Morton, D.L. Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol. Immunother. 2004, 53, 110–117. [Google Scholar] [CrossRef]
- Zhang, P.; Fu, C.; Hu, Y.; Dong, C.; Song, Y.; Song, E. C6-ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCzeta tumor-suppressive activities and regulating integrin affinity modulation. Sci. Rep. 2015, 5, 1–16. [Google Scholar] [CrossRef]
- Tran, M.A.; Smith, C.D.; Kester, M.; Robertson, G.P. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin. Cancer Res. 2008, 14, 3571–3581. [Google Scholar] [CrossRef][Green Version]
- Riscal, R.; Skuli, N.; Simon, M.C. Even cancer cells watch their cholesterol. Mol. Cell 2019, 76, 220–231. [Google Scholar] [CrossRef]
- Sharma, B.; Agnihotri, N. Role of cholesterol homeostasis and its efflux pathways in cancer progression. J. Steroid. Biochem. Mol. Biol. 2019, 191, 1–11. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Hasse, S.; Rokos, H.; Chavan, B.; Shalbaf, M.; Spencer, J.D.; Wood, J.M. Cholesterol regulates melanogenesis in human epidermal melanocytes and melanoma cells. Exp. Dermatol. 2009, 18, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E.; Jansen, M. Cellular sterol trafficking and metabolism: Spotlight on structure. Curr. Opin. Cell Biol. 2008, 20, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Furukawa, K.; Hamamura, K.; Furukawa, K. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: Induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011, 71, 4989–4997. [Google Scholar] [CrossRef]
- Tian, W.; Pang, W.; Ge, Y.; He, X.; Wang, D.; Li, X.; Hou, H.; Zhou, D.; Feng, S.; Chen, Z.; et al. Hepatocyte-generated 27-hydroxycholesterol promotes the growth of melanoma by activation of estrogen receptor alpha. J. Cell Biochem. 2018, 119, 2929–2938. [Google Scholar] [CrossRef]
- Pencheva, N.; Buss, C.G.; Posada, J.; Merghoub, T.; Tavazoie, S.F. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell 2014, 156, 986–1001. [Google Scholar] [CrossRef]
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. Low neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Lee, C.S.; Park, M.; Han, J.; Lee, J.H.; Bae, I.H.; Choi, H.; Son, E.D.; Park, Y.H.; Lim, K.M. Liver X receptor activation inhibits melanogenesis through the acceleration of ERK-mediated MITF degradation. J. Invest. Dermatol. 2013, 133, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Kinslechner, K.; Schorghofer, D.; Schutz, B.; Vallianou, M.; Wingelhofer, B.; Mikulits, W.; Rohrl, C.; Hengstschlager, M.; Moriggl, R.; Stangl, H.; et al. Malignant phenotypes in metastatic melanoma are governed by SR-BI and its association with glycosylation and STAT5 activation. Mol. Cancer Res. 2018, 16, 135–146. [Google Scholar] [CrossRef]
- Kinslechner, K.; Schutz, B.; Pistek, M.; Rapolter, P.; Weitzenbock, H.P.; Hundsberger, H.; Mikulits, W.; Grillari, J.; Rohrl, C.; Hengstschlager, M.; et al. Loss of SR-BI down-regulates MITF and suppresses extracellular vesicle release in human melanoma. Int. J. Mol. Sci. 2019, 20, 1063. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Schlegel, N.C.; Eichhoff, O.M.; Widmer, D.S.; Praetorius, C.; Einarsson, S.O.; Valgeirsdottir, S.; Bergsteinsdottir, K.; Schepsky, A.; Dummer, R.; et al. Novel MITF targets identified using a two-step DNA microarray strategy. Pigment Cell Melanoma Res. 2008, 21, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, Y.; He, X.; He, Z.; Wang, T.; Wan, L.; Chen, L.; Yan, N. Hydroxypropylbetacyclodextrin attenuates the epithelialtomesenchymal transition via endoplasmic reticulum stress in MDAMB231 breast cancer cells. Mol. Med. Rep. 2020, 21, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fedida-Metula, S.; Feldman, B.; Koshelev, V.; Levin-Gromiko, U.; Voronov, E.; Fishman, D. Lipid rafts couple store-operated Ca2+ entry to constitutive activation of PKB/Akt in a Ca2+/calmodulin-, Src- and PP2A-mediated pathway and promote melanoma tumor growth. Carcinogenesis 2012, 33, 740–750. [Google Scholar] [CrossRef]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef]
