Prognostic Significance of Oxidation Pathway Mutations in Recurrent Laryngeal Squamous Cell Carcinoma

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
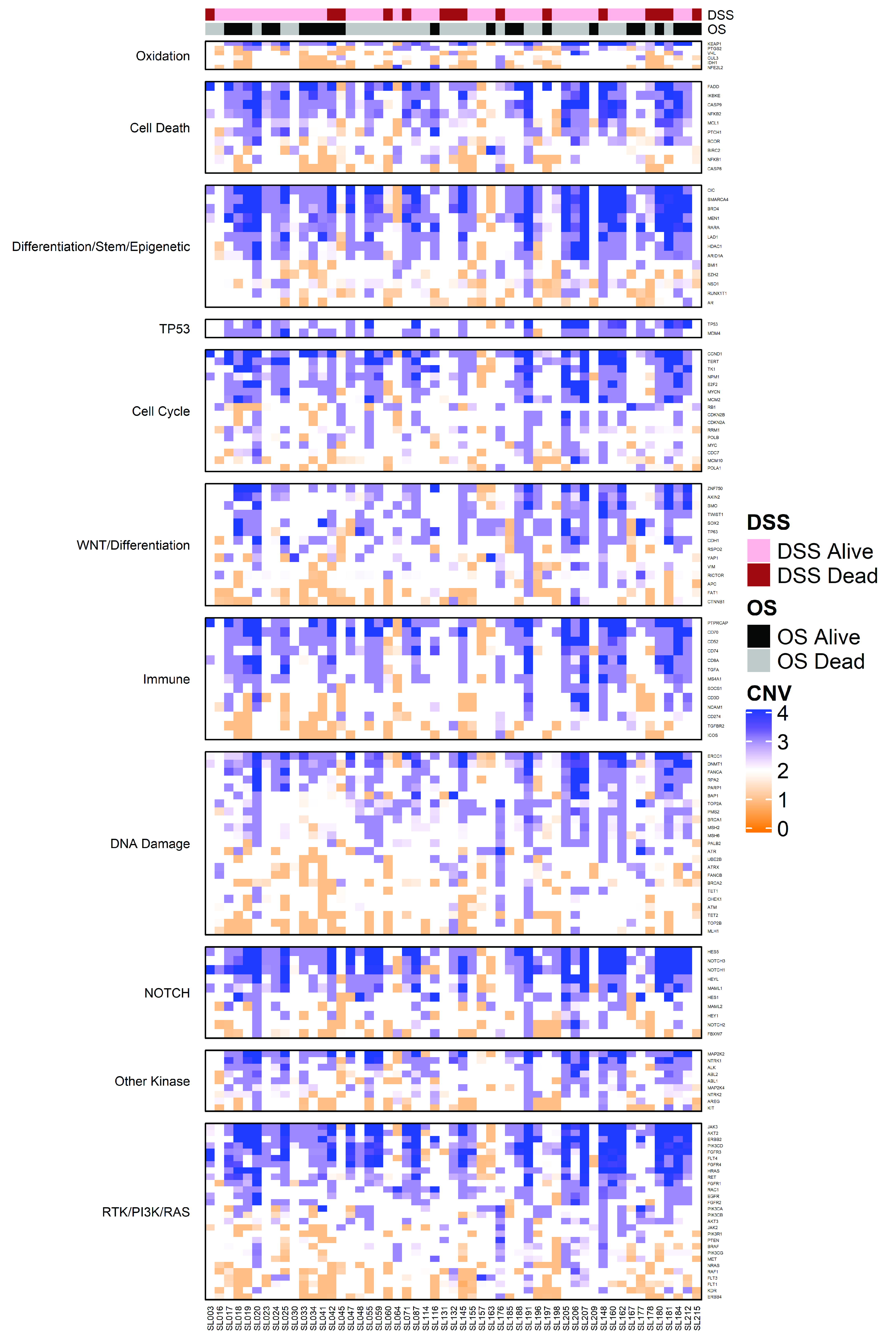
2. Results
2.1. Cohort Demographics and Clinical Variables Predictive of Survival
2.2. Characterization of High Frequency Genomic Alterations
2.3. Predictive Pathways: Mutational Signatures
2.4. Transcriptome Signature Analysis Supports Possible Functional Role of Oxidation Pathway
2.5. High NRF2 Protein Expression in Recurrent Larynx Specimens
2.6. Validation of Oxidation Pathway in TCGA
3. Discussion
4. Materials and Methods
4.1. Cohort Selection
4.2. DNA Extraction
4.3. Targeted DNA Sequencing
4.4. Exome Variant Calling
4.5. Copy Number Analysis
4.6. Transcriptome Sequencing
4.7. Transcriptome Quantification
4.8. TCGA Analysis
4.9. Immunohistochemistry
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forastiere, A.A.; Goepfert, H.; Maor, M.; Pajak, T.F.; Weber, R.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; Chao, C.; et al. Concurrent Chemotherapy and Radiotherapy for Organ Preservation in Advanced Laryngeal Cancer. N. Engl. J. Med. 2003, 349, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Department of Veterans Affairs Laryngeal Cancer Study Group; Wolf, G.T.; Fisher, S.G.; Hong, W.K.; Hillman, R.; Spaulding, M.; Laramore, G.E.; Endicott, J.W.; McClatchey, K.; Henderson, W.G. Induction Chemotherapy plus Radiation Compared with Surgery plus Radiation in Patients with Advanced Laryngeal Cancer. N. Engl. J. Med. 1991, 324, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- Denaro, N.; Russi, E.G.; Lefebvre, J.L.; Merlano, M.C. A systematic review of current and emerging approaches in the field of larynx preservation. Radiother. Oncol. 2014, 110, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.A.; Puram, S.V.; Rocco, J.W.; Old, M.O.; Kang, S. Salvage laryngectomy following organ-preservation therapy—An evidence-based review. Oral Oncol. 2019, 88, 137–144. [Google Scholar] [CrossRef]
- Birkeland, A.C.; Beesley, L.J.; Bellile, E.; Rosko, A.J.; Hoesli, R.; Chinn, S.B.; Shuman, A.G.; Prince, M.E.; Wolf, G.T.; Bradford, C.R.; et al. Predictors of survival after total laryngectomy for recurrent/persistent laryngeal squamous cell carcinoma. Head Neck 2017, 39, 2512–2518. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Bs, L.J.V.; Skinner, H.D.; Cata, J.; Hutcheson, K.; Fuller, C.D.; Phan, J.; Ms, Z.S.; Lai, S.Y.; Weber, R.S.; et al. Salvage total laryngectomy after external-beam radiotherapy: A 20-year experience. Head Neck 2016, 38, E1962–E1968. [Google Scholar] [CrossRef]
