Cutting the Brakes on Ras—Cytoplasmic GAPs as Targets of Inactivation in Cancer


Simple Summary
Abstract
1. Introduction
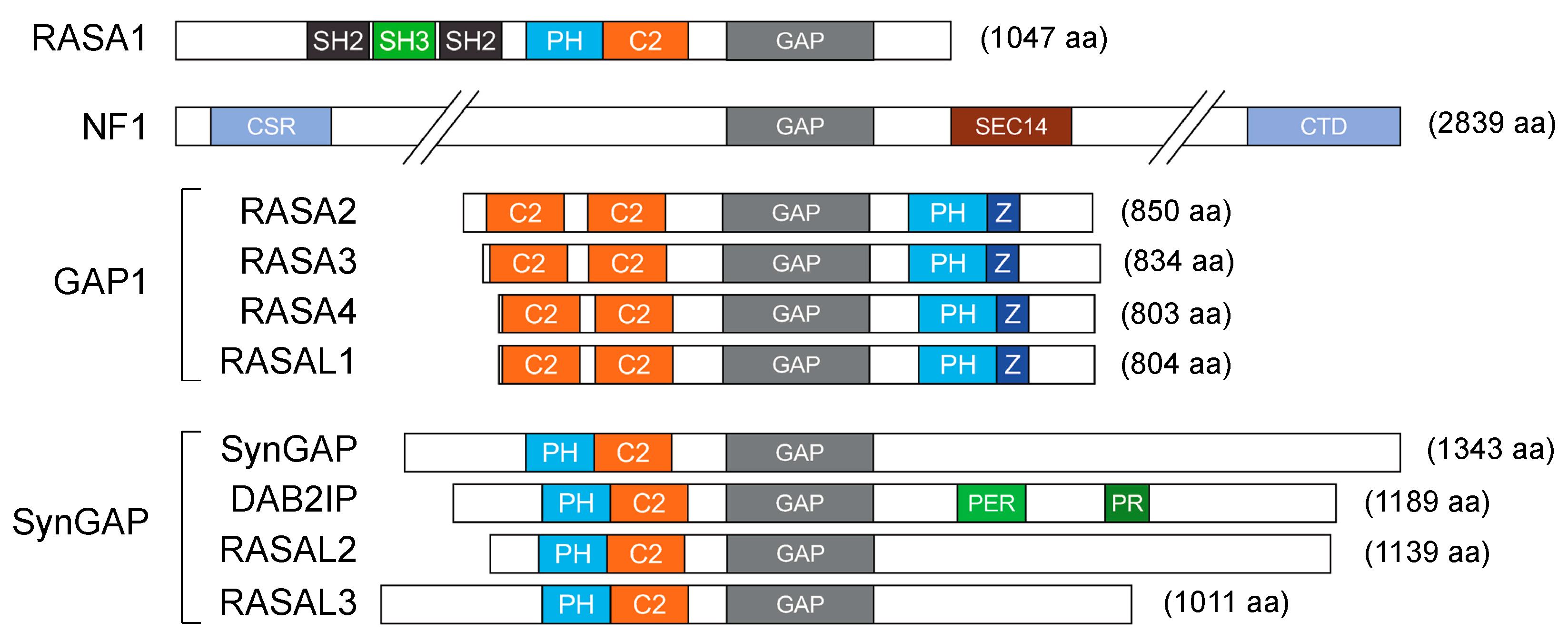
2. The Brakes: GAPs as Negative Regulators of Ras Activity
- RASA1 (Ras p21 protein activator 1)/p120GAP was the first RasGAP to be identified. In early studies, RASA1 appeared to be essential for embryonic blood vessel development [13]; subsequently, a germline mutation in RASA1 was associated with vascular malformations [14]. Although RASA1 can mitigate Ras activity, its role in cancer has remained unclear for a long time; recent works described RASA1 inhibition by non-coding RNA in multiple aggressive tumors, supportive of a tumor-suppressive activity [15,16,17,18,19].
- Neurofibromin (NF1) is perhaps the most extensively studied RasGAP. Germline mutations of NF1 are associated with the familiar disorder Neurofibromatosis Type 1. Germline NF1 mutations cause tumors along the nervous system, including neurofibromas and malignant peripheral nerve sheath tumors (MPNSTs). NF1 patients are also predisposed to develop gliomas, melanoma, and myeloid leukemia [20]. NF1 is also somatically altered in multiple sporadic tumors, including skin, lung, breast, and ovarian cancers [21], where its tumor-suppressive function seems to be linked primarily to Ras inhibition [11].
- The GAP1 family includes RASA2 (Ras p21 protein activator 2)/GAP1m, RASA3 (Ras p21 protein activator 3)/GAPIP4BP, RASA4 (Ras p21 protein activator 4)/CAPRI, and RASAL1 (Ras protein activator like 1). Members of this group share large significant sequence homology and act as dual GAPs, binding and inactivating Ras and Rap1 GTPases. All members of this group are clearly implicated in cancer. For example, the recurrent inactivation of RASA2 in melanoma favors constitutive activation of Ras signaling [22], and the frequent downregulation of RASA4 in myelomonocytic leukemia correlates with poor prognosis and higher risk of relapse after therapy [23]. Furthermore, the recurrent deletion of chromosome 13, which includes the RASA3 gene, in Burkitt and T-cell lymphomas and in acute myeloid leukemia (AML) suggests a potential role of RASA3 in these blood tumors [24]. Finally, RASAL1 displays MAPK- and PI3K-suppressing activities in multiple tumors, including thyroid cancer [25,26].
- The SynGAP family includes SynGAP (synaptic Ras GTPase-Activating Protein 1), DAB2IP (Dab2 interacting protein), RASAL2 (Ras protein activator like 2), and RASAL3 (Ras protein activator like 3). The founding member SynGAP is expressed only in neuronal tissue; germline SynGAP mutations have been associated with intellectual disabilities and autism [27]. RASAL2 functions as a tumor suppressor in a broad range of human tumors, including lung, ovarian, breast, and bladder cancer; low RASAL2 expression often correlates with aberrant Ras-ERK activation and worst prognosis [28,29]. Curiously, RASAL2 has also been described as an oncogene, based on its ability to promote EMT and metastasis in several tumors, fostering YAP (Yes1 associated transcriptional regulator), Wnt/β-catenin, PI3K/Akt, and Rac1 signaling [28]. DAB2IP is a tumor suppressor whose expression and function are altered in multiple human malignancies, causing uncontrolled activation of multiple oncogenic pathways, including Ras, NF-κB, PI3K/Akt, and Wnt/β-catenin [30,31]. Much less studied, RASAL3 epigenetic silencing in fibroblasts was linked to reprogramming of the tumor stroma in prostate cancer patients failing androgen deprivation therapy [32].
- The IQGAPs (IQ motif containing GTPase-Activating Protein) are cytoskeletal scaffold proteins with high sequence homology but variable tissue distribution. Despite the presence of a GAP domain, the IQGAPs are not Ras inhibitors, since the GAP domain lacks an arginine essential to assist Ras in GTP hydrolysis [11]. For this reason, they will not be considered here.