- Wang, R.; Bi, J.; Ampah, K.K.; Ba, X.; Liu, W.; Zeng, X. Lipid rafts control human melanoma cell migration by regulating focal adhesion disassembly. Biochim. Biophys. Acta. 2013, 1833, 3195–3205. [Google Scholar] [CrossRef]
- Costa, G.A.; de Souza, S.B.; da Silva Teixeira, L.R.; Okorokov, L.A.; Arnholdt, A.C.V.; Okorokova-Facanha, A.L.; Facanha, A.R. Tumor cell cholesterol depletion and V-ATPase inhibition as an inhibitory mechanism to prevent cell migration and invasiveness in melanoma. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Caldieri, G.; Giacchetti, G.; Beznoussenko, G.; Attanasio, F.; Ayala, I.; Buccione, R. Invadopodia biogenesis is regulated by caveolin-mediated modulation of membrane cholesterol levels. J. Cell. Mol. Med. 2009, 13, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Ayee, M.A.; Levitan, I. Paradoxical impact of cholesterol on lipid packing and cell stiffness. Front. Biosci. (Landmark Ed.) 2016, 21, 1245–1259. [Google Scholar] [CrossRef]
- Zalba, S.; Ten Hagen, T.L. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef]
- Zhao, Z.; Hao, D.; Wang, L.; Li, J.; Meng, Y.; Li, P.; Wang, Y.; Zhang, C.; Zhou, H.; Gardner, K.; et al. CtBP promotes metastasis of breast cancer through repressing cholesterol and activating TGF-beta signaling. Oncogene 2019, 38, 2076–2091. [Google Scholar] [CrossRef]
- Zhao, W.; Prijic, S.; Urban, B.C.; Tisza, M.J.; Zuo, Y.; Li, L.; Tan, Z.; Chen, X.; Mani, S.A.; Chang, J.T. Candidate antimetastasis drugs suppress the metastatic capacity of breast cancer cells by reducing membrane fluidity. Cancer Res. 2016, 76, 2037–2049. [Google Scholar] [CrossRef]
- Solomon, K.R.; Freeman, M.R. Do the cholesterol-lowering properties of statins affect cancer risk? Trends Endocrinol. Metab. 2008, 19, 113–121. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Newton, C.C.; Thun, M.J.; Gapstur, S.M. Long-term use of cholesterol-lowering drugs and cancer incidence in a large United States cohort. Cancer Res. 2011, 71, 1763–1771. [Google Scholar] [CrossRef]
- Farwell, W.R.; Scranton, R.E.; Lawler, E.V.; Lew, R.A.; Brophy, M.T.; Fiore, L.D.; Gaziano, J.M. The association between statins and cancer incidence in a veterans population. J. Natl. Cancer Inst. 2008, 100, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.R.; Drake, A.L.; Heilig, L.F.; Graber, M.; McNealy, K.; Schilling, L.M.; Dellavalle, R.P. Statins, fibrates, and melanoma risk: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2006, 98, 1538–1546. [Google Scholar] [CrossRef]
- Farooqi, M.A.M.; Malhotra, N.; Mukherjee, S.D.; Sanger, S.; Dhesy-Thind, S.K.; Ellis, P.; Leong, D.P. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2018, 13, e0209486. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Kim, H.S.; Kim, J.H.; Lee, J. The effect of statin added to systemic anticancer therapy: A meta-analysis of randomized, controlled trials. J. Clin. Med. 2018, 7, 325. [Google Scholar] [CrossRef]
- Koomen, E.R.; Joosse, A.; Herings, R.M.; Casparie, M.K.; Bergman, W.; Nijsten, T.; Guchelaar, H.J. Is statin use associated with a reduced incidence, a reduced Breslow thickness or delayed metastasis of melanoma of the skin? Eur. J. Cancer 2007, 43, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- von Schuckmann, L.A.; Khosrotehrani, K.; Ghiasvand, R.; Hughes, M.C.B.; van der Pols, J.C.; Malt, M.; Smithers, B.M.; Green, A.C. Statins may reduce disease recurrence in patients with ulcerated primary melanoma. Br. J. Dermatol. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Saito, N.; Mol, W.; Furukawa, H.; Tsutsumida, A.; Oyama, A.; Sekido, M.; Sasaki, S.; Yamamoto, Y. Simvastatin inhibits growth via apoptosis and the induction of cell cycle arrest in human melanoma cells. Melanoma Res. 2008, 18, 85–94. [Google Scholar] [CrossRef]
- Favero, G.M.; Otuki, M.F.; Oliveira, K.A.; Bohatch, M.S.J.; Borelli, P.; Barros, F.E.; Maria, D.A.; Fernandes, D.; Bydlowski, S.P. Simvastatin impairs murine melanoma growth. Lipids Health Dis. 2010, 9, 2–8. [Google Scholar] [CrossRef]
