Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded

 and
and 
Simple Summary
Abstract
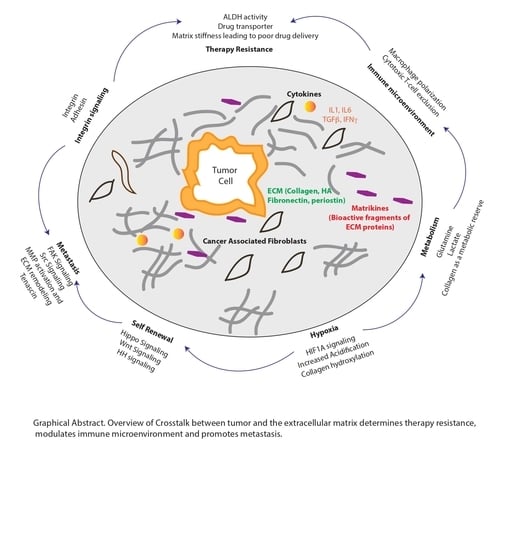
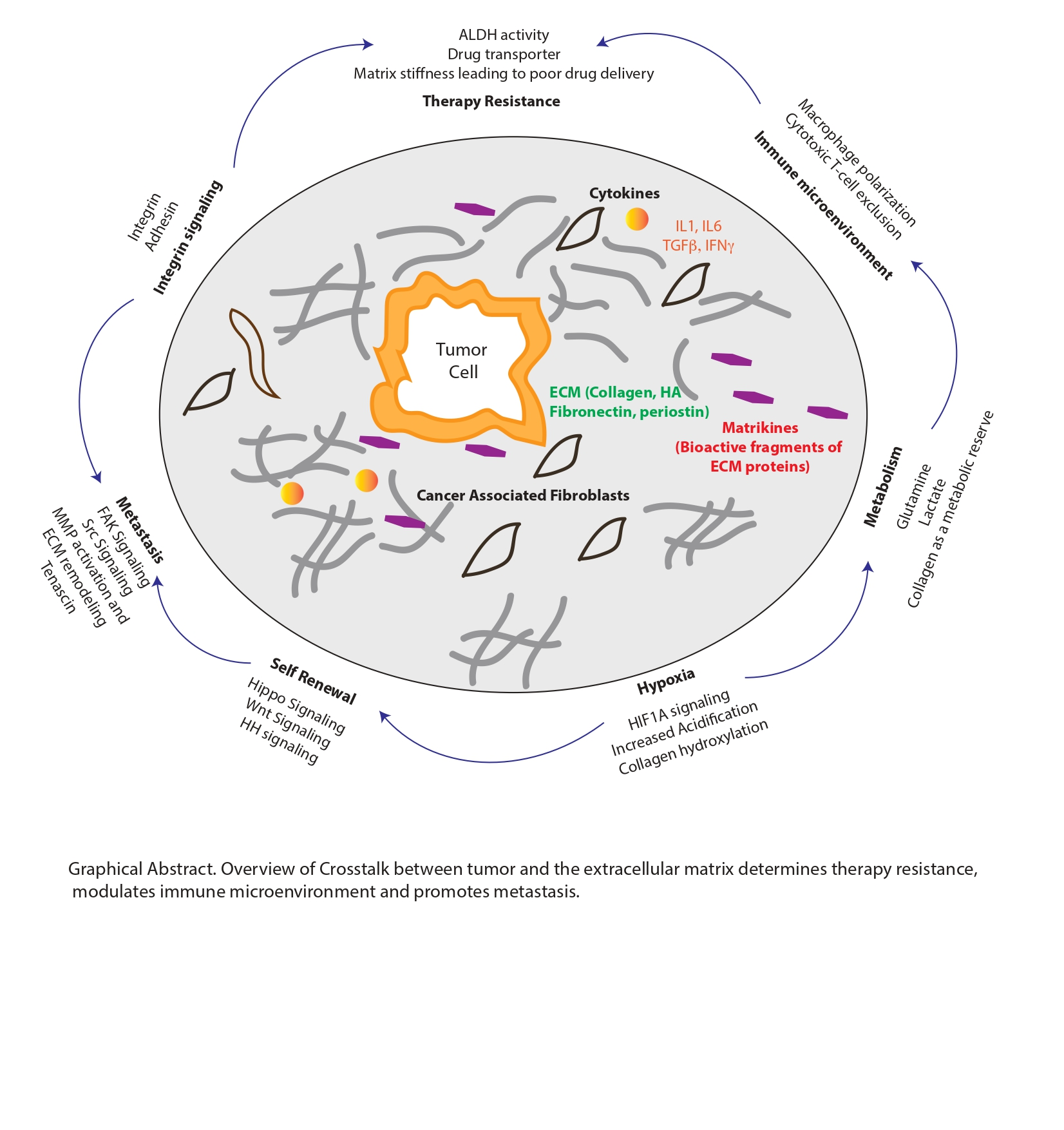{kind=link}
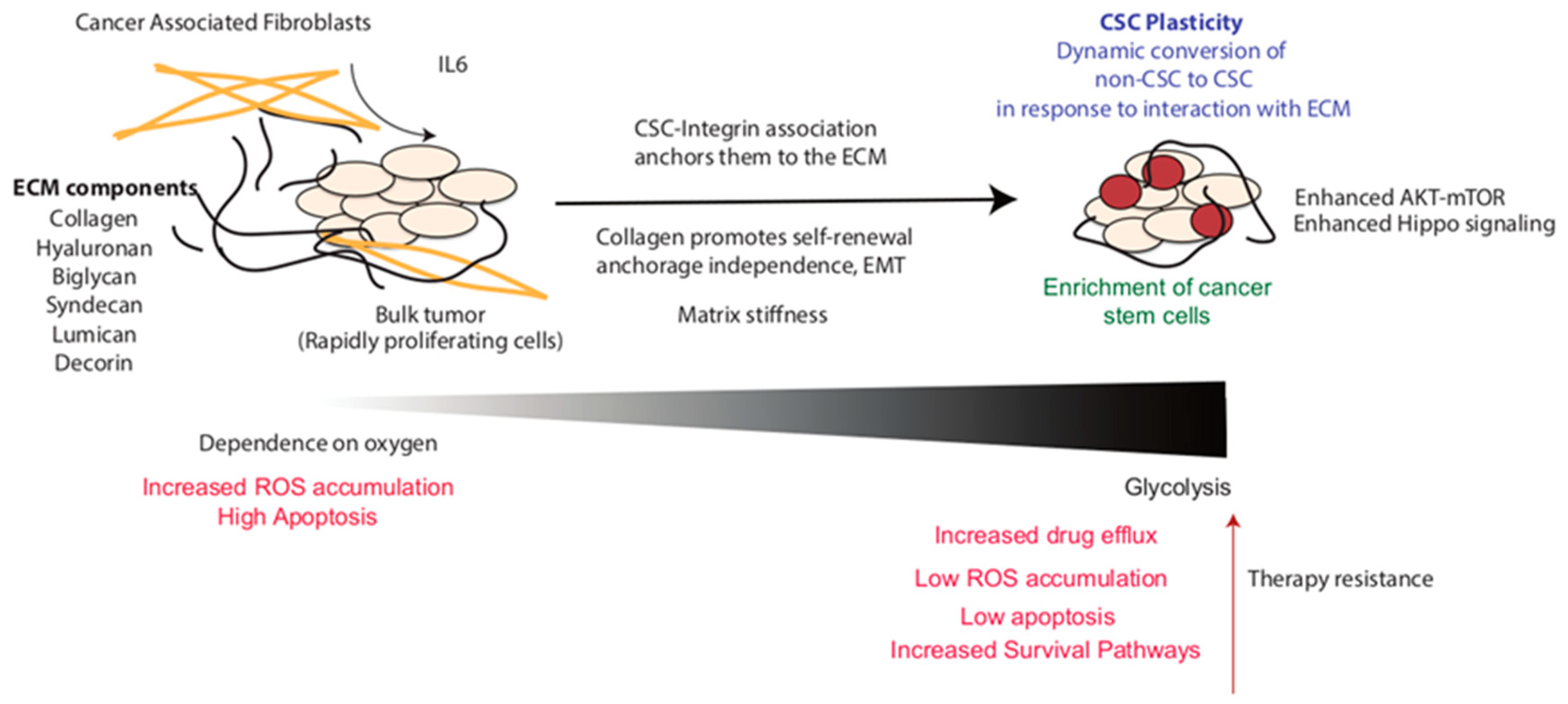{kind=link}
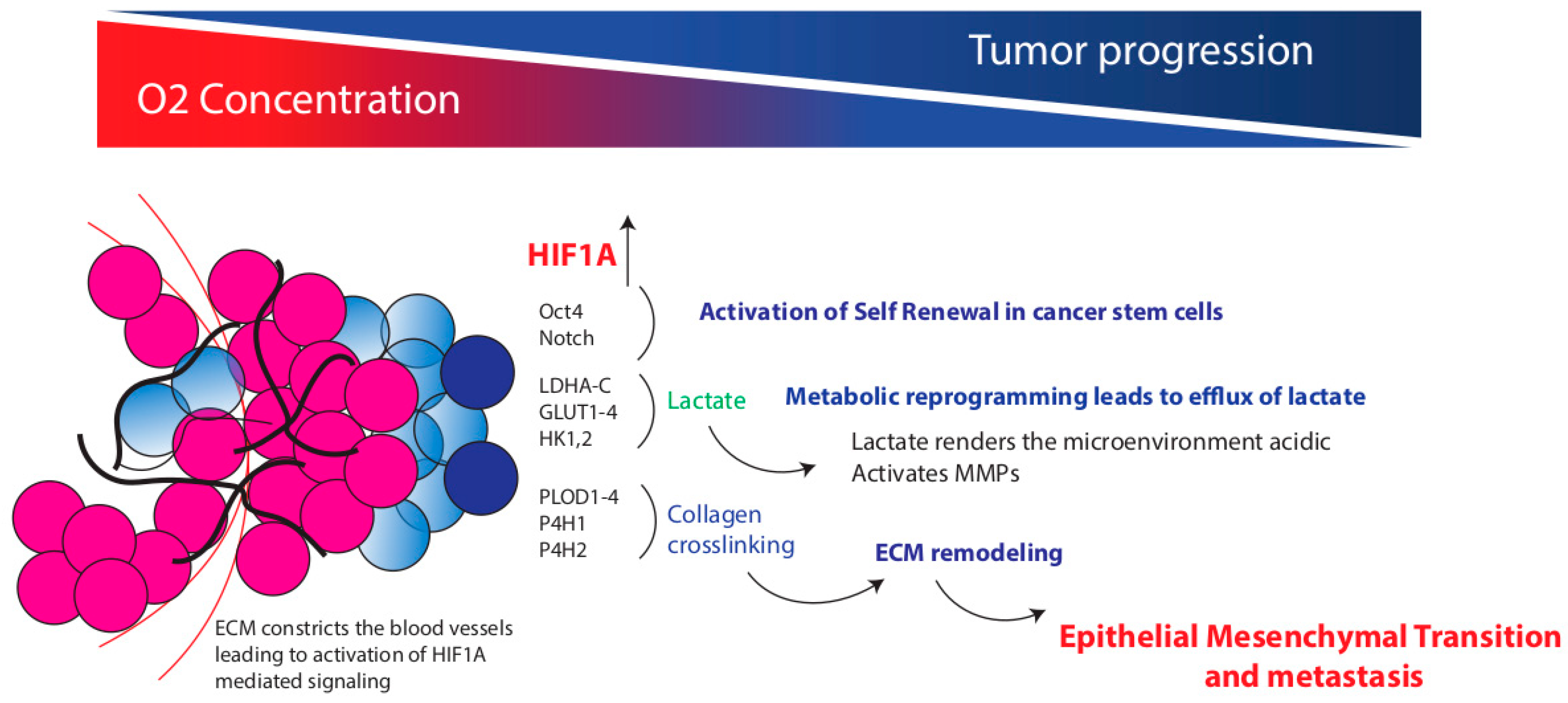{kind=link}
{kind=link}
1. Introduction
2. Extracellular Matrix as a Niche for Cancer Stem Cell Formation and Maintenance
2.1. Enrichment of CSC Population within the Tumor
2.2. ECM and Stem Cell Homeostasis
2.3. ECM–Integrin Interaction and Stemness
2.4. Role of Collagen in CSC Enrichment and Function
2.5. Proteoglycans and CSC
2.6. Hyaluronan and CSCs
3. Extracellular Matrix in Cancer Stem Cell Migration and Metastatic Niche Regulation
4. Regulation of Cancer Stem Cell Metabolism by ECM
4.1. Hypoxia in Regulation of ECM and CSC Metabolism
4.2. ECM Regulated-Metastasis in CSCs
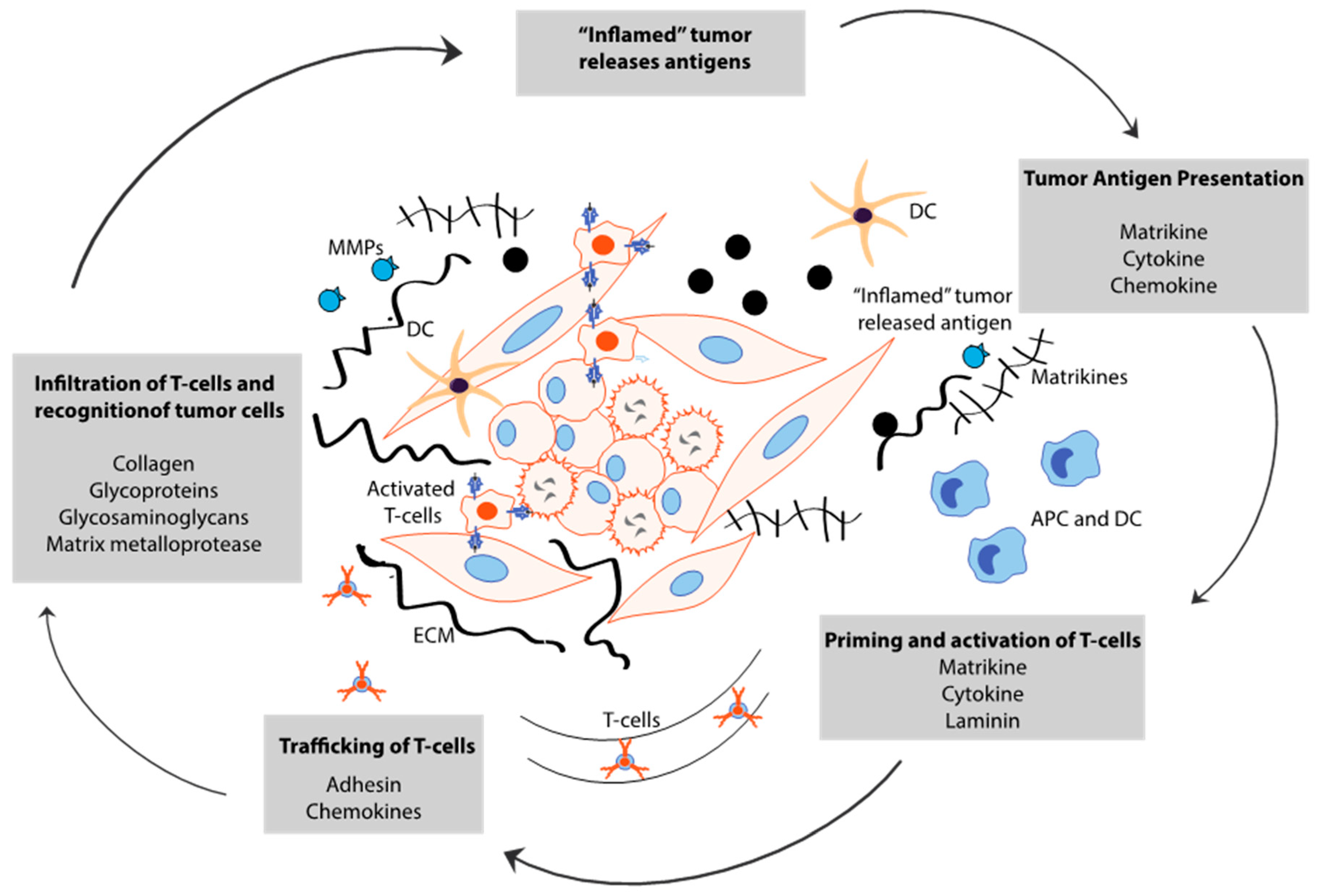
5. Cancer Stem Cell-Derived ECM Maintains Immune Evasion
6. Targeting Extracellular Matrix to Remodel CSC Niche
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123 Pt 24, 4195–4200. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef]
- Stromnes, I.M.; DelGiorno, K.E.; Greenberg, P.D.; Hingorani, S.R. Stromal reengineering to treat pancreas cancer. Carcinogenesis 2014, 35, 1451–1460. [Google Scholar] [CrossRef]
- McGinn, O.; Gupta, V.K.; Dauer, P.; Arora, N.; Sharma, N.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Nomura, A.; Dauer, P.; Gupta, V.; McGinn, O.; Arora, N.; Majumdar, K.; Iii, C.U.; Dalluge, J.; Dudeja, V.; Saluja, A.; et al. Microenvironment mediated alterations to metabolic pathways confer increased chemo-resistance in CD133+ tumor initiating cells. Oncotarget 2016, 7, 56324–56337. [Google Scholar] [CrossRef]
- Crawford, H.C.; Di Magliano, M.P.; Banerjee, S. Signaling networks that control cellular plasticity in pancreatic tumorigenesis, progression, and metastasis. Gastroenterology 2019, 156, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, Y.; Lu, Y.; Liu, M.; Li, M.; Li, J.; Wu, L. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Sainz, B. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Biol. 2018, 53, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Hall, R.R. Cancer Stem Cells: Cellular Plasticity, Niche, and its Clinical Relevance. J. Stem Cell Res. Ther. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Gnamlin, P.; Durden, B.; Gupta, V.K.; Kesh, K.; Garrido, V.T.; Dudeja, V.; Saluja, A.; Banerjee, S. Long non-coding RNA GAS5 acts as proliferation “brakes” in CD133+ cells responsible for tumor recurrence. Oncogenesis 2019, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.H.; Batchelder, C.A.; Lee, C.I.; Tarantal, A.F. Decellularized rhesus monkey kidney as a three-dimensional scaffold for renal tissue engineering. Tissue Eng. Part A 2010, 16, 2207–2216. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Anderson, L.R.; Owens, T.W.; Naylor, M.J. Integrins in development and cancer. Biophys. Rev. 2013, 6, 191–202. [Google Scholar] [CrossRef]
- Moon, J.H.; Rho, Y.S.; Lee, S.H.; Koo, B.S.; Lee, H.J.; Do, S.I.; Cho, J.H.; Eun, Y.G.; Park, M.W.; Shin, H.A.; et al. Role of integrin β1 as a biomarker of stemness in head and neck squamous cell carcinoma. Oral Oncol. 2019, 96, 34–41. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The role of the extracellular matrix in cancer stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef]
- De Toni, F.; Racaud-Sultan, C.; Chicanne, G.; Mas, V.M.-D.; Cariven, C.; Mesange, F.; Salles, J.-P.; Demur, C.; Allouche, M.; Payrastre, B.; et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene 2006, 25, 3113–3122. [Google Scholar] [CrossRef]
- Brown, Y.; Hua, S.; Tanwar, P.S. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int. J. Biochem. Cell Biol. 2019, 109, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, A.M.; Verhoef, E.I.; Roobol-Bouts, M.; Schröder, F.H.; Wildhagen, M.F.; Van Der Kwast, T.H.; Jenster, G.; Van Leenders, G.J. Validation of stem cell markers in clinical prostate cancer: α6-Integrin is predictive for non-aggressive disease. Prostate 2013, 74, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF- and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar] [CrossRef]
- Martin, T.A.; Jiang, W.G. Evaluation of the expression of stem cell markers in human breast cancer reveals a correlation with clinical progression and metastatic disease in ductal carcinoma. Oncol. Rep. 2013, 31, 262–272. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Ohta, K.; Uemura, M.; Nishimura, J.; Hata, T.; Takemasa, I.; Mizushima, T.; Yamamoto, H.; et al. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int. J. Oncol. 2013, 43, 425–430. [Google Scholar] [CrossRef]
- Zheng, Y.; De La Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A Rare Population of CD24+ITGB4+Notchhi Cells Drives Tumor Propagation in NSCLC and Requires Notch3 for Self-Renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef]
- Goel, H.L.; Pursell, B.; Chang, C.; Shaw, L.M.; Mao, J.; Simin, K.; Kumar, P.; Kooi, C.W.V.; Shultz, L.D.; Greiner, D.L.; et al. GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol. Med. 2013, 5, 488–508. [Google Scholar] [CrossRef]