- Van Der Putten, L.; De Bree, R.; Kuik, D.; Rietveld, D.; Buter, J.; Eerenstein, S.; Leemans, C. Salvage laryngectomy: Oncological and functional outcome. Oral Oncol. 2011, 47, 296–301. [Google Scholar] [CrossRef]
- Goodwin, W.J. Salvage Surgery for Patients With Recurrent Squamous Cell Carcinoma of the Upper Aerodigestive Tract: When Do the Ends Justify the Means? Laryngoscope 2000, 110, 1–18. [Google Scholar] [CrossRef]
- Fletcher, K.T.; Gal, T.J.; Ebelhar, A.J.; Valentino, J.; Brill, Y.M.; Dressler, E.V.; Aouad, R.K. Prognostic indicators and survival in salvage surgery for laryngeal cancer. Head Neck 2017, 39, 2021–2026. [Google Scholar] [CrossRef]
- Scharpf, J.; Ward, M.; Adelstein, D.; Koyfman, S.; Li, M. Elucidation of salvage laryngectomy pathologic and clinical variables to guide further treatment intensification investigation. Laryngoscope 2017, 128, 823–830. [Google Scholar] [CrossRef]
- Stankovic, M.; Milisavljevic, D.; Zivic, M.; Stojanov, D.; Stankovic, P. Primary and salvage total laryngectomy. Influential factors, complications, and survival. J. BUON Off. J. Balk. Union Oncol. 2015, 20, 527–539. [Google Scholar]
- Wulff, N.B.; Andersen, E.; Kristensen, C.; Sørensen, C.; Charabi, B.; Homøe, P. Prognostic factors for survival after salvage total laryngectomy following radiotherapy or chemoradiation failure: A 10-year retrospective longitudinal study in eastern Denmark. Clin. Otolaryngol. 2016, 42, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Birkeland, A.C.; Rosko, A.J.; Hoesli, R.C.; Foltin, S.K.; Swiecicki, P.; Mierzwa, M.; Chinn, S.B.; Shuman, A.G.; Malloy, K.M.; et al. Mutational profiles of persistent/recurrent laryngeal squamous cell carcinoma. Head Neck 2018, 41, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-J.; Wang, S.-G.; Roh, H.-J.; Goh, E.-K.; Chon, K.-M.; Park, D.-Y. Changes in expression of p53, proliferating cell nuclear antigen and bcl-2 in recurrent laryngeal cancer after radiotherapy. J. Laryngol. Otol. 2006, 120, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Shi, E.; Wang, Q.; Dong, Z.; Xu, P. A potential panel of two-long non-coding RNA signature to predict recurrence of patients with laryngeal cancer. Oncotarget 2017, 8, 69641–69650. [Google Scholar] [CrossRef]
- Lee, M.; Ryu, C.H.; Chang, H.W.; Kim, G.C.; Kim, S.W.; Kim, S.Y. Radiotherapy-associated Furin Expression and Tumor Invasiveness in Recurrent Laryngeal Cancer. Anticancer. Res. 2016, 36, 5117–5126. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wei, G.; Zhang, L.; Xu, Z. Signature microRNAs and long noncoding RNAs in laryngeal cancer recurrence identified using a competing endogenous RNA network. Mol. Med. Rep. 2019, 19, 4806–4818. [Google Scholar] [CrossRef]