- Glynn, S.A.; O’Sullivan, D.; Eustace, A.J.; Clynes, M.; O’Donovan, N. The 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, simvastatin, lovastatin and mevastatin inhibit proliferation and invasion of melanoma cells. BMC Cancer 2008, 8, 1–9. [Google Scholar] [CrossRef]
- Depasquale, I.; Wheatley, D.N. Action of Lovastatin (Mevinolin) on an in vitro model of angiogenesis and its co-culture with malignant melanoma cell lines. Cancer Cell Int. 2006, 6, 1–12. [Google Scholar] [CrossRef]
- Collisson, E.A.; Kleer, C.; Wu, M.; De, A.; Gambhir, S.S.; Merajver, S.D.; Kolodney, M.S. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol. Cancer Ther. 2003, 2, 941–948. [Google Scholar] [PubMed]
- Kidera, Y.; Tsubaki, M.; Yamazoe, Y.; Shoji, K.; Nakamura, H.; Ogaki, M.; Satou, T.; Itoh, T.; Isozaki, M.; Kaneko, J.; et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J. Exp. Clin. Cancer Res. 2010, 29, 1–11. [Google Scholar] [CrossRef]
- Pich, C.; Teiti, I.; Rochaix, P.; Mariame, B.; Couderc, B.; Favre, G.; Tilkin-Mariame, A.F. Statins reduce melanoma development and metastasis through MICA overexpression. Front. Immunol. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Warita, K.; Warita, T.; Beckwitt, C.H.; Schurdak, M.E.; Vazquez, A.; Wells, A.; Oltvai, Z.N. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci. Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G. Targeting the mevalonate pathway for improved anticancer therapy. Curr. Cancer Drug Targets 2009, 9, 626–638. [Google Scholar] [CrossRef]
- Sarrabayrouse, G.; Pich, C.; Teiti, I.; Tilkin-Mariame, A.F. Regulatory properties of statins and rho gtpases prenylation inhibitiors to stimulate melanoma immunogenicity and promote anti-melanoma immune response. Int. J. Cancer 2017, 140, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Tilkin-Mariame, A.F.; Cormary, C.; Ferro, N.; Sarrabayrouse, G.; Lajoie-Mazenc, I.; Faye, J.C.; Favre, G. Geranylgeranyl transferase inhibition stimulates anti-melanoma immune response through MHC class I and costimulatory molecule expression. FASEB J. 2005, 19, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Sarrabayrouse, G.; Pich, C.; Moriez, R.; Armand-Labit, V.; Rochaix, P.; Favre, G.; Tilkin-Mariame, A.F. Melanoma cells treated with GGTI and IFN-gamma allow murine vaccination and enhance cytotoxic response against human melanoma cells. PLoS ONE 2010, 5, e9043. [Google Scholar] [CrossRef]
- de Medina, P.; Paillasse, M.R.; Segala, G.; Voisin, M.; Mhamdi, L.; Dalenc, F.; Lacroix-Triki, M.; Filleron, T.; Pont, F.; Saati, T.A.; et al. Dendrogenin a arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin a drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Gowda, R.; Madhunapantula, S.V.; Kuzu, O.F.; Sharma, A.; Robertson, G.P. Targeting multiple key signaling pathways in melanoma using leelamine. Mol. Cancer Ther. 2014, 13, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Gowda, R.; Sharma, A.; Robertson, G.P. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol. Cancer Ther. 2014, 13, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.; Inamdar, G.S.; Kuzu, O.; Dinavahi, S.S.; Krzeminski, J.; Battu, M.B.; Voleti, S.R.; Amin, S.; Robertson, G.P. Identifying the structure-activity relationship of leelamine necessary for inhibiting intracellular cholesterol transport. Oncotarget 2017, 8, 28260–28277. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.; Madhunapantula, S.V.; Sharma, A.; Kuzu, O.F.; Robertson, G.P. Nanolipolee-007, a novel nanoparticle-based drug containing leelamine for the treatment of melanoma. Mol. Cancer Ther. 2014, 13, 2328–2340. [Google Scholar] [CrossRef]
- Chen, Y.C.; Gowda, R.; Newswanger, R.K.; Leibich, P.; Fell, B.; Rosenberg, G.; Robertson, G.P. Targeting cholesterol transport in circulating melanoma cells to inhibit metastasis. Pigment Cell Melanoma Res. 2017, 30, 541–552. [Google Scholar] [CrossRef]
- Lambeau, G.; Lazdunski, M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 1999, 20, 162–170. [Google Scholar] [CrossRef]