- Plexins are transmembrane receptors that bind semaphorins and regulate cell migration and angiogenesis. Plexins display GAP activity to Ras and Rap (a sub-family of Ras homologs) so they have a dual specificity [33]. Plexins also bind and modulate cytosolic tyrosine kinases, serine/threonine kinases, and other adaptors and scaffolding proteins, orchestrating complex signaling networks. Functional alterations in plexins cause cardiovascular and neuronal disorders, but their role in cancer is unclear: loss of plexins, specially plexin B1, was correlated with aggressive tumors [34]. However, there is also evidence of an oncogenic role for plexins in multiple malignancies [35,36,37]. Given their role as receptors and their complex Ras-independent functions, plexins will not be considered in this review.
3. Mutation of RasGAPs in Cancer
4. Transcriptional Repression of RasGAPs in Cancer
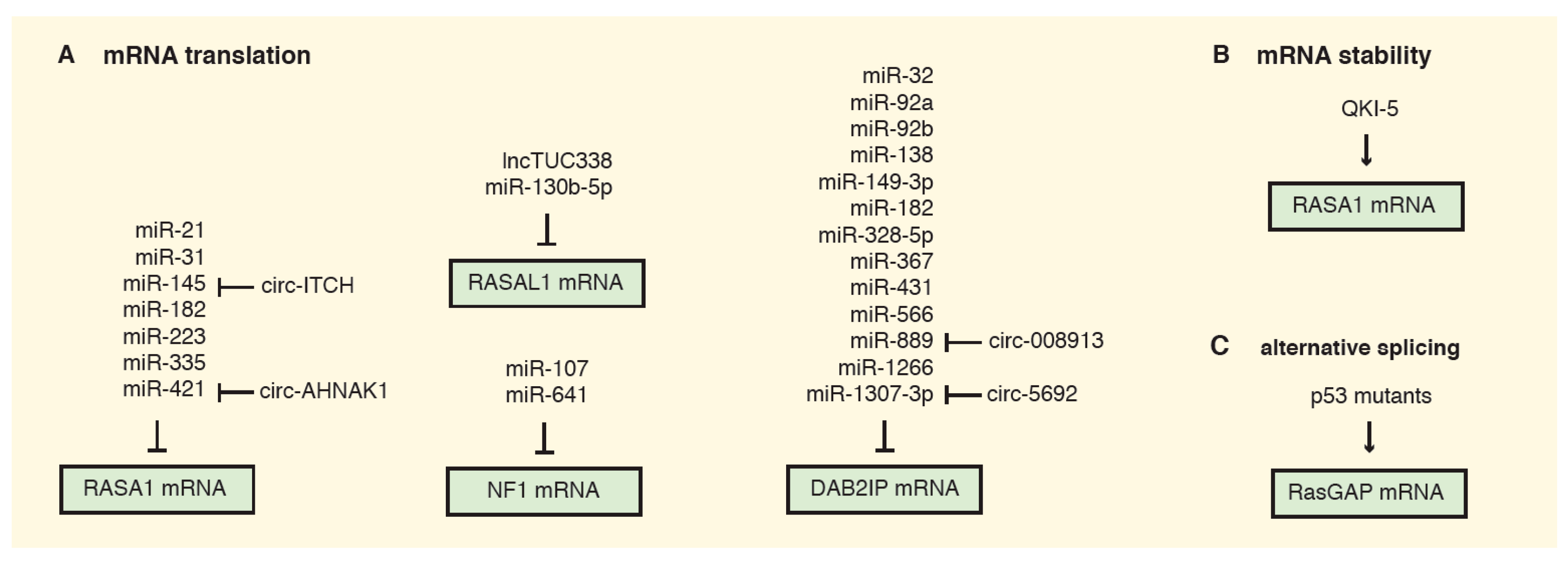
5. Post-Transcriptional Inhibition of RasGAPs in Cancer
6. Post-Translational Inhibition of RasGAPs in Cancer
6.1. Phosphorylation
6.2. Protein–Protein Interactions
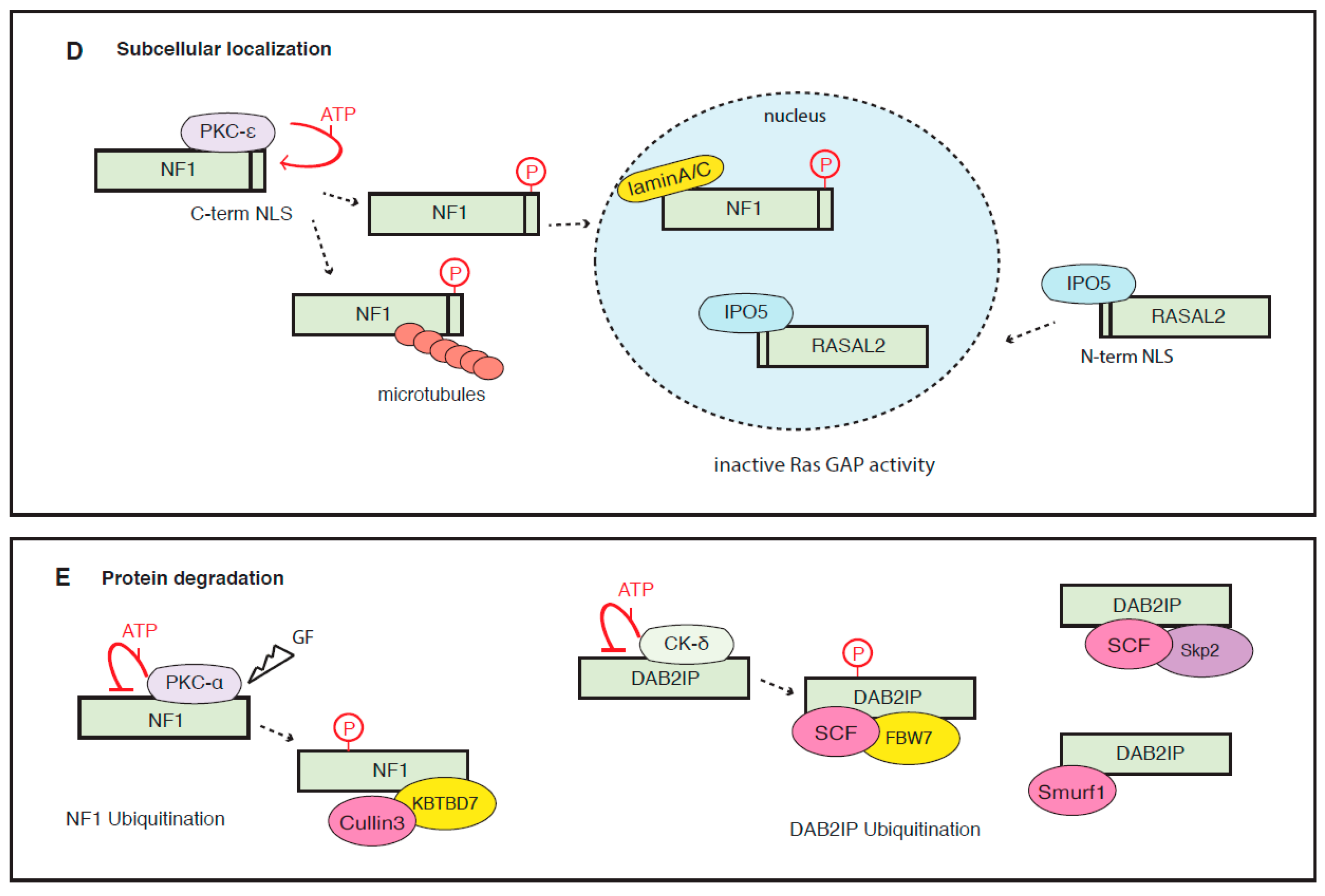
6.3. Subcellular Localization
6.4. Protein Degradation
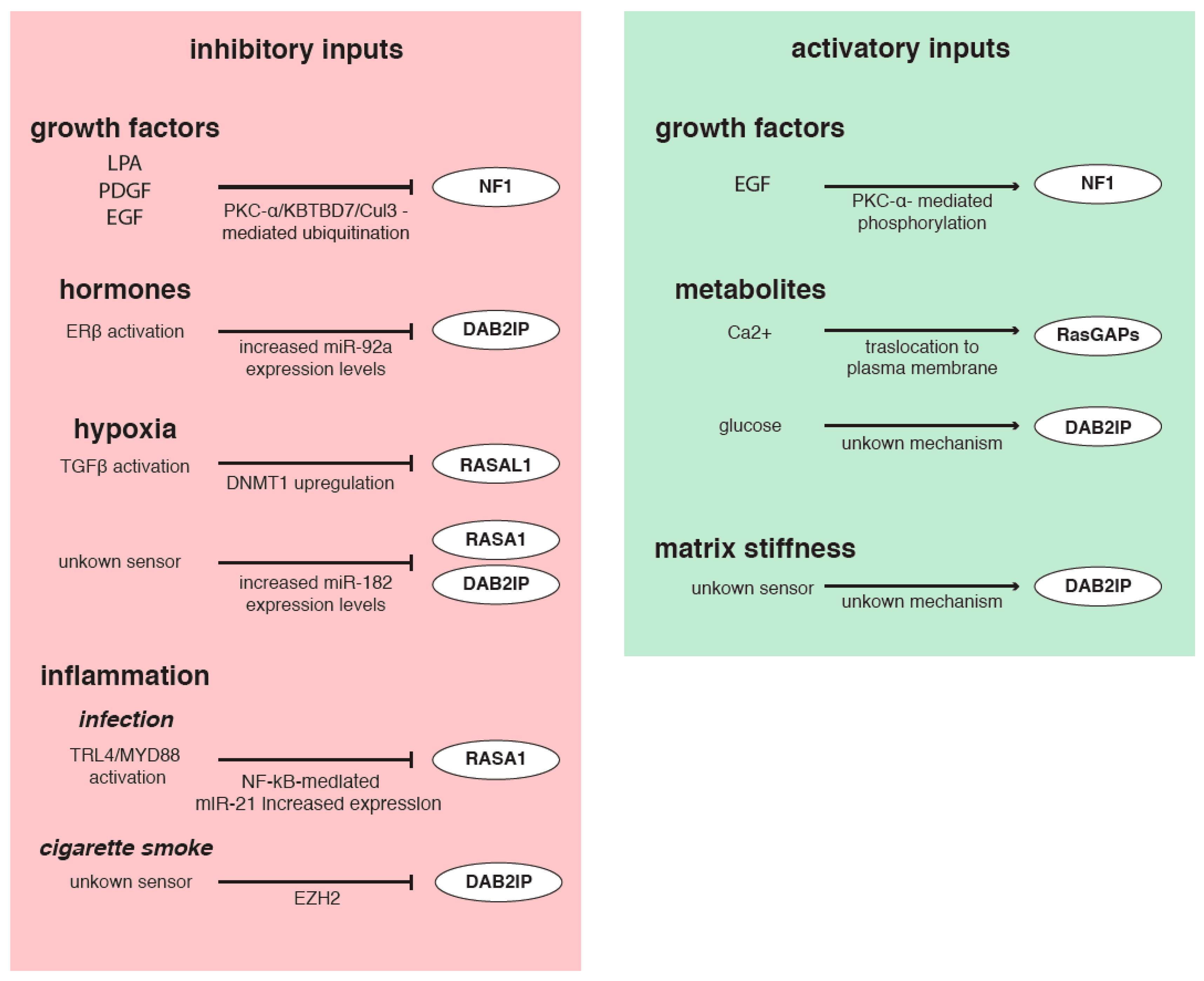
7. Control of RasGAPs Levels and Functions by Extracellular Inputs
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Sanchez-Vega, F.; La, K.; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G.; et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018, 23, 172–180.e173. [Google Scholar] [CrossRef]
- Sears, R.; Gray, J.W. Epigenomic Inactivation of RasGAPs Activates RAS Signaling in a Subset of Luminal B Breast Cancers. Cancer Discov. 2017, 7, 131–133. [Google Scholar] [CrossRef]
- Siewertsz van Reesema, L.L.; Lee, M.P.; Zheleva, V.; Winston, J.S.; O’Connor, C.F.; Perry, R.R.; Hoefer, R.A.; Tang, A.H. RAS pathway biomarkers for breast cancer prognosis. Clin. Lab. Int. 2016, 40, 18–23. [Google Scholar]
- Calvisi, D.F.; Ladu, S.; Conner, E.A.; Seo, D.; Hsieh, J.T.; Factor, V.M.; Thorgeirsson, S.S. Inactivation of Ras GTPase-activating proteins promotes unrestrained activity of wild-type Ras in human liver cancer. J. Hepatol. 2011, 54, 311–319. [Google Scholar] [CrossRef]
- De Smedt, E.; Maes, K.; Verhulst, S.; Lui, H.; Kassambara, A.; Maes, A.; Robert, N.; Heirman, C.; Cakana, A.; Hose, D.; et al. Loss of RASSF4 Expression in Multiple Myeloma Promotes RAS-Driven Malignant Progression. Cancer Res. 2018, 78, 1155–1168. [Google Scholar] [CrossRef]
- Maertens, O.; Cichowski, K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef]
- Scheffzek, K.; Shivalingaiah, G. Ras-Specific GTPase-Activating Proteins-Structures, Mechanisms, and Interactions. Cold Spring Harb. Perspect. Med. 2019, 9, a031500. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Rossi, D.J.; Holmyard, D.P.; Puri, M.C.; Mbamalu, G.; Harpal, K.; Shih, T.S.; Jacks, T.; Pawson, T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 1995, 377, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Eerola, I.; Boon, L.M.; Mulliken, J.B.; Burrows, P.E.; Dompmartin, A.; Watanabe, S.; Vanwijck, R.; Vikkula, M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 2003, 73, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yu, F.; Ma, Y.; Zhao, R.; Chen, X.; Zhu, J.; Zhang, C.Y.; Chen, J.; Zhang, J. MicroRNA-31 activates the RAS pathway and functions as an oncogenic MicroRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). J. Biol. Chem. 2013, 288, 9508–9518. [Google Scholar] [CrossRef]
- Sun, D.; Wang, C.; Long, S.; Ma, Y.; Guo, Y.; Huang, Z.; Chen, X.; Zhang, C.; Chen, J.; Zhang, J. C/EBP-beta-activated microRNA-223 promotes tumour growth through targeting RASA1 in human colorectal cancer. Br. J. Cancer 2015, 112, 1491–1500. [Google Scholar] [CrossRef]
- Sharma, S.B.; Lin, C.C.; Farrugia, M.K.; McLaughlin, S.L.; Ellis, E.J.; Brundage, K.M.; Salkeni, M.A.; Ruppert, J.M. MicroRNAs 206 and 21 cooperate to promote RAS-extracellular signal-regulated kinase signaling by suppressing the translation of RASA1 and SPRED1. Mol. Cell. Biol. 2014, 34, 4143–4164. [Google Scholar] [CrossRef]
- Xiao, W.; Zheng, S.; Zou, Y.; Yang, A.; Xie, X.; Tang, H.; Xie, X. CircAHNAK1 inhibits proliferation and metastasis of triple-negative breast cancer by modulating miR-421 and RASA1. Aging 2019, 11, 12043–12056. [Google Scholar] [CrossRef]
- Chen, X.; Cai, S.; Li, B.; Zhang, X.; Li, W.; Liang, H.; Cao, X.; Wang, L.; Wu, Z. MicroRNA21 regulates the biological behavior of esophageal squamous cell carcinoma by targeting RASA1. Oncol. Rep. 2019, 41, 1627–1637. [Google Scholar] [CrossRef]