- Network, T.C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nat. Cell Biol. 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Hammad, A.; Namani, A.; Elshaer, M.; Wang, X.J.; Tang, X. “NRF2 addiction” in lung cancer cells and its impact on cancer therapy. Cancer Lett. 2019, 467, 40–49. [Google Scholar] [CrossRef]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef]
- Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.-A.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 2014, 112, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Marshak, G.; Sulkes, A.; Rakowsky, E. Prognosis of patients with recurrent laryngeal carcinoma. Head Neck 2001, 23, 531–535. [Google Scholar] [CrossRef]
- Davidson, J.; Keane, T.; Brown, D.; Freeman, J.; Gullane, P.; Irish, J.; Rotstein, L.; Pintilie, M.; Cummings, B. Surgical salvage after radiotherapy for advanced laryngopharyngeal carcinoma. Arch. Otolaryngol. Head Neck Surg. 1997, 123, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, W.H.; Min, Y.J.; Cha, H.J.; Han, M.W.; Chang, H.W.; A Kim, S.; Choi, S.H.; Kim, S.W.; Kim, S.Y. Development of TRAIL Resistance by Radiation-Induced Hypermethylation of DR4 CpG Island in Recurrent Laryngeal Squamous Cell Carcinoma. Int. J. Radiat. Oncol. 2014, 88, 1203–1211. [Google Scholar] [CrossRef]
- Li, M.; Lorenz, R.R.; Khan, M.J.; Burkey, B.B.; Adelstein, D.J.; Greskovich, J.F.; Koyfman, S.A.; Scharpf, J. Salvage Laryngectomy in Patients with Recurrent Laryngeal Cancer in the Setting of Nonoperative Treatment Failure. Otolaryngol. Neck Surg. 2013, 149, 245–251. [Google Scholar] [CrossRef]
- Bednarek, K.; Kiwerska, K.; Szaumkessel, M.; Bodnar, M.; Kostrzewska-Poczekaj, M.; Marszalek, A.; Janiszewska, J.; Bartochowska, A.; Jackowska, J.; Wierzbicka, M.; et al. Recurrent CDK1 overexpression in laryngeal squamous cell carcinoma. Tumor Biol. 2016, 37, 11115–11126. [Google Scholar] [CrossRef]
- Bradford, C.R.; Kumar, B.; Bellile, E.; Lee, J.; Taylor, J.; D’Silva, N.; Cordell, K.; Kleer, C.; Kupfer, R.; Kumar, P.; et al. Biomarkers in advanced larynx cancer. Laryngoscope 2013, 124, 179–187. [Google Scholar] [CrossRef]
- Byzia, E.; Soloch, N.; Bodnar, M.; Szaumkessel, M.; Kiwerska, K.; Kostrzewska-Poczekaj, M.; Jarmuz-Szymczak, M.; Szylberg, Ł.; Wierzbicka, M.; Anna, B.; et al. Recurrent transcriptional loss of the PCDH17 tumor suppressor in laryngeal squamous cell carcinoma is partially mediated by aberrant promoter DNA methylation. Mol. Carcinog. 2018, 57, 878–885. [Google Scholar] [CrossRef]