- Falchi, M.; Bataille, V.; Hayward, N.K.; Duffy, D.L.; Bishop, J.A.; Pastinen, T.; Cervino, A.; Zhao, Z.Z.; Deloukas, P.; Soranzo, N.; et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat. Genet. 2009, 41, 915–919. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Meyer, S.; Vogt, T.; Landthaler, M.; Berand, A.; Reichle, A.; Bataille, F.; Marx, A.H.; Menz, A.; Hartmann, A.; Kunz-Schughart, L.A.; et al. Cyclooxygenase 2 (COX2) and peroxisome proliferator-activated receptor gamma (PPARG) are stage-dependent prognostic markers of malignant melanoma. PPAR Res. 2009, 2009, 1–11. [Google Scholar] [CrossRef][Green Version]
- Minisini, A.M.; Pascoletti, G.; Intersimone, D.; Poletto, E.; Driol, P.; Spizzo, R.; Scott, C.A.; Puglisi, F.; Fasola, G.; Di Loreto, C. Expression of thymidine phosphorylase and cyclooxygenase-2 in melanoma. Melanoma Res. 2013, 23, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Tudor, D.V.; Baldea, I.; Lupu, M.; Kacso, T.; Kutasi, E.; Hopartean, A.; Stretea, R.; Gabriela Filip, A. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol. Med. 2020, 17, 20–31. [Google Scholar] [CrossRef]
- Cahlin, C.; Gelin, J.; Delbro, D.; Lonnroth, C.; Doi, C.; Lundholm, K. Effect of cyclooxygenase and nitric oxide synthase inhibitors on tumor growth in mouse tumor models with and without cancer cachexia related to prostanoids. Cancer Res. 2000, 60, 1742–1749. [Google Scholar] [PubMed]
- Denkert, C.; Kobel, M.; Berger, S.; Siegert, A.; Leclere, A.; Trefzer, U.; Hauptmann, S. Expression of cyclooxygenase 2 in human malignant melanoma. Cancer Res. 2001, 61, 303–308. [Google Scholar] [PubMed]
- Vogt, T.; McClelland, M.; Jung, B.; Popova, S.; Bogenrieder, T.; Becker, B.; Rumpler, G.; Landthaler, M.; Stolz, W. Progression and NSAID-induced apoptosis in malignant melanomas are independent of cyclooxygenase II. Melanoma Res. 2001, 11, 587–599. [Google Scholar] [CrossRef]
- Goulet, A.C.; Einsphar, J.G.; Alberts, D.S.; Beas, A.; Burk, C.; Bhattacharyya, A.; Bangert, J.; Harmon, J.M.; Fujiwara, H.; Koki, A.; et al. Analysis of cyclooxygenase 2 (COX-2) expression during malignant melanoma progression. Cancer Biol. Ther. 2003, 2, 713–718. [Google Scholar] [CrossRef]
- Panza, E.; De Cicco, P.; Ercolano, G.; Armogida, C.; Scognamiglio, G.; Anniciello, A.M.; Botti, G.; Cirino, G.; Ianaro, A. Differential expression of cyclooxygenase-2 in metastatic melanoma affects progression free survival. Oncotarget 2016, 7, 57077–57085. [Google Scholar] [CrossRef]
- Inada, M.; Takita, M.; Yokoyama, S.; Watanabe, K.; Tominari, T.; Matsumoto, C.; Hirata, M.; Maru, Y.; Maruyama, T.; Sugimoto, Y.; et al. Direct melanoma cell contact induces stromal cell autocrine prostaglandin E2-EP4 receptor signaling that drives tumor growth, angiogenesis, and metastasis. J. Biol. Chem. 2015, 290, 29781–29793. [Google Scholar] [CrossRef]
- Vo, B.T.; Morton, D.J.; Komaragiri, S.; Millena, A.C.; Leath, C.; Khan, S.A. TGF-beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology 2013, 154, 1768–1779. [Google Scholar] [CrossRef]
- Neil, J.R.; Johnson, K.M.; Nemenoff, R.A.; Schiemann, W.P. Cox-2 inactivates smad signaling and enhances EMT stimulated by TGF-beta through a PGE2-dependent mechanisms. Carcinogenesis 2008, 29, 2227–2235. [Google Scholar] [CrossRef]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q.; et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/beta-catenin signalling pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef]