- Riccardi, V.M. Type 1 neurofibromatosis and the pediatric patient. Curr. Probl. Pediatrics 1992, 22, 66–106. [Google Scholar] [CrossRef]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Qutob, N.; Emmanuel, R.; Keren-Paz, A.; Madore, J.; Elkahloun, A.; Wilmott, J.S.; Gartner, J.J.; Di Pizio, A.; Winograd-Katz, S.; et al. Recurrent inactivating RASA2 mutations in melanoma. Nat. Genet. 2015, 47, 1408–1410. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.R.; Lipka, D.B.; Witte, T.; Claus, R.; Nollke, P.; Zucknick, M.; Olk-Batz, C.; Fluhr, S.; Dworzak, M.; De Moerloose, B.; et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics 2014, 9, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Schurmans, S.; Polizzi, S.; Scoumanne, A.; Sayyed, S.; Molina-Ortiz, P. The Ras/Rap GTPase activating protein RASA3: From gene structure to in vivo functions. Adv. Biol. Regul. 2015, 57, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. 2013, 105, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, X.; Ying, J.; Wong, A.H.; Cui, Y.; Srivastava, G.; Shen, Z.Y.; Li, E.M.; Zhang, Q.; Jin, J.; et al. Epigenetic silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 12353–12358. [Google Scholar] [CrossRef] [PubMed]
- Jeyabalan, N.; Clement, J.P. SYNGAP1: Mind the Gap. Front. Cell. Neurosci. 2016, 10, 32. [Google Scholar] [CrossRef]
- Zhou, B.; Zhu, W.; Jiang, X.; Ren, C. RASAL2 Plays Inconsistent Roles in Different Cancers. Front. Oncol. 2019, 9, 1235. [Google Scholar] [CrossRef]
- Wang, X.; Qian, C.; Yang, Y.; Liu, M.Y.; Ke, Y.; Qian, Z.M. Phosphorylated Rasal2 facilitates breast cancer progression. EBioMedicine 2019, 50, 144–155. [Google Scholar] [CrossRef]
- Bellazzo, A.; Di Minin, G.; Collavin, L. Block one, unleash a hundred. Mechanisms of DAB2IP inactivation in cancer. Cell Death Differ. 2017, 24, 15–25. [Google Scholar] [CrossRef]
- Liu, L.; Xu, C.; Hsieh, J.T.; Gong, J.; Xie, D. DAB2IP in cancer. Oncotarget 2016, 7, 3766–3776. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, H.G.; Wang, Y.; Zhang, X. Structural mechanisms of plexin signaling. Prog. Biophys. Mol. Biol. 2015, 118, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Rody, A.; Holtrich, U.; Gaetje, R.; Gehrmann, M.; Engels, K.; von Minckwitz, G.; Loibl, S.; Diallo-Danebrock, R.; Ruckhaberle, E.; Metzler, D.; et al. Poor outcome in estrogen receptor-positive breast cancers predicted by loss of plexin B1. Clin. Cancer Res. 2007, 13, 1115–1122. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Piperi, C. Emerging role of plexins signaling in glioma progression and therapy. Cancer Lett. 2018, 414, 81–87. [Google Scholar] [CrossRef]
- Gurrapu, S.; Tamagnone, L. Semaphorins as Regulators of Phenotypic Plasticity and Functional Reprogramming of Cancer Cells. Trends Mol. Med. 2019, 25, 303–314. [Google Scholar] [CrossRef]
- Worzfeld, T.; Offermanns, S. Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov. 2014, 13, 603–621. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Wiesner, T.; Kiuru, M.; Scott, S.N.; Arcila, M.; Halpern, A.C.; Hollmann, T.; Berger, M.F.; Busam, K.J. NF1 Mutations Are Common in Desmoplastic Melanoma. Am. J. Surg. Pathol. 2015, 39, 1357–1362. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Frampton, G.M.; Ferrer-Lozano, J.; Yelensky, R.; Perez-Fidalgo, J.A.; Wang, Y.; Palmer, G.A.; Ross, J.S.; Miller, V.A.; Su, X.; et al. Concordance of genomic alterations between primary and recurrent breast cancer. Mol. Cancer Ther. 2014, 13, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Sangha, N.; Wu, R.; Kuick, R.; Powers, S.; Mu, D.; Fiander, D.; Yuen, K.; Katabuchi, H.; Tashiro, H.; Fearon, E.R.; et al. Neurofibromin 1 (NF1) defects are common in human ovarian serous carcinomas and co-occur with TP53 mutations. Neoplasia 2008, 10, 1362–1372. [Google Scholar] [CrossRef]
- Holzel, M.; Huang, S.; Koster, J.; Ora, I.; Lakeman, A.; Caron, H.; Nijkamp, W.; Xie, J.; Callens, T.; Asgharzadeh, S.; et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 2010, 142, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Otto, G.A.; He, J.; Palmer, G.; Yelensky, R.; Lipson, D.; et al. Advanced urothelial carcinoma: Next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod. Pathol. 2014, 27, 271–280. [Google Scholar] [CrossRef]