- Kumar, B.; Cordell, K.G.; D’Silva, N.; Prince, M.E.; Adams, M.E.; Fisher, S.G.; Wolf, G.T.; Carey, T.E.; Bradford, C.R. Expression of p53 and Bcl-xL as Predictive Markers for Larynx Preservation in Advanced Laryngeal Cancer. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Nakayama, K.; Razia, S.; Nakamura, K.; Ishikawa, M.; Minamoto, T.; Ishibashi, T.; Yamashita, H.; Iida, K.; Kyo, S. ARID1B as a Potential Therapeutic Target for ARID1A-Mutant Ovarian Clear Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 1710. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, A.; Weller, P.; Kanaan, O.; Nel, I.; Heusgen, L.; Höing, B.; Haßkamp, P.; Zander, S.; Mandapathil, M.; Dominas, N.; et al. CD31 and VEGF are prognostic biomarkers in early-stage, but not in late-stage, laryngeal squamous cell carcinoma. BMC Cancer 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Starska, K.; Forma, E.; Lewy-Trenda, I.; Stasikowska-Kanicka, O.; Skóra, M.; Brys, M. Fibroblast growth factor receptor 1 and 3 expression is associated with regulatory PI3K/AKT kinase activity, as well as invasion and prognosis, in human laryngeal cancer. Cell. Oncol. 2018, 41, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Sutherland, K.D. “Keaping” a lid on lung cancer: The Keap1-Nrf2 pathway. Cell Cycle 2018, 17, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; König, K.; Michels, S.; Fischer, R.; et al. Clinical and Pathological Characteristics ofKEAP1- andNFE2L2-Mutated Non–Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nat. Cell Biol. 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Wolf, B.; Göbel, G.; Hackl, H.; Fiegl, H. Reduced mRNA expression levels of NFE2L2 are associated with poor outcome in breast cancer patients. BMC Cancer 2016, 16, 821. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nat. Cell Biol. 2014, 511, 543–550. [CrossRef]
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nat. Cell Biol. 2012, 489, 519–525. [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2009, 220, 446–451. [Google Scholar] [CrossRef]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [PubMed]
- Aso, T.; Uozaki, H.; Morita, S.; Kumagai, A.; Watanabe, M. Loss of ARID1A, ARID1B, and ARID2 Expression During Progression of Gastric Cancer. Anticancer. Res. 2015, 35, 6819–6827. [Google Scholar] [PubMed]
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.C.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Meersma, G.J.; Lieftink, C.; Beijersbergen, R.L.; et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef] [PubMed]