- Pang, M.-F.; Siedlik, M.J.; Han, S.; Stallings-Mann, M.; Radisky, D.C.; Nelson, C.M. Tissue stiffness and hypoxia modulate the integrin-linked kinase ilk to control breast cancer stem-like cells. Cancer Res. 2016, 76, 5277–5287. [Google Scholar] [CrossRef]
- Motegi, H.; Kamoshima, Y.; Terasaka, S.; Kobayashi, H.; Houkin, K. Type 1 collagen as a potential niche component for CD133-positive glioblastoma cells. Neuropathology 2014, 34, 378–385. [Google Scholar] [CrossRef]
- Kirkland, S.C. Type I collagen inhibits differentiation and promotes a stem cell-like phenotype in human colorectal carcinoma cells. Br. J. Cancer 2009, 101, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Cai, J.; Zuo, Z.; Li, J. Collagen facilitates the colorectal cancer stemness and metastasis through an integrin/PI3K/AKT/Snail signaling pathway. Biomed. Pharmacother. 2019, 114, 108708. [Google Scholar] [CrossRef] [PubMed]
- Shea, M.P.; O’Leary, K.A.; Wegner, K.A.; Vezina, C.M.; Schuler, L.A. High collagen density augments mTOR-dependent cancer stem cells in ERα+ mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett. 2018, 433, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Huang, Y.; Pei, Q.; Liu, H.; Pei, H.; Hong, Z. Matrix stiffness mediates stemness characteristics via activating the Yes-associated protein in colorectal cancer cells. J. Cell. Biochem. 2018, 120, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef]
- Sasisekharan, R.; Shriver, Z.; Venkataraman, G.; Narayanasami, U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat. Rev. Cancer 2002, 2, 521–528. [Google Scholar] [CrossRef]
- Vitale, D.; Katakam, S.K.; Greve, B.; Jang, B.; Oh, E.; Alaniz, L.; Götte, M. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. 2019, 286, 2870–2882. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Götte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 1–19. [Google Scholar] [CrossRef]
- Hu, P.; Ke, C.; Guo, X.; Ren, P.; Tong, Y.; Luo, S.; He, Y.; Wei, Z.; Cheng, B.; Li, R.; et al. Both glypican-3/Wnt/β-catenin signaling pathway and autophagy contributed to the inhibitory effect of curcumin on hepatocellular carcinoma. Dig. Liver Dis. 2019, 51, 120–126. [Google Scholar] [CrossRef]
- Cao, J.; Ma, J.; Sun, L.; Li, J.; Qin, T.; Zhou, C.; Cheng, L.; Chen, K.; Qian, W.; Duan, W.; et al. Targeting glypican-4 overcomes 5-FU resistance and attenuates stem cell-like properties via suppression of Wnt/β-catenin pathway in pancreatic cancer cells. J. Cell. Biochem. 2018, 119, 9498–9512. [Google Scholar] [CrossRef]