- Boudry-Labis, E.; Roche-Lestienne, C.; Nibourel, O.; Boissel, N.; Terre, C.; Perot, C.; Eclache, V.; Gachard, N.; Tigaud, I.; Plessis, G.; et al. Neurofibromatosis-1 gene deletions and mutations in de novo adult acute myeloid leukemia. Am. J. Hematol. 2013, 88, 306–311. [Google Scholar] [CrossRef]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Kanchi, K.L.; Wang, X.; Hill, K.S.; Messina, J.L.; Lee, J.H.; Kim, Y.; Dees, N.D.; Ding, L.; Teer, J.K.; et al. Inactivation of RASA1 promotes melanoma tumorigenesis via R-Ras activation. Oncotarget 2016, 7, 23885–23896. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Cabrera, C.; Quintana, R.M.; Bravo, A.; Casanova, M.L.; Page, A.; Alameda, J.P.; Paramio, J.M.; Maroto, A.; Salamanca, J.; Dupuy, A.J.; et al. A Transposon-based Analysis Reveals RASA1 Is Involved in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.; Gejman, P.V.; Martin, G.A.; McCormick, F. Nonsense mutations in the C-terminal SH2 region of the GTPase activating protein (GAP) gene in human tumours. Nat. Genet. 1993, 5, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Desmeules, P.; Smith, R.S.; Drilon, A.; Somwar, R.; Ladanyi, M. RASA1 and NF1 are Preferentially Co-Mutated and Define A Distinct Genetic Subset of Smoking-Associated Non-Small Cell Lung Carcinomas Sensitive to MEK Inhibition. Clin. Cancer Res. 2018, 24, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Olsen, S.N.; Wronski, A.; Castano, Z.; Dake, B.; Malone, C.; De Raedt, T.; Enos, M.; DeRose, Y.S.; Zhou, W.; Guerra, S.; et al. Loss of RasGAP Tumor Suppressors Underlies the Aggressive Nature of Luminal B Breast Cancers. Cancer Discov. 2017, 7, 202–217. [Google Scholar] [CrossRef]
- McLaughlin, S.K.; Olsen, S.N.; Dake, B.; De Raedt, T.; Lim, E.; Bronson, R.T.; Beroukhim, R.; Polyak, K.; Brown, M.; Kuperwasser, C.; et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell 2013, 24, 365–378. [Google Scholar] [CrossRef]
- Son, H.J.; Jo, Y.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. DAB2IP with tumor-inhibiting activities exhibits frameshift mutations in gastrointestinal cancers. Pathol. Res. Pract. 2018, 214, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, F.; Irnaten, M.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Hypoxia-Induced Changes in DNA Methylation Alter RASAL1 and TGFbeta1 Expression in Human Trabecular Meshwork Cells. PLoS ONE 2016, 11, e0153354. [Google Scholar] [CrossRef]
- Chen, H.; Tu, S.W.; Hsieh, J.T. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J. Biol. Chem. 2005, 280, 22437–22444. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; De Raedt, T.; Guney, I.; Strochlic, D.E.; Macconaill, L.E.; Beroukhim, R.; et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Wang, W.; Ozes, A.; Fang, F.; Sandusky, G.E.; Nephew, K.P. EZH2-mediated Downregulation of the Tumor Suppressor DAB2IP Maintains Ovarian Cancer Stem Cells. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Zhu, J.; Jiang, Z.; Gao, F.; Hu, X.; Zhou, L.; Chen, J.; Luo, H.; Sun, J.; Wu, S.; Han, Y.; et al. A systematic analysis on DNA methylation and the expression of both mRNA and microRNA in bladder cancer. PLoS ONE 2011, 6, e28223. [Google Scholar] [CrossRef]
- Grewal, T.; Koese, M.; Tebar, F.; Enrich, C. Differential Regulation of RasGAPs in Cancer. Genes Cancer 2011, 2, 288–297. [Google Scholar] [CrossRef]
- Hui, K.; Yue, Y.; Wu, S.; Gu, Y.; Guan, B.; Wang, X.; Hsieh, J.T.; Chang, L.S.; He, D.; Wu, K. The expression and function of RASAL2 in renal cell carcinoma angiogenesis. Cell Death Dis. 2018, 9, 881. [Google Scholar] [CrossRef]
- Kolfschoten, I.G.; van Leeuwen, B.; Berns, K.; Mullenders, J.; Beijersbergen, R.L.; Bernards, R.; Voorhoeve, P.M.; Agami, R. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell 2005, 121, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Liu, W.W.; Nie, W.J.; Li, D.F.; Xie, Z.J.; Liu, C.; Liu, Y.H.; Mei, P.; Li, Z.J. MiR-21/RASA1 axis affects malignancy of colon cancer cells via RAS pathways. World J. Gastroenterol. 2015, 21, 1488–1497. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, H.; Yuan, L.; Liu, G.; Zhang, C.; Hong, M.; Liu, Y.; Zhou, M.; Chen, F.; Li, X. Overexpression of miR-335 confers cell proliferation and tumour growth to colorectal carcinoma cells. Mol. Cell. Biochem. 2016, 412, 235–245. [Google Scholar] [CrossRef]
- Kent, O.A.; Mendell, J.T.; Rottapel, R. Transcriptional Regulation of miR-31 by Oncogenic KRAS Mediates Metastatic Phenotypes by Repressing RASA1. Mol. Cancer Res. 2016, 14, 267–277. [Google Scholar] [CrossRef]
- Zhang, L.; Zhan, X.; Yan, D.; Wang, Z. Circulating MicroRNA-21 Is Involved in Lymph Node Metastasis in Cervical Cancer by Targeting RASA1. Int. J. Gynecol. Cancer 2016, 26, 810–816. [Google Scholar] [CrossRef]