- Cajuso, T.; Hänninen, U.A.; Kondelin, J.; Gylfe, A.E.; Tanskanen, T.; Katainen, R.; Pitkänen, E.; Ristolainen, H.; Kaasinen, E.; Taipale, M.; et al. Exome sequencing reveals frequent inactivating mutations inARID1A, ARID1B, ARID2andARID4Ain microsatellite unstable colorectal cancer. Int. J. Cancer 2014, 135, 611–623. [Google Scholar] [CrossRef]
- Ho, A.S.; Ochoa, A.; Jayakumaran, G.; Zehir, A.; Mayor, C.V.; Tepe, J.; Makarov, V.; Dalin, M.G.; He, J.; Bailey, M.; et al. Genetic hallmarks of recurrent/metastatic adenoid cystic carcinoma. J. Clin. Investig. 2019, 129, 4276–4289. [Google Scholar] [CrossRef]
- Vasileiou, G.; Ekici, A.B.; Uebe, S. Chromatin-Remodeling-Factor ARID1B Represses Wnt/beta-Catenin Signaling. Am. J. Human Genet. 2015, 97, 445–456. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; A Boroevich, K.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef]
- Niedermaier, B.; Sak, A.; Zernickel, E.; Xu, S.; Groneberg, M.; Stuschke, M. Targeting ARID1A-mutant colorectal cancer: Depletion of ARID1B increases radiosensitivity and modulates DNA damage response. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Franchi, A.; Gallo, O.; Sardi, I.; Santucci, M. Downregulation of transforming growth factor beta type II receptor in laryngeal carcinogenesis. J. Clin. Pathol. 2001, 54, 201–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nadauld, L.D.; Garcia, S.; Natsoulis, G.; Bell, J.M.; Miotke, L.; Hopmans, E.S.; Xu, H.; Pai, R.K.; Palm, C.; Regan, J.F.; et al. Metastatic tumor evolution and organoid modeling implicate TGFBR2as a cancer driver in diffuse gastric cancer. Genome Biol. 2014, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2017, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, G.; Ma, X.; Xiao, J.; Yu, G.; Yang, C.; Xu, N.; Zhang, B.; Zhou, J.; Ye, Z.; et al. Attenuation of TGFBR2 expression and tumour progression in prostate cancer involve diverse hypoxia-regulated pathways. J. Exp. Clin. Cancer Res. 2018, 37, 89. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.T.; Clavijo, P.E.; Van Waes, C.; Chen, Z. Anti-Tumor Immunity in Head and Neck Cancer: Understanding the Evidence, How Tumors Escape and Immunotherapeutic Approaches. Cancers 2015, 7, 2397–2414. [Google Scholar] [CrossRef]
- Wojtowicz-Praga, S. Reversal of Tumor-Induced Immunosuppression: A New Approach to Cancer Therapy. J. Immunother. 1997, 20, 165–177. [Google Scholar] [CrossRef]
- Hoesli, R.; Birkeland, A.C.; Rosko, A.J.; Issa, M.; Chow, K.L.; Michmerhuizen, N.L.; Mann, J.E.; Chinn, S.B.; Shuman, A.G.; Prince, M.E.; et al. Proportion of CD4 and CD8 tumor infiltrating lymphocytes predicts survival in persistent/recurrent laryngeal squamous cell carcinoma. Oral Oncol. 2018, 77, 83–89. [Google Scholar] [CrossRef]