- Farace, C.; Oliver, J.A.; Melguizo, C.; Alvarez, P.; Bandiera, P.; Rama, A.R.; Malaguarnera, G.; Ortiz, R.; Madeddu, R.; Prados, J. Microenvironmental Modulation of Decorin and Lumican in Temozolomide-Resistant Glioblastoma and Neuroblastoma Cancer Stem-Like Cells. PLoS ONE 2015, 10, e0134111. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, T.; Xu, X.; Cui, Y.; Xing, X. Biglycan promotes the chemotherapy resistance of colon cancer by activating NF-κB signal transduction. Mol. Cell. Biochem. 2018, 449, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, S.S.; Karalis, T.T.; Chatzopoulos, A.; Karamanos, N.K. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell. Signal. 2019, 63, 109377. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K.; et al. Hyaluronan Synthase HAS2 Promotes Tumor Progression in Bone by Stimulating the Interaction of Breast Cancer Stem-Like Cells with Macrophages and Stromal Cells. Cancer Res. 2011, 72, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Mochizuki, N.; Kongtawelert, P.; Konno, K.; Itano, N. Excessive Hyaluronan Production Promotes Acquisition of Cancer Stem Cell Signatures through the Coordinated Regulation of Twist and the Transforming Growth Factor β (TGF-β)-Snail Signaling Axis. J. Biol. Chem. 2014, 289, 26038–26056. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Wong, G.; Earle, C.; Chen, L. Hyaluronan-CD44v3 Interaction with Oct4-Sox2-Nanog Promotes miR-302 Expression Leading to Self-renewal, Clonal Formation, and Cisplatin Resistance in Cancer Stem Cells from Head and Neck Squamous Cell Carcinoma. J. Biol. Chem. 2012, 287, 32800–32824. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Savic, D.; Dudás, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.-I. Cancer stem cells and their unique role in metastatic spread. Semin. Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef]
- Alvarez, M.V.; Gutierrez, L.M.; Correa, A.; Lazarowski, A.; Bolontrade, M.F. Metastatic Niches and the Modulatory Contribution of Mesenchymal Stem Cells and Its Exosomes. Int. J. Mol. Sci. 2019, 20, 1946. [Google Scholar] [CrossRef]
- Borovski, T.; Melo, F.D.S.E.; Vermeulen, L.; Medema, J.P. Cancer Stem Cell Niche: The Place to Be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol. 2018, 20, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adhes. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Ungewiss, C.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Uraoka, N.; Mino, B.; Behrens, C.; et al. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 2016, 36, 1925–1938. [Google Scholar] [CrossRef]