- Kim, J.Y.; Shin, J.Y.; Kim, M.R.; Hann, S.K.; Oh, S.H. siRNA-mediated knock-down of COX-2 in melanocytes suppresses melanogenesis. Exp. Dermatol. 2012, 21, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Eo, S.H.; Kim, S.J. Resveratrol-mediated inhibition of cyclooxygenase-2 in melanocytes suppresses melanogenesis through extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/Akt signalling. Eur. J. Pharmacol. 2019, 860, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; De Cicco, P.; Rubino, V.; Terrazzano, G.; Ruggiero, G.; Carriero, R.; Kunderfranco, P.; Ianaro, A. Knockdown of PTGS2 by CRISPR/CAS9 system designates a new potential gene target for melanoma treatment. Front. Pharmacol. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Roszik, J.; Cho, S.N.; Ogata, D.; Milton, D.R.; Peng, W.; Menter, D.G.; Ekmekcioglu, S.; Grimm, E.A. The COX2 effector microsomal PGE2 synthase 1 is a regulator of immunosuppression in cutaneous melanoma. Clin. Cancer Res. 2019, 25, 1650–1663. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe-Donk, J.; Namboodiri, K.K. Inverse association of non-steroidal anti-inflammatory drugs and malignant melanoma among women. Oncol. Rep. 2001, 8, 655–657. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe-Donk, J.; Doss, H.; Burr Doss, D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: A critical review of non-selective COX-2 blockade (review). Oncol. Rep. 2005, 13, 559–583. [Google Scholar] [CrossRef]
- Lejeune, F.J.; Monnier, Y.; Ruegg, C. Complete and long-lasting regression of disseminated multiple skin melanoma metastases under treatment with cyclooxygenase-2 inhibitor. Melanoma Res. 2006, 16, 263–265. [Google Scholar] [CrossRef]
- Duff, M.; Stapleton, P.P.; Mestre, J.R.; Maddali, S.; Smyth, G.P.; Yan, Z.; Freeman, T.A.; Daly, J.M. Cyclooxygenase-2 inhibition improves macrophage function in melanoma and increases the antineoplastic activity of interferon gamma. Ann. Surg. Oncol. 2003, 10, 305–313. [Google Scholar] [CrossRef]
- Pritchard, R.; Rodriguez-Enriquez, S.; Pacheco-Velazquez, S.C.; Bortnik, V.; Moreno-Sanchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Gobel, C.; Breitenbuecher, F.; Kalkavan, H.; Hahnel, P.S.; Kasper, S.; Hoffarth, S.; Merches, K.; Schild, H.; Lang, K.S.; Schuler, M. Functional expression cloning identifies COX-2 as a suppressor of antigen-specific cancer immunity. Cell Death Dis. 2014, 5, e1568. [Google Scholar] [CrossRef] [PubMed]
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; Plaen, E.; Van den Eynde, B.J. Constitutive IDO1 Expression in human tumors is driven by cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol. Res. 2017, 5, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Katz, J.B.; Muller, A.J.; Prendergast, G.C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol. Rev. 2008, 222, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Ellebaek, E.; Engell-Noerregaard, L.; Iversen, T.Z.; Froesig, T.M.; Munir, S.; Hadrup, S.R.; Andersen, M.H.; Svane, I.M. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: Results from a phase II trial. Cancer Immunol. Immunother. 2012, 61, 1791–1804. [Google Scholar] [CrossRef]
- Neumann, S.; Shirley, S.A.; Kemp, R.A.; Hook, S.M. Improved antitumor activity of a therapeutic melanoma vaccine through the use of the dual COX-2/5-LO inhibitor licofelone. Front. Immunol. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Nath, A.; Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep. 2016, 6, 18669. [Google Scholar] [CrossRef]
- Warner, A.B.; McQuade, J.L. Modifiable host factors in melanoma: Emerging evidence for obesity, diet, exercise, and the microbiome. Curr. Oncol. Rep. 2019, 21, 72. [Google Scholar] [CrossRef]
- Sergentanis, T.N.; Antoniadis, A.G.; Gogas, H.J.; Antonopoulos, C.N.; Adami, H.O.; Ekbom, A.; Petridou, E.T. Obesity and risk of malignant melanoma: A meta-analysis of cohort and case-control studies. Eur. J. Cancer 2013, 49, 642–657. [Google Scholar] [CrossRef]
- Skowron, F.; Berard, F.; Balme, B.; Maucort-Boulch, D. Role of obesity on the thickness of primary cutaneous melanoma. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 262–269. [Google Scholar] [CrossRef]
- Stenehjem, J.S.; Veierod, M.B.; Nilsen, L.T.; Ghiasvand, R.; Johnsen, B.; Grimsrud, T.K.; Babigumira, R.; Stoer, N.C.; Rees, J.R.; Robsahm, T.E. Anthropometric factors and Breslow thickness: Prospective data on 2570 cases of cutaneous melanoma in the population-based janus cohort. Br. J. Dermatol. 2018, 179, 632–641. [Google Scholar] [CrossRef]