- Mann, J.E.; Smith, J.D.; Birkeland, A.C.; Bellile, E.; Swiecicki, P.; Mierzwa, M.; Chinn, S.B.; Shuman, A.G.; Malloy, K.M.; Casper, K.A.; et al. Analysis of tumor-infiltrating CD103 resident memory T-cell content in recurrent laryngeal squamous cell carcinoma. Cancer Immunol. Immunother. 2018, 68, 213–220. [Google Scholar] [CrossRef]
- Spector, M.E.; Bellile, E.; Amlani, L.; Zarins, K.; Smith, J.; Brenner, J.C.; Rozek, L.; Nguyen, A.; Thomas, D.; McHugh, J.B.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol. Neck Surg. 2019, 145, 1012–1019. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Tillman, B.N.; Yanik, M.; Birkeland, A.C.; Liu, C.-J.; Ms, D.H.H.; Cani, A.K.; Palanisamy, N.; Ms, S.C.; Carey, T.E.; Bradford, C.R.; et al. Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low-risk patient as defined by targeted sequencing. Head Neck 2016, 38, E1646–E1652. [Google Scholar] [CrossRef]
- Jeddi, F.; Soozangar, N.; Sadeghi, M.R.; Somi, M.H.; Shirmohamadi, M.; Eftekhar-Sadat, A.-T.; Samadi, N. Nrf2 overexpression is associated with P-glycoprotein upregulation in gastric cancer. Biomed. Pharmacother. 2018, 97, 286–292. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Count (%), n = 62 | |
|---|---|---|
| Age (yrs) | 59 (range 39–85) | |
| Gender | Male | 53 (85) |
| Female | 9 (15) | |
| Race | Caucasian | 52 (84) |
| Black | 3 (5) | |
| Other | 7 (11) | |
| Primary Subsite | Supraglottis | 36 (58) |
| Glottis | 25 (40) | |
| Subglottis | 1 (2) | |
| Primary Overall Stage | I | 16 (26) |
| II | 23 (37) | |
| III | 13 (21) | |
| IV | 7 (11) | |
| Primary T Stage | I | 16 (26) |
| II | 27 (44) | |
| III | 11 (18) | |
| IV | 4 (6) | |
| Primary N Stage | N0 | 51 (82) |
| N+ | 8 (13) | |
| Treatment | Radiation | 45 (73) |
| C/RT | 17 (27) | |
| Time to recurrence (mo) | 14 (2–158) | |
| Recurrent Overall Stage | I | 2 (3) |
| II | 24 (39) | |
| III | 14 (23) | |
| IV | 22 (35) | |
| Recurrent T Stage | I | 2 (3) |
| II | 28 (45) | |
| III | 15 (24) | |
| IV | 17 (27) | |
| Recurrent N Stage | N0 | 52 (84) |
| N+ | 10 (16) | |
| Recurrent Subsite | Hypopharynx | 1 (20) |
| Supraglottis | 28 (45) | |
| Glottis | 32 (52) | |
| Subglottis | 1 (2) | |
| Tobacco | Never | 2 (3) |
| Current | 35 (56) | |
| Former | 25 (40) | |
| RTK/PI3K/RAS | Other Kinase | NOTCH | DNA Damage | WNT/ Diff. | Immune | Oxidation | Cell Cycle | Cell Death | TP53 | Differentiation/ Stem/ Epigenetic | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AKT1 | IGF1R | ABL1 | DTX1 | ATM | MSH2 | APC | SMAD2 | CUL3 | AURKA | MYC | BCL2 | HPV16 | AR | KMT2C |
| AKT2 | JAK2 | ABL2 | FBXW7 | ATR | MSH6 | AXIN2 | TGFA | HIF1A | CCND1 | MYCL | BCOR | MDM2 | ARID1A | KMT2D |