- Du, C.; Weng, X.; Hu, W.; Lv, Z.; Xiao, H.; Ding, C.; Gyabaah, O.A.; Xie, H.; Zhou, L.; Wu, J.; et al. Hypoxia-inducible MiR-182 promotes angiogenesis by targeting RASA1 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Zhang, Q.; Lin, R. miR-182 contributes to cell proliferation, invasion and tumor growth in colorectal cancer by targeting DAB2IP. Int. J. Biochem. Cell Biol. 2019, 111, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Li, J.; Wu, L.; Song, M.; Meng, Q. miR182 activates the Ras-MEK-ERK pathway in human oral cavity squamous cell carcinoma by suppressing RASA1 and SPRED1. Oncotargets Ther. 2017, 10, 667–679. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, S.; Ma, G.; Zhu, H.; Lv, C.; Chu, H.; Tong, N.; Wu, D.; Qiang, F.; Gong, W.; Zhao, Q.; et al. miR-107 regulates tumor progression by targeting NF1 in gastric cancer. Sci. Rep. 2016, 6, 36531. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Y.; Wang, J.; Shen, D.; Zhao, J.; Yu, Q. miR-130b-5p promotes proliferation, migration and invasion of gastric cancer cells via targeting RASAL1. Oncol. Lett. 2018, 15, 6361–6367. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, J.D.; Guo, X.T.; Cao, X.; Li, Q. Increased expression of miR-641 contributes to erlotinib resistance in non-small-cell lung cancer cells by targeting NF1. Cancer Med. 2018, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Xiao, Y.; Hu, Y.; Xiao, Y.; Yin, Z.; Liu, L. microRNA-32 induces radioresistance by targeting DAB2IP and regulating autophagy in prostate cancer cells. Oncol. Lett. 2015, 10, 2055–2062. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, J.; Wang, L.; Chen, J.; Gao, H.; Zhao, W.; Huang, Y.; Jiang, T.; Zhou, J.; Chen, Y. The circular RNA circ-ITCH suppresses ovarian carcinoma progression through targeting miR-145/RASA1 signaling. Biochem. Biophys. Res. Commun. 2018, 505, 222–228. [Google Scholar] [CrossRef]
- Jin, W.; Chen, L.; Cai, X.; Zhang, Y.; Zhang, J.; Ma, D.; Cai, X.; Fu, T.; Yu, Z.; Yu, F.; et al. Long non-coding RNA TUC338 is functionally involved in sorafenib-sensitized hepatocarcinoma cells by targeting RASAL1. Oncol. Rep. 2017, 37, 273–280. [Google Scholar] [CrossRef]
- Bellazzo, A.; Di Minin, G.; Valentino, E.; Sicari, D.; Torre, D.; Marchionni, L.; Serpi, F.; Stadler, M.B.; Taverna, D.; Zuccolotto, G.; et al. Cell-autonomous and cell non-autonomous downregulation of tumor suppressor DAB2IP by microRNA-149-3p promotes aggressiveness of cancer cells. Cell Death Differ. 2018, 25, 1224–1238. [Google Scholar] [CrossRef]
- Xu, Y.; He, J.; Wang, Y.; Zhu, X.; Pan, Q.; Xie, Q.; Sun, F. miR-889 promotes proliferation of esophageal squamous cell carcinomas through DAB2IP. FEBS Lett. 2015, 589, 1127–1135. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, Z.H.; Bi, Y.H. MicroRNA-889 promotes cell proliferation in colorectal cancer by targeting DAB2IP. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3326–3334. [Google Scholar] [CrossRef]
- Xiao, T.; Xue, J.; Shi, M.; Chen, C.; Luo, F.; Xu, H.; Chen, X.; Sun, B.; Sun, Q.; Yang, Q.; et al. Circ008913, via miR-889 regulation of DAB2IP/ZEB1, is involved in the arsenite-induced acquisition of CSC-like properties by human keratinocytes in carcinogenesis. Metallomics 2018, 10, 1328–1338. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, Y.; Huang, Z.; Kong, Y.; Hu, X.; Xiao, W.; Quan, J.; Fan, X. CircRNA-5692 inhibits the progression of hepatocellular carcinoma by sponging miR-328-5p to enhance DAB2IP expression. Cell Death Dis. 2019, 10, 900. [Google Scholar] [CrossRef]
- Huang, J.; Wang, B.; Hui, K.; Zeng, J.; Fan, J.; Wang, X.; Hsieh, J.T.; He, D.; Wu, K. miR-92b targets DAB2IP to promote EMT in bladder cancer migration and invasion. Oncol. Rep. 2016, 36, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.F.; Zhang, Y.; Yu, J.W.; Hua, R.H.; Wang, Q.H.; Zhu, J.W. miR-92b promotes gastric cancer growth by activating the DAB2IP-mediated PI3K/AKT signalling pathway. Cell Prolif. 2020, 53, e12630. [Google Scholar] [CrossRef]
- Ou, Z.; Wang, Y.; Chen, J.; Tao, L.; Zuo, L.; Sahasrabudhe, D.; Joseph, J.; Wang, L.; Yeh, S. Estrogen receptor beta promotes bladder cancer growth and invasion via alteration of miR-92a/DAB2IP signals. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.B. MicroRNA-556-3p promotes the progression of esophageal cancer via targeting DAB2IP. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6816–6823. [Google Scholar] [CrossRef]