- Fang, S.; Wang, Y.; Dang, Y.; Gagel, A.; Ross, M.I.; Gershenwald, J.E.; Cormier, J.N.; Wargo, J.; Haydu, L.E.; Davies, M.A.; et al. Association between body mass index, C-Reactive protein levels, and melanoma patient outcomes. J. Invest. Dermatol. 2017, 137, 1792–1795. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Muller, C.; Nieto, L. Obesity and melanoma: Could fat be fueling malignancy? Pigment. Cell Melanoma. Res. 2017, 30, 294–306. [Google Scholar] [CrossRef]
- Smith, L.K.; Arabi, S.; Lelliott, E.J.; McArthur, G.A.; Sheppard, K.E. Obesity and the impact on cutaneous melanoma: Friend or foe? Cancers 2020, 12, 1583. [Google Scholar] [CrossRef]
- Dobbins, M.; Decorby, K.; Choi, B.C. The association between obesity and cancer risk: A meta-analysis of observational studies from 1985 to 2011. ISRN Prev. Med. 2013, 2013, 680536. [Google Scholar] [CrossRef]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D.; Million Women Study Collaboration. Cancer incidence and mortality in relation to body mass index in the million women study: Cohort study. BMJ 2007, 335, 1134. [Google Scholar] [CrossRef]
- Gallus, S.; Naldi, L.; Martin, L.; Martinelli, M.; La Vecchia, C.; Oncology Study Group of the Italian Group for Epidemiologic Research in Dermatology. Anthropometric measures and risk of cutaneous malignant melanoma: A case-control study from Italy. Melanoma Res. 2006, 16, 83–87. [Google Scholar] [CrossRef]
- Rousseau, M.C.; Parent, M.E.; Siemiatycki, J. Comparison of self-reported height and weight by cancer type among men from Montreal, Canada. Eur. J. Cancer Prev. 2005, 14, 431–438. [Google Scholar] [CrossRef]
- Shors, A.R.; Solomon, C.; McTiernan, A.; White, E. Melanoma risk in relation to height, weight, and exercise (United States). Cancer Causes Control 2001, 12, 599–606. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet. Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, inflammation, and cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Attane, C.; Muller, C.; Nieto, L. A new role for extracellular vesicles: How small vesicles can feed tumors’ big appetite. J. Lipid Res. 2018, 59, 1793–1804. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef]
- Wang, J.; Badeanlou, L.; Bielawski, J.; Ciaraldi, T.P.; Samad, F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E756–E768. [Google Scholar] [CrossRef]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS ONE 2013, 8, e72449. [Google Scholar] [CrossRef]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of obesity and metabolic syndrome on immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Clements, V.K.; Long, T.; Long, R.; Figley, C.; Smith, D.M.C.; Ostrand-Rosenberg, S. Frontline science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef]
- Ko, J.H.; Um, J.Y.; Lee, S.G.; Yang, W.M.; Sethi, G.; Ahn, K.S. Conditioned media from adipocytes promote proliferation, migration, and invasion in melanoma and colorectal cancer cells. J. Cell Physiol. 2019, 234, 18249–18261. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Mergian, T.A.; Cho, K.W.; Martinez-Santibanez, G.; Luan, D.; Singer, K.; DelProposto, J.L.; Geletka, L.M.; Muir, L.A.; Lumeng, C.N. Macrophage proliferation sustains adipose tissue inflammation in formerly obese mice. Diabetes 2017, 66, 392–406. [Google Scholar] [CrossRef]
- Zecchin, K.G.; Rossato, F.A.; Raposo, H.F.; Melo, D.R.; Alberici, L.C.; Oliveira, H.C.; Castilho, R.F.; Coletta, R.D.; Vercesi, A.E.; Graner, E. Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab. Invest. 2011, 91, 232–240. [Google Scholar] [CrossRef]
- Ji, C.; Yang, Y.L.; He, L.; Gu, B.; Xia, J.P.; Sun, W.L.; Su, Z.L.; Chen, B.; Bi, Z.G. Increasing ceramides sensitizes genistein-induced melanoma cell apoptosis and growth inhibition. Biochem. Biophys. Res. Commun. 2012, 421, 462–467. [Google Scholar] [CrossRef]