| AKT3 | JAK3 | ALK | HES1 | ATRX | PALB2 | CDH1 | TGFBR2 | IDH1 | CDC7 | MYCN | BIRC2 | MDM4 | ARID1B | LAD1 |
| BRAF | KDR | DDR1 | HES5 | BAP1 | PARP1 | CTNNB1 | TGIF1 | IDH2 | CDKN1A | NPM1 | CASP8 | TP53 | ASXL1 | MEN1 |
| EGFR | KRAS | KIT | HEY1 | BRCA1 | PMS2 | FAT1 | TNF | KEAP1 | CDKN1B | POLA1 | CASP9 | BMI1 | NSD1 | |
| ERBB2 | MET | MAP2K1 | HEYL | BRCA2 | RPA2 | RICTOR | ADA | NRF2 | CDKN2A | POLB | FADD | BRD1 | RARA | |
| ERBB3 | NRAS | MAP2K2 | MAML1 | CHEK1 | TET1 | SMO | CD274 | PTGS2 | CDKN2B | RB1 | IKBKE | BRD4 | RUNX1 | |
| ERBB4 | PIK3CA | MAP2K4 | MAML2 | CHEK2 | TET2 | SOX2 | HLA-A | VHL | E2F1 | RRM1 | MCL1 | CIC | RUNX1T1 | |
| FGFR1 | PIK3CB | NTRK1 | NOTCH1 | DNMT1 | TOP1 | SRC | HLA-B | E2F2 | TERT | NFKB1 | EZH2 | RXRB | ||
| FGFR2 | PIK3CD | NTRK2 | NOTCH2 | ERCC1 | TOP2A | TP63 | HLA-DRA | KNSTRN | TERT_Prom | NFKB2 | HDAC1 | SMARCA4 | ||
| FGFR3 | PIK3CG | NTRK3 | NOTCH3 | FANCA | TOP2B | TWIST1 | ICOS | MCM10 | TK1 | PTCH1 | KMT2A | |||
| FGFR4 | PIK3R1 | PTPN11 | NOTCH4 | FANCB | TRAF3 | VIM | LAG3 | MCM2 | TYMS | KMT2B | ||||
| FLT1 | PTEN | MLH1 | UBE2B | YAP1 | MS4A1 | MIRLET7C | ||||||||
| FLT3 | RAC1 | ZNF750 | NCAM1 | |||||||||||
| FLT4 | RAF1 | PTPRCAP | ||||||||||||
| HRAS | RET | SOCS1 | ||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heft Neal, M.E.; Bhangale, A.D.; Birkeland, A.C.; McHugh, J.B.; Shuman, A.G.; Rosko, A.J.; Swiecicki, P.L.; Spector, M.E.; Brenner, J.C. Prognostic Significance of Oxidation Pathway Mutations in Recurrent Laryngeal Squamous Cell Carcinoma. Cancers 2020, 12, 3081. https://doi.org/10.3390/cancers12113081
Heft Neal ME, Bhangale AD, Birkeland AC, McHugh JB, Shuman AG, Rosko AJ, Swiecicki PL, Spector ME, Brenner JC. Prognostic Significance of Oxidation Pathway Mutations in Recurrent Laryngeal Squamous Cell Carcinoma. Cancers. 2020; 12(11):3081. https://doi.org/10.3390/cancers12113081
Chicago/Turabian StyleHeft Neal, Molly E., Apurva D. Bhangale, Andrew C. Birkeland, Jonathan B. McHugh, Andrew G. Shuman, Andrew J. Rosko, Paul L. Swiecicki, Matthew E. Spector, and J. Chad Brenner. 2020. "Prognostic Significance of Oxidation Pathway Mutations in Recurrent Laryngeal Squamous Cell Carcinoma" Cancers 12, no. 11: 3081. https://doi.org/10.3390/cancers12113081
APA StyleHeft Neal, M. E., Bhangale, A. D., Birkeland, A. C., McHugh, J. B., Shuman, A. G., Rosko, A. J., Swiecicki, P. L., Spector, M. E., & Brenner, J. C. (2020). Prognostic Significance of Oxidation Pathway Mutations in Recurrent Laryngeal Squamous Cell Carcinoma. Cancers, 12(11), 3081. https://doi.org/10.3390/cancers12113081