- Du, Q.; Wang, W.; Liu, T.; Shang, C.; Huang, J.; Liao, Y.; Qin, S.; Chen, Y.; Liu, P.; Liu, J.; et al. High Expression of Integrin α3 Predicts Poor Prognosis and Promotes Tumor Metastasis and Angiogenesis by Activating the c-Src/Extracellular Signal-Regulated Protein Kinase/Focal Adhesion Kinase Signaling Pathway in Cervical Cancer. Front. Oncol. 2020, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, Y.; Kane, K.T.; Collins, M.A.; Simeone, D.M.; Di Magliano, M.P.; Nguyen, K.T. CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol. Cancer Res. 2015, 13, 9–15. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; Van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nat. Cell Biol. 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Grassian, A.R.; Coloff, J.L.; Brugge, J.S. Extracellular Matrix Regulation of Metabolism and Implications for Tumorigenesis. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 313–324. [Google Scholar] [CrossRef]
- Colpaert, C.G.; Vermeulen, P.B.; Fox, S.B.; Harris, A.L.; Dirix, L.Y.; Van Marck, E.A. The Presence of a Fibrotic Focus in Invasive Breast Carcinoma Correlates with the Expression of Carbonic Anhydrase IX and is a Marker of Hypoxia and Poor Prognosis. Breast Cancer Res. Treat. 2003, 81, 137–147. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Aro, E.; Khatri, R.; Gerard-O’Riley, R.; Mangiavini, L.; Myllyharju, J.; Schipani, E. Hypoxia-inducible Factor-1 (HIF-1) but Not HIF-2 Is Essential for Hypoxic Induction of Collagen Prolyl 4-Hydroxylases in Primary Newborn Mouse Epiphyseal Growth Plate Chondrocytes. J. Biol. Chem. 2012, 287, 37134–37144. [Google Scholar] [CrossRef]
- Gupta, V.K.; Sharma, N.S.; Kesh, K.; Dauer, P.; Nomura, A.; Giri, B.; Dudeja, V.; Banerjee, S.; Bhattacharya, S.; Saluja, A.; et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018, 439, 101–112. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-Inducible Factor 1α Induces Fibrosis and Insulin Resistance in White Adipose Tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-O.; Welch, T.P.; Gonzalez, F.J.; Copple, B.L. Reduced liver fibrosis in hypoxia-inducible factor-1α-deficient mice. Am. J. Physiol. Liver Physiol. 2009, 296, G582–G592. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V.; Martin, T.A.; Takagi, H.; Kirsner, R.S.; Helfman, T.; Pardes, J.; Ochoa, M.S. Low oxygen tension increases mRNA levels of alpha 1 (I) procollagen in human dermal fibroblasts. J. Cell. Physiol. 1993, 157, 408–412. [Google Scholar] [CrossRef]