- Kumar, D.; Rahman, H.; Tyagi, E.; Liu, T.; Li, C.; Lu, R.; Lum, D.; Holmen, S.L.; Maschek, J.A.; Cox, J.E.; et al. Aspirin suppresses PGE2 and activates AMP kinase to inhibit melanoma cell motility, pigmentation, and selective tumor growth in vivo. Cancer Prev. Res. (Phila.) 2018, 11, 629–642. [Google Scholar] [CrossRef]
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J. Transl. Med. 2017, 15, 1–12. [Google Scholar] [CrossRef]
- Gowda, R.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Simultaneous targeting of COX-2 and AKT using selenocoxib-1-GSH to inhibit melanoma. Mol. Cancer Ther. 2013, 12, 3–15. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Lipid Classes | Genes | Expression in Melanoma | Outcome | References |
|---|---|---|---|---|
| FA | ACLY | Overexpressed | Worse prognosis | [5,6] |
| ACSL3 | Overexpressed | Worse prognosis | [15] | |
| CD36 | Amplified in metastasis | Worse prognosis | [210] | |
| FAO (3 genes) | Overexpressed | Worse prognosis | [25] | |
| FASN | Overexpressed | Worse prognosis | [5] | |
| SCD | Overexpressed | Worse prognosis | [5] | |
| SL | SGMS1 | Downregulated | Worse prognosis | [83] |
| SPHK1 | Overexpressed | Shorter survival after anti-PD1 | [89] | |
| Sterols | Cholesterol synthesis (7 genes) | Overexpressed | Worse prognosis | [22] |
| Eicosanoids | COX-2 | Overexpressed in primary melanoma | Decreased PFS; poor prognosis factors (thicker melanoma, high mitotic count) | [182,183] |
| PTGES | Overexpressed | Worse prognosis (stage III melanoma) | [198] |
| Targeted Enzyme/Receptor | Agent | Melanoma Cells/Models | Effects | Reference |
|---|---|---|---|---|
| FASN | Orlistat | B16F10 | Reduced number of metastasis in mice | [14] |
| Cerulenin | Reduced proliferation. Increased apoptosis | [234] | ||
| MAGL | JZL184 | C8161 | Decreased cell migration | [50] |
| SCD1 | A939572 | IGR37 501mel IGR39 A375M | IGR37, 501mel: Decreased cell proliferation Increased apoptosis IGR39, A375M: No effect | [49] |
| CAY10566 | M381 | Apoptosis | [53] | |
| FATP2 | Lipofermata | WM793 1205Lu Yumm 1.7 | Sensitization of melanoma cells in an aged TME to BRAF and MEK inhibitors Increased survival in old animals | [31] |
| FATP1 | Zebrafish | Reduced melanoma growth and invasion | [28] | |
| CPT1/ECHA (FAO) | Etomoxir/Trimetazidine | SK-Mel-28 1205Lu | Reduced melanoma migration | [29] |
| AC | ARN14988 ARN398 | G361 A375 | Sensitization of proliferative but not invasive cells to 5-FU | [84] |
| SphK1 | FTY720 | SK-Mel-28 A375 | Increased cisplatin-induced apoptosis | [113] |
| WM115 SK-Mel-28 | Increased vemurafenib-induced apoptosis | [114] | ||
| SKI-I | WM9 | Reduced proliferation of vemurafenib-resistant cells | [115] | |
| Yumm 1.7 | Increased efficacy of anti-PD1 and anti-CTLA-4 in mice | [89] | ||
| PF-543 | B16F10 | Increased efficacy of anti-PD1 in mice | [116] | |
| GCS | PDMP | B16F10 | Increased genistein-induced apoptosis | [235] |
| OGT2378 | MEB4 | Reduced tumor growth in mice | [117] | |
| LXRβ | GW3965 T0901317 | SK-Mel-2 SK-Mel-334.2 B16F10 | Reduced cell invasion and sensitization to vemurafenib in vitro. Reduced tumor growth, angiogenesis and metastasis in vivo | [131] |
| ChEH LXRβ | DDA | B16F10, SK-MEL-28 | Increased autophagy. Reduced tumor growth in mice | [172] |
| HMGCR | Simvastatin | A375M, G361, C8161, GAK, MMAc | Cell cycle arrest and increased apoptosis | [159] |
| B16F10 | Dose-dependent cell cycle arrest. Reduced tumor growth in mice | [160] | ||
| Lovastatin, Mevastatin, Simvastatin | HT144, M14, SK-MEL-28 | Reduced cell growth, migration and invasion | [161] | |
| Fluvastatin, Simvastatin | B16BL6 | Reduced cell migration, adhesion and invasion in vitro and metastasis in mice | [164] | |
| Lovastatin | LB1319-MEL, BB74-MEL, LB2033-MEL, LB583-MEL | Increased expression of MHC class I Chain-related protein A (MICA) | [165] | |
| A375 and G361 | Reduced cell growth and angiogenesis and increased apoptosis | [162] | ||
| Atorvastatin | A375M, SK-MEL-28, WM-266-4 | Reduced invasion in vitro and in mice | [163] | |
| NPC1 | Leelamine | Nine human melanoma cell lines | Reduced cell proliferation in vitro and tumor growth in mice | [173] |