- Feng, C.; Sun, P.; Hu, J.; Feng, H.; Li, M.; Liu, G.; Pan, Y.; Feng, Y.; Xu, Y.; Feng, K.; et al. miRNA-556-3p promotes human bladder cancer proliferation, migration and invasion by negatively regulating DAB2IP expression. Int. J. Oncol. 2017, 50, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Zhou, J.; Lin, C.J.; Xu, S.; Santoyo, J.; Hernandez, E.; Lai, C.H.; Lin, H.; He, D.; Hsieh, J.T. The network of DAB2IP-miR-138 in regulating drug resistance of renal cell carcinoma associated with stem-like phenotypes. Oncotarget 2017, 8, 66975–66986. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Jiang, H.; Yu, Y.; Xu, Y.; Zuo, W.; Wang, S.; Su, Z. miR-367 regulation of DOC-2/DAB2 interactive protein promotes proliferation, migration and invasion of osteosarcoma cells. Biomed. Pharmacother. 2017, 95, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Wang, X.; Li, J.; Wei, J.; Wang, Y.; Song, W.; Zhang, Z. MiR-1266 promotes cell proliferation, migration and invasion in cervical cancer by targeting DAB2IP. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3623–3630. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Yao, B.; Liu, Q.; Guo, C. miR-1307-3p promotes tumor growth and metastasis of hepatocellular carcinoma by repressing DAB2 interacting protein. Biomed. Pharmacother. 2019, 117, 109055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Choi, S.; Zhang, T.; Chen, Z.; Chi, Y.; Huang, S.; Xiang, J.Z.; Du, Y.N. miR-431 Promotes Metastasis of Pancreatic Neuroendocrine Tumors by Targeting DAB2 Interacting Protein, a Ras GTPase Activating Protein Tumor Suppressor. Am. J. Pathol. 2020, 190, 689–701. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, W169–W173. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.L.; Yang, J.P.; Peng, L.X.; Zheng, L.S.; Xie, P.; Wang, M.Y.; Cao, Y.; Zhang, Z.L.; Zhou, F.J.; Qian, C.N.; et al. RNA-binding protein QKI-5 inhibits the proliferation of clear cell renal cell carcinoma via post-transcriptional stabilization of RASA1 mRNA. Cell Cycle 2016, 15, 3094–3104. [Google Scholar] [CrossRef]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020. [Google Scholar] [CrossRef]
- Yi, C.; Troutman, S.; Fera, D.; Stemmer-Rachamimov, A.; Avila, J.L.; Christian, N.; Persson, N.L.; Shimono, A.; Speicher, D.W.; Marmorstein, R.; et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 2011, 19, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Park, S.; Jove, R. Tyrosine phosphorylation of Ras GTPase-activating protein stabilizes its association with p62 at membranes of v-Src transformed cells. J. Biol. Chem. 1993, 268, 25728–25734. [Google Scholar]
- Feng, L.; Yunoue, S.; Tokuo, H.; Ozawa, T.; Zhang, D.; Patrakitkomjorn, S.; Ichimura, T.; Saya, H.; Araki, N. PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett. 2004, 557, 275–282. [Google Scholar] [CrossRef]
- Izawa, I.; Tamaki, N.; Saya, H. Phosphorylation of neurofibromatosis type 1 gene product (neurofibromin) by cAMP-dependent protein kinase. FEBS Lett. 1996, 382, 53–59. [Google Scholar] [CrossRef]
- Mangoura, D.; Sun, Y.; Li, C.; Singh, D.; Gutmann, D.H.; Flores, A.; Ahmed, M.; Vallianatos, G. Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 2006, 25, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, P.E.; Cichowski, K. Identifying the Ubiquitin Ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 2013, 3, 880–893. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, L.T.; Fromm, J.A.; Hollstein, P.E.; Kubek, S.; Beroukhim, R.; De Raedt, T.; Johnson, B.W.; Williams, S.M.; Nghiemphu, P.; Liau, L.M.; et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 2009, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; North, B.J.; Inuzuka, H. Negative regulation of DAB2IP by Akt and SCFFbw7 pathways. Oncotarget 2014, 5, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, H.; Lin, Y.; Li, J.; Pober, J.S.; Min, W. RIP1-mediated AIP1 phosphorylation at a 14-3-3-binding site is critical for tumor necrosis factor-induced ASK1-JNK/p38 activation. J. Biol. Chem. 2007, 282, 14788–14796. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Stowe, I.B.; Mercado, E.L.; Stowe, T.R.; Bell, E.L.; Oses-Prieto, J.A.; Hernandez, H.; Burlingame, A.L.; McCormick, F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012, 26, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Markegard, E.; Dharmaiah, S.; Urisman, A.; Drew, M.; Esposito, D.; Scheffzek, K.; Nissley, D.V.; McCormick, F.; Simanshu, D.K. Structural Insights into the SPRED1-Neurofibromin-KRAS Complex and Disruption of SPRED1-Neurofibromin Interaction by Oncogenic EGFR. Cell Rep. 2020, 32, 107909. [Google Scholar] [CrossRef]
- Davis, A.J.; Butt, J.T.; Walker, J.H.; Moss, S.E.; Gawler, D.J. The Ca2+-dependent lipid binding domain of P120GAP mediates protein-protein interactions with Ca2+-dependent membrane-binding proteins. Evidence for a direct interaction between annexin VI and P120GAP. J. Biol. Chem. 1996, 271, 24333–24336. [Google Scholar] [CrossRef]