- Tamamori, M.; Ito, H.; Hiroe, M.; Marumo, F.; Hata, R. Stimulation of collagen synthesis in rat cardiac fibroblasts by exposure to hypoxic culture conditions and suppression of the effect by natriuretic peptides. Cell Biol. Int. 1997, 21, 175–180. [Google Scholar] [CrossRef]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000, 58, 2351–2366. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.-L.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen Prolyl Hydroxylases Are Essential for Breast Cancer Metastasis. Cancer Res. 2013, 73, 3285–3296. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible Factor 1 (HIF-1) Promotes Extracellular Matrix Remodeling under Hypoxic Conditions by InducingP4HA1, P4HA2,andPLOD2Expression in Fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar] [PubMed]
- Muñoz-Nájar, U.M.; Neurath, K.M.; Vumbaca, F.; Claffey, K.P. Hypoxia stimulates breast carcinoma cell invasion through MT1-MMP and MMP-2 activation. Oncogene 2005, 25, 2379–2392. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1α in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.R.; Barcaroli, D.; Volpe, S.L.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; Dagostino, D.M.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Rinkes, I.H.B.; Jimenez, C.R.; Kranenburg, O. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Liao, J.; Tang, Z.-J.; Wu, W.-J.; Yang, J.; Zeng, Z.-L.; Hu, Y.; Wang, P.; Ju, H.-Q.; Xu, R.-H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2013, 21, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Yamawaki, K.; Ishiguro, T.; Yoshihara, K.; Ueda, H.; Sato, A.; Ohata, H.; Yoshida, Y.; Minamino, T.; Okamoto, K.; et al. Aldh-dependent glycolytic activation mediates stemness and paclitaxel resistance in patient-derived spheroid models of uterine endometrial cancer. Stem Cell Rep. 2019, 13, 730–746. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Doglioni, G.; Fendt, S.-M. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef]
- Attanasio, F.; Caldieri, G.; Giacchetti, G.; Van Horssen, R.; Wieringa, B.; Buccione, R. Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol. 2011, 90, 115–127. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Nokin, M.-J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5. [Google Scholar] [CrossRef]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Rodrigues, M.F.; Obre, E.; De Melo, F.H.; Santos, J.G.C.; Galina, A.; Jasiulionis, M.G.; Rossignol, R.; Rumjanek, F.D.; Amoêdo, N.D. Enhanced OXPHOS, glutaminolysis and β-oxidation constitute the metastatic phenotype of melanoma cells. Biochem. J. 2016, 473, 703–715. [Google Scholar] [CrossRef]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, L.M. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages as Incessant Builders and Destroyers of the Cancer Stroma. Cancers 2011, 3, 3740–3761. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Esbona, K.; Inman, D.; Saha, S.; Jeffery, J.; Schedin, P.; Wilke, L.; Keely, P. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016, 18, 1–15. [Google Scholar] [CrossRef]