| COX-2 | NS-398 | B16F10 | Reduced cell growth and improved survival in mice | [202] |
| Aspirin | Melanoma PDX cell lines A375, B16F10 | Reduced cell motility, pigmentation in vitro, and tumor growth in immunodeficient mice | [236] | |
| Celecoxib | SK-Mel-5 | Reduced cell proliferation | [189] | |
| B16F10 | Increased ROS-dependent apoptosis | [203] | ||
| KUL98-MELA | Rejection of IDO1-expressing human tumor xenografts in modified immunodeficient mice | [205] | ||
| A375, SK-MEL-2 | Reduced PD-L1 expression and cell growth | [237] | ||
| Selenocoxib-1-GSH (analog of celecoxib) | WM35, WM115, WM278.1, A375M, 1205 Lu | Cell cycle arrest and increased apoptosis Reduced tumor growth in mice | [238] | |
| Celecoxib (+cyclophosphamide) | 28 patients with metastatic melanoma | Six-month increase in survival | [208] | |
| COX-2/5-lipoxygenase inhibitor | Licofelone | B16F10 | Improved antitumor activity of a therapeutic melanoma vaccine | [209] |
| Agent | Clinical Trial | Title | Posting Year | Status |
|---|---|---|---|---|
| Lovastatin | NCT00963664 | Evaluation of interferon–lovastatin therapy for malignant melanoma | 2009 | Withdrawn |
| NCT00462280 | Lovastatin in treating patients at high risk of melanoma | 2007 | Completed (with results) | |
| Fluvastatin | NCT04285749 | Prevention of recurrence and metastasis in genetically high-risk melanomas | 2020 | Withdrawn |
| Aspirin | NCT04062032 | Metabolomic and inflammatory effects of oral aspirin (ASA) in subjects at risk for melanoma | 2019 | Recruiting |
| NCT04066725 | Aspirin as an ultraviolet (UV) protectant in human subjects at risk for melanoma | 2019 | Recruiting | |
| NCT03396952 | Prostaglandin inhibition and immune checkpoint blockade in melanoma | 2018 | Active, not recruiting | |
| NCT01753089 | Dendritic-cell-activating scaffold in melanoma | 2012 | Active, not recruiting | |
| Celecoxib | NCT04093323 | Polarized dendritic cell (aDC1) vaccine, interferon alpha-2, rintatolimod, and celecoxib for the treatment of HLA-A2+ refractory melanoma | 2019 | Not yet recruiting |
| NCT01313429 | Tumor-cell vaccine for patients undergoing surgery for sarcomas, melanomas, germ cell tumors or malignancies that have metastasized to the lungs, pleura or mediastinum | 2011 | Recruitment terminated | |
| NCT01341496 | Tumor-cell vaccines and iscomatrix with chemotherapy after tumor removal | 2011 | Recruitment terminated | |
| NCT00197912 | Dendritic-cell-based therapy of malignant melanoma | 2005 | Completed | |
| NCT00093678 | Celecoxib in managing pain, weight loss and weakness in patients with advanced cancer | 2004 | Withdrawn | |
| NCT02839694 | Adjuvant oral decitabine and tetrahydrouridine, with or without celecoxib, in people undergoing pulmonary metastasectomy | 2016 | Withdrawn | |
| NCT02054104 | Adjuvant tumor lysate vaccine and iscomatrix with or without metronomic oral cyclophosphamide and celecoxib in patients with malignancies involving lungs, esophagus, pleura or mediastinum | 2014 | Suspended |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellerin, L.; Carrié, L.; Dufau, C.; Nieto, L.; Ségui, B.; Levade, T.; Riond, J.; Andrieu-Abadie, N. Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives. Cancers 2020, 12, 3147. https://doi.org/10.3390/cancers12113147
Pellerin L, Carrié L, Dufau C, Nieto L, Ségui B, Levade T, Riond J, Andrieu-Abadie N. Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives. Cancers. 2020; 12(11):3147. https://doi.org/10.3390/cancers12113147
Chicago/Turabian StylePellerin, Laurence, Lorry Carrié, Carine Dufau, Laurence Nieto, Bruno Ségui, Thierry Levade, Joëlle Riond, and Nathalie Andrieu-Abadie. 2020. "Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives" Cancers 12, no. 11: 3147. https://doi.org/10.3390/cancers12113147
APA StylePellerin, L., Carrié, L., Dufau, C., Nieto, L., Ségui, B., Levade, T., Riond, J., & Andrieu-Abadie, N. (2020). Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives. Cancers, 12(11), 3147. https://doi.org/10.3390/cancers12113147