Cx43 Present at the Leading Edge Membrane Governs Promigratory Effects of Osteoblast-Conditioned Medium on Human Prostate Cancer Cells in the Context of Bone Metastasis

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Cx43 Expression Varies with PCa Progression and Exhibits a Specific Pattern in Bone Secondary Sites
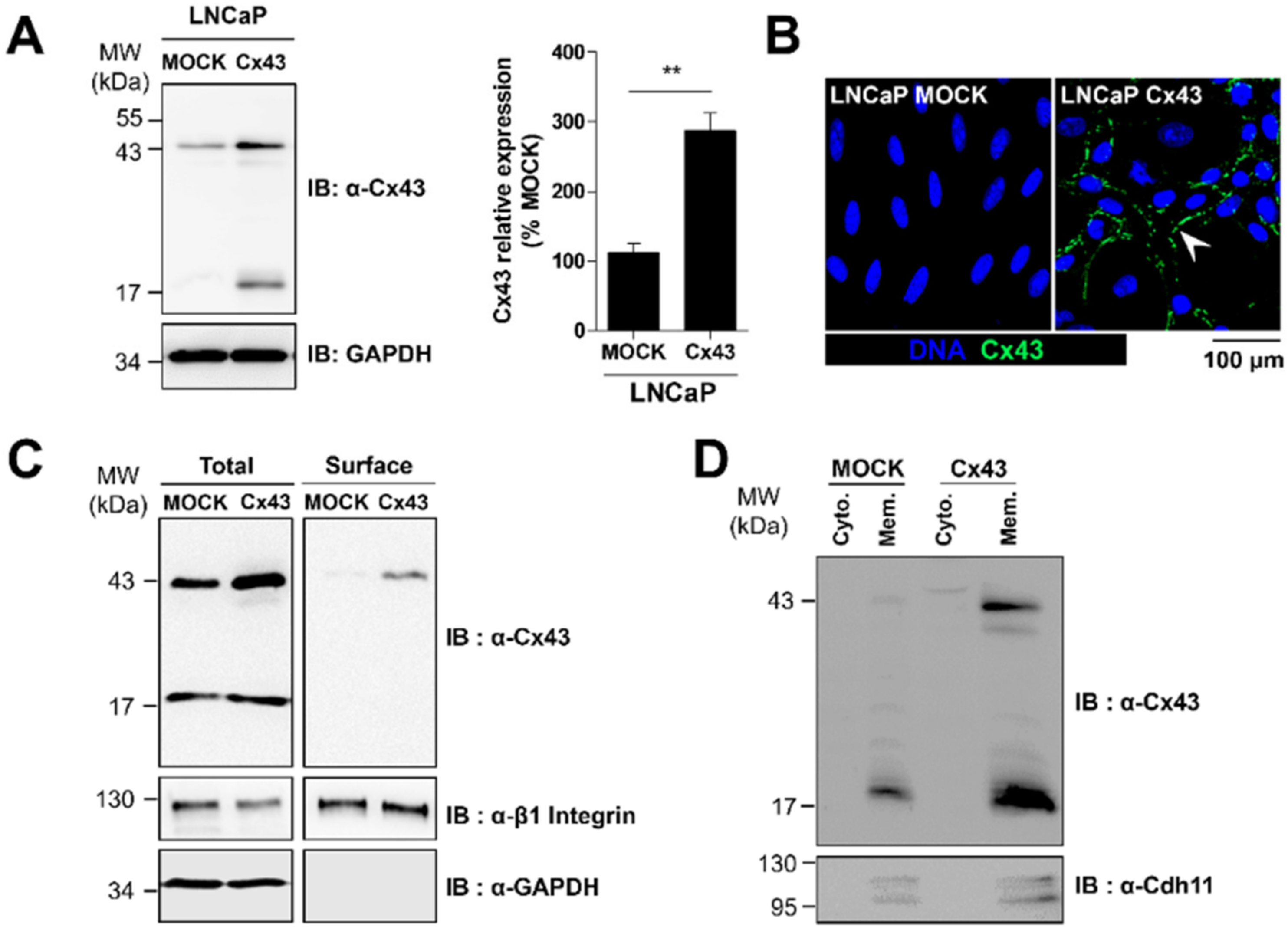
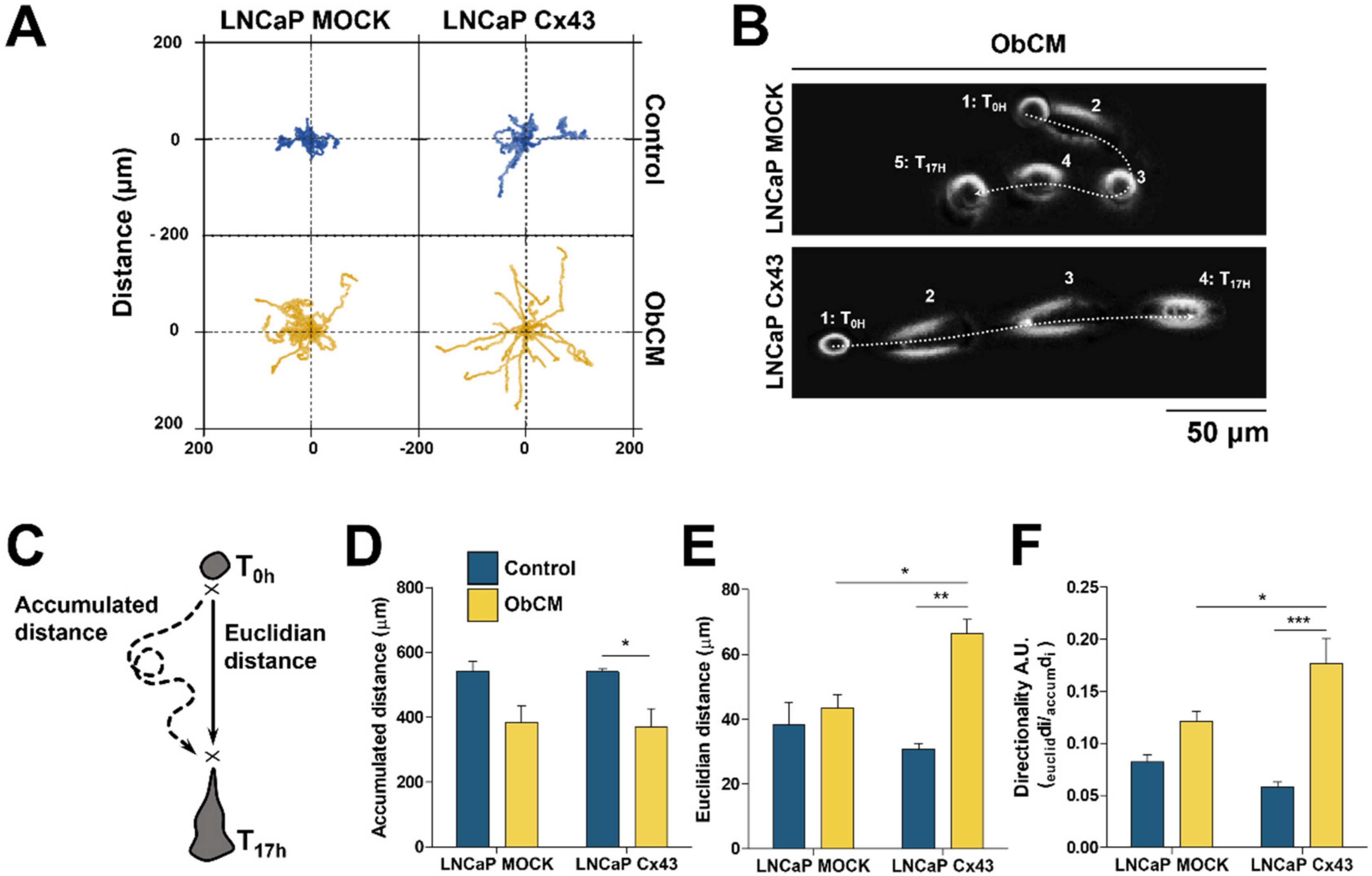
2.2. Cx43 Enhances the Migration Ability of LNCaP Cells and the Sensitivity to Bone Conditioned Medium