- Weathington, N.M.; Van Houwelingen, A.H.; Noerager, B.D.; Jackson, P.L.; Kraneveld, A.D.; Galin, F.S.; Folkerts, G.; Nijkamp, F.P.; Blalock, J.E. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef]
- Hunninghake, G.; Davidson, J.; Rennard, S.; Szapiel, S.; Gadek, J.; Crystal, R. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science 1981, 212, 925–927. [Google Scholar] [CrossRef]
- Boyd, D.F.; Thomas, P.G. Towards integrating extracellular matrix and immunological pathways. Cytokine 2017, 98, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Adair-Kirk, T.L.; Senior, R.M. Fragments of extracellular matrix as mediators of inflammation. Int. J. Biochem. Cell Biol. 2008, 40, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Enikitovic, D.; Etzardi, M.; Eberdiaki, A.; Etsatsakis, A.; Tzanakakis, G.N. Cancer Microenvironment and Inflammation: Role of Hyaluronan. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican—A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Takahashi, H.; Lin, W.-W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.-L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nat. Cell Biol. 2009, 457, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Tunger, A.; Plesca, I.; Wehner, R.; Temme, A.; Westphal, D.; Meier, F.; Bachmann, M.; Schmitz, M. Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Jachetti, E.; Caputo, S.; Mazzoleni, S.; Brambillasca, C.S.; Parigi, S.M.; Grioni, M.; Piras, I.S.; Restuccia, U.; Calcinotto, A.; Freschi, M.; et al. Tenascin-C Protects Cancer Stem-like Cells from Immune Surveillance by Arresting T-cell Activation. Cancer Res. 2015, 75, 2095–2108. [Google Scholar] [CrossRef]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. OncoImmunology 2018, 7, e1478647. [Google Scholar] [CrossRef]
- VanDyke, K.; Zannettino, A.C.W.; Noll, J.E. The Role of the “Cancer Stem Cell Niche” in Cancer Initiation and Progression. In Adult Stem Cell Niches; IntechOpen: Rijeka, Croatia, 2014. [Google Scholar]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic Stem Cells: Sources, Niches, and Vital Pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef]
- Karalis, T.; Heldin, P.; Vynios, D.H.; Neill, T.; Buraschi, S.; Iozzo, R.V.; Karamanos, N.K.; Skandalis, S.S. Tumor-suppressive functions of 4-MU on breast cancer cells of different ER status: Regulation of hyaluronan/HAS2/CD44 and specific matrix effectors. Matrix Biol. 2019, 78–79, 118–138. [Google Scholar] [CrossRef]