- Zhou, X.; Agazie, Y.M. Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. J. Biol. Chem. 2009, 284, 12226–12234. [Google Scholar] [CrossRef]
- Hartman, Z.; Geldenhuys, W.J.; Agazie, Y.M. A specific amino acid context in EGFR and HER2 phosphorylation sites enables selective binding to the active site of Src homology phosphatase 2 (SHP2). J. Biol. Chem. 2020, 295, 3563–3575. [Google Scholar] [CrossRef]
- Tokuo, H.; Yunoue, S.; Feng, L.; Kimoto, M.; Tsuji, H.; Ono, T.; Saya, H.; Araki, N. Phosphorylation of neurofibromin by cAMP-dependent protein kinase is regulated via a cellular association of N(G),N(G)-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001, 494, 48–53. [Google Scholar] [CrossRef]
- Li, L.; Zeng, T.T.; Zhang, B.Z.; Li, Y.; Zhu, Y.H.; Guan, X.Y. Overexpression of HN1L promotes cell malignant proliferation in non-small cell lung cancer. Cancer Biol. Ther. 2017, 18, 904–915. [Google Scholar] [CrossRef]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Valentino, E.; Bellazzo, A.; Di Minin, G.; Sicari, D.; Apollonio, M.; Scognamiglio, G.; Di Bonito, M.; Botti, G.; Del Sal, G.; Collavin, L. Mutant p53 potentiates the oncogenic effects of insulin by inhibiting the tumor suppressor DAB2IP. Proc. Natl. Acad. Sci. USA 2017, 114, 7623–7628. [Google Scholar] [CrossRef] [PubMed]
- Koliou, X.; Fedonidis, C.; Kalpachidou, T.; Mangoura, D. Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J. Neurochem. 2016, 136, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; McCormick, F.; Clark, R. Characterization of full-length neurofibromin: Tubulin inhibits Ras GAP activity. EMBO J. 1993, 12, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.Y.; Anurag, M.; Lei, J.T.; Cao, J.; Singh, P.; Peng, J.; Kennedy, H.; Nguyen, N.C.; Chen, Y.; Lavere, P.; et al. Neurofibromin Is an Estrogen Receptor-alpha Transcriptional Co-repressor in Breast Cancer. Cancer Cell 2020, 37, 387–402.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, Y.; Li, X.; Zhang, J.; Lin, W.; Zhang, W.; Zheng, L.; Li, X. IPO5 promotes the proliferation and tumourigenicity of colorectal cancer cells by mediating RASAL2 nuclear transportation. J. Exp. Clin. Cancer Res. 2019, 38, 296. [Google Scholar] [CrossRef]
- Tsai, Y.S.; Lai, C.L.; Lai, C.H.; Chang, K.H.; Wu, K.; Tseng, S.F.; Fazli, L.; Gleave, M.; Xiao, G.; Gandee, L.; et al. The role of homeostatic regulation between tumor suppressor DAB2IP and oncogenic Skp2 in prostate cancer growth. Oncotarget 2014, 5, 6425–6436. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, X.; Wan, L.; Inuzuka, H.; Sun, L.; North, B.J. Smurf1 regulation of DAB2IP controls cell proliferation and migration. Oncotarget 2016, 7, 26057–26069. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Fan, J.; Chen, R. Deletion of SMURF 1 represses ovarian cancer invasion and EMT by modulating the DAB2IP/AKT/Skp2 feedback loop. J. Cell. Biochem. 2019, 120, 10643–10651. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Santiago, S.; Jardim, M.; Johnson, B.W.; Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 2003, 17, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-kappaB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e824. [Google Scholar] [CrossRef]
- Yang, F.; Xu, Y.; Liu, C.; Ma, C.; Zou, S.; Xu, X.; Jia, J.; Liu, Z. NF-kappaB/miR-223-3p/ARID1A axis is involved in Helicobacter pylori CagA-induced gastric carcinogenesis and progression. Cell Death Dis. 2018, 9, 12. [Google Scholar] [CrossRef]
- Chamorro-Jorganes, A.; Araldi, E.; Rotllan, N.; Cirera-Salinas, D.; Suarez, Y. Autoregulation of glypican-1 by intronic microRNA-149 fine tunes the angiogenic response to FGF2 in human endothelial cells. J. Cell Sci. 2014, 127, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Capponi, S.; Geroldi, A.; Mura, M.; Mandich, P.; Palombo, D. TNFalpha induces the expression of genes associated with endothelial dysfunction through p38MAPK-mediated down-regulation of miR-149. Biochem. Biophys. Res. Commun. 2014, 443, 246–251. [Google Scholar] [CrossRef]
- Anzalone, G.; Arcoleo, G.; Bucchieri, F.; Montalbano, A.M.; Marchese, R.; Albano, G.D.; Di Sano, C.; Moscato, M.; Gagliardo, R.; Ricciardolo, F.L.M.; et al. Cigarette smoke affects the onco-suppressor DAB2IP expression in bronchial epithelial cells of COPD patients. Sci. Rep. 2019, 9, 15682. [Google Scholar] [CrossRef]
- Lockyer, P.J.; Kupzig, S.; Cullen, P.J. CAPRI regulates Ca(2+)-dependent inactivation of the Ras-MAPK pathway. Curr. Biol. 2001, 11, 981–986. [Google Scholar] [CrossRef]
- Walker, S.A.; Kupzig, S.; Bouyoucef, D.; Davies, L.C.; Tsuboi, T.; Bivona, T.G.; Cozier, G.E.; Lockyer, P.J.; Buckler, A.; Rutter, G.A.; et al. Identification of a Ras GTPase-activating protein regulated by receptor-mediated Ca2+ oscillations. EMBO J. 2004, 23, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Walker, S.A.; de Vet, E.; Cook, S.; Welch, H.C.; Lockyer, P.J. Ca2+-dependent monomer and dimer formation switches CAPRI Protein between Ras GTPase-activating protein (GAP) and RapGAP activities. J. Biol. Chem. 2011, 286, 19905–19916. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Q.; Yu, W.; Xiao, Q. High glucose and/or high insulin affects HIF-1 signaling by regulating AIP1 in human umbilical vein endothelial cells. Diabetes Res. Clin. Pract. 2015, 109, 48–56. [Google Scholar] [CrossRef]
- Bandara, K.V.; Michael, M.Z.; Gleadle, J.M. MicroRNA Biogenesis in Hypoxia. Microrna 2017, 6, 80–96. [Google Scholar] [CrossRef]
- Xu, H.; Gutmann, D.H. Mutations in the GAP-related domain impair the ability of neurofibromin to associate with microtubules. Brain Res. 1997, 759, 149–152. [Google Scholar] [CrossRef]
- Endo, M.; Yamashita, T. Inactivation of Ras by p120GAP via focal adhesion kinase dephosphorylation mediates RGMa-induced growth cone collapse. J. Neurosci. 2009, 29, 6649–6662. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, C.; Wang, H.Z.; Peng, Y.N.; Li, H.O.; Zhou, Y.J.; Liu, S.; Wang, F.; Liu, L.; Chang, Y.; et al. Soft fibrin matrix downregulates DAB2IP to promote Nanog-dependent growth of colon tumor-repopulating cells. Cell Death Dis. 2019, 10, 151. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Tumor | References |
|---|---|---|
| NF1 | Hepatocellular carcinoma | [9] |
| RASA4 | Myelomonocytic leukemia | [23] |
| RASAL1 | Hepatocellular and nasopharyngeal carcinoma; breast, thyroid, and bladder cancer; NK/T-cell lymphoma | [9,25,26,63,68], reviewed in [69] |
| RASAL2 | Renal carcinoma, breast cancer | [60,70] |
| RASAL3 | Prostate CAFs | [32] |
| DAB2IP | Hepatocellular carcinoma; lung, breast, prostate, and gastrointestinal cancer; ovarian carcinoma | [9,67], reviewed in [30] |
| (a) number of cancer-related miRNAs predicted to bind a single RasGAP | ||
| RasGAP | TargetScan conserved sites | DIANA microT-CDS |
| RASA1 | 42 (53) | 111 (350) |
| NF1 | 33 (42) | 150 (388) |
| RASA2 | 51 (57) | 102 (288) |
| RASA3 | 0 (0) | 15 (39) |
| RASA4 | 0 (0) | 24 (121) |
| RASAL1 | 11 (11) | 13 (34) |
| RASAL2 | 57 (79) | 91 (354) |
| RASAL3 | 0 (0) | 15 (58) |
| SYNGAP1 | 24 (34) | 56 (257) |
| DAB2IP | 31 (41) | 41 (132) |
| (b) cancer-related miRNAs predicted to bind at least four RasGAPs | ||
| Targeted RasGAPs | onco-miRNAs | |
| NF1, RASA1, RASA2, RASA3, RASAL1, RASAL2, SYNGAP1 | hsa-miR-3163 | |
| DAB2IP, NF1, RASA1, RASA2, RASA3, RASAL2 | hsa-miR-548c-3p | |
| DAB2IP, NF1, RASA1, RASA2, RASAL1, RASAL2 | hsa-miR-5692a | |
| NF1, RASA1, RASA2, RASA4, RASAL2 | hsa-miR-5590-3p | |
| DAB2IP, NF1, RASA1, RASA2, RASAL2 | hsa-miR-582-5p | |
| DAB2IP, NF1, RASA2, RASA3, RASAL2 | hsa-miR-27b-3p, hsa-miR-27a-3p | |
| NF1, RASA2, RASAL1, RASAL2, SYNGAP1 | hsa-miR-4282 | |
| NF1, RASA1, RASA2, RASA4 | hsa-miR-129-5p | |
| NF1, RASA1, RASA2, RASAL2 | hsa-miR-30d-5p, hsa-miR-30b-5p, hsa-miR-320c, hsa-miR-4429, hsa-miR-30a-5p, hsa-miR-30e-5p, hsa-miR-320b, hsa-miR-495-3p, hsa-miR-33a-3p, hsa-miR-320d | |
| NF1, RASA1, RASA2, SYNGAP1 | hsa-miR-224-3p | |
| DAB2IP, NF1, RASA,1, RASA3 | hsa-miR-588 | |
| NF1, RASA2, RASA3, RASAL2 | hsa-miR-543 | |
| NF1, RASA2, RASA4, RASAL2 | hsa-miR-4775 | |
| DAB2IP, NF1, RASA2, RASAL2 | hsa-miR-126-5p | |
| DAB2IP, NF1, RASA2, SYNGAP1 | hsa-miR-130a-5p | |
| DAB2IP, RASA4, RASAL1, RASAL3 | hsa-miR-1285-3p | |
| RASA4, RASAL2, RASAL3, SYNGAP1 | hsa-miR-1275 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellazzo, A.; Collavin, L. Cutting the Brakes on Ras—Cytoplasmic GAPs as Targets of Inactivation in Cancer. Cancers 2020, 12, 3066. https://doi.org/10.3390/cancers12103066
Bellazzo A, Collavin L. Cutting the Brakes on Ras—Cytoplasmic GAPs as Targets of Inactivation in Cancer. Cancers. 2020; 12(10):3066. https://doi.org/10.3390/cancers12103066
Chicago/Turabian StyleBellazzo, Arianna, and Licio Collavin. 2020. "Cutting the Brakes on Ras—Cytoplasmic GAPs as Targets of Inactivation in Cancer" Cancers 12, no. 10: 3066. https://doi.org/10.3390/cancers12103066
APA StyleBellazzo, A., & Collavin, L. (2020). Cutting the Brakes on Ras—Cytoplasmic GAPs as Targets of Inactivation in Cancer. Cancers, 12(10), 3066. https://doi.org/10.3390/cancers12103066