- Lompardía, S.; Díaz, M.; Papademetrio, D.L.; Pibuel, M.; Álvarez, É.; Hajos, S.E. 4-methylumbelliferone and imatinib combination enhances senescence induction in chronic myeloid leukemia cell lines. Investig. New Drugs 2016, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2012, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Xiong, G.; Deng, L.; Zhu, J.; Rychahou, P.G.; Xu, R. Prolyl-4-hydroxylase α subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer 2014, 14, 1. [Google Scholar] [CrossRef]
- Rossow, L.; Veitl, S.; Vorlová, S.; Wax, J.K.; Kuhn, A.E.; Maltzahn, V.; Upcin, B.; Karl, F.; Hoffmann, H.; Gätzner, S.; et al. LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene 2018, 37, 4921–4940. [Google Scholar] [CrossRef]
- Benson, A.B.; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241-e15. [Google Scholar] [CrossRef]
- Molnar, J.; Fong, K.; He, Q.; Hayashi, K.; Kim, Y.; Fong, S.; Fogelgren, B.; Szauter, K.M.; Mink, M.; Csiszar, K. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2003, 1647, 220–224. [Google Scholar] [CrossRef]
- Pak, S.; Park, S.; Kim, Y.; Park, J.-H.; Park, C.-H.; Lee, K.-J.; Kim, C.-S.; Ahn, H. The small molecule WNT/β-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef]
- Semba, T.; Sammons, R.M.; Wang, X.; Xie, X.; Dalby, K.N.; Ueno, N.T. JNK Signaling in Stem Cell Self-Renewal and Differentiation. Int. J. Mol. Sci. 2020, 21, 2613. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Gupta, V.K.; Garrido, V.T.; Hadad, R.; Durden, B.C.; Kesh, K.; Giri, B.; Ferrantella, A.; Dudeja, V.; Saluja, A.; et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J. Clin. Investig. 2019, 130, 451–465. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesh, K.; Gupta, V.K.; Durden, B.; Garrido, V.; Mateo-Victoriano, B.; Lavania, S.P.; Banerjee, S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers 2020, 12, 3067. https://doi.org/10.3390/cancers12103067
Kesh K, Gupta VK, Durden B, Garrido V, Mateo-Victoriano B, Lavania SP, Banerjee S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers. 2020; 12(10):3067. https://doi.org/10.3390/cancers12103067
Chicago/Turabian StyleKesh, Kousik, Vineet K. Gupta, Brittany Durden, Vanessa Garrido, Beatriz Mateo-Victoriano, Shweta P. Lavania, and Sulagna Banerjee. 2020. "Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded" Cancers 12, no. 10: 3067. https://doi.org/10.3390/cancers12103067
APA StyleKesh, K., Gupta, V. K., Durden, B., Garrido, V., Mateo-Victoriano, B., Lavania, S. P., & Banerjee, S. (2020). Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers, 12(10), 3067. https://doi.org/10.3390/cancers12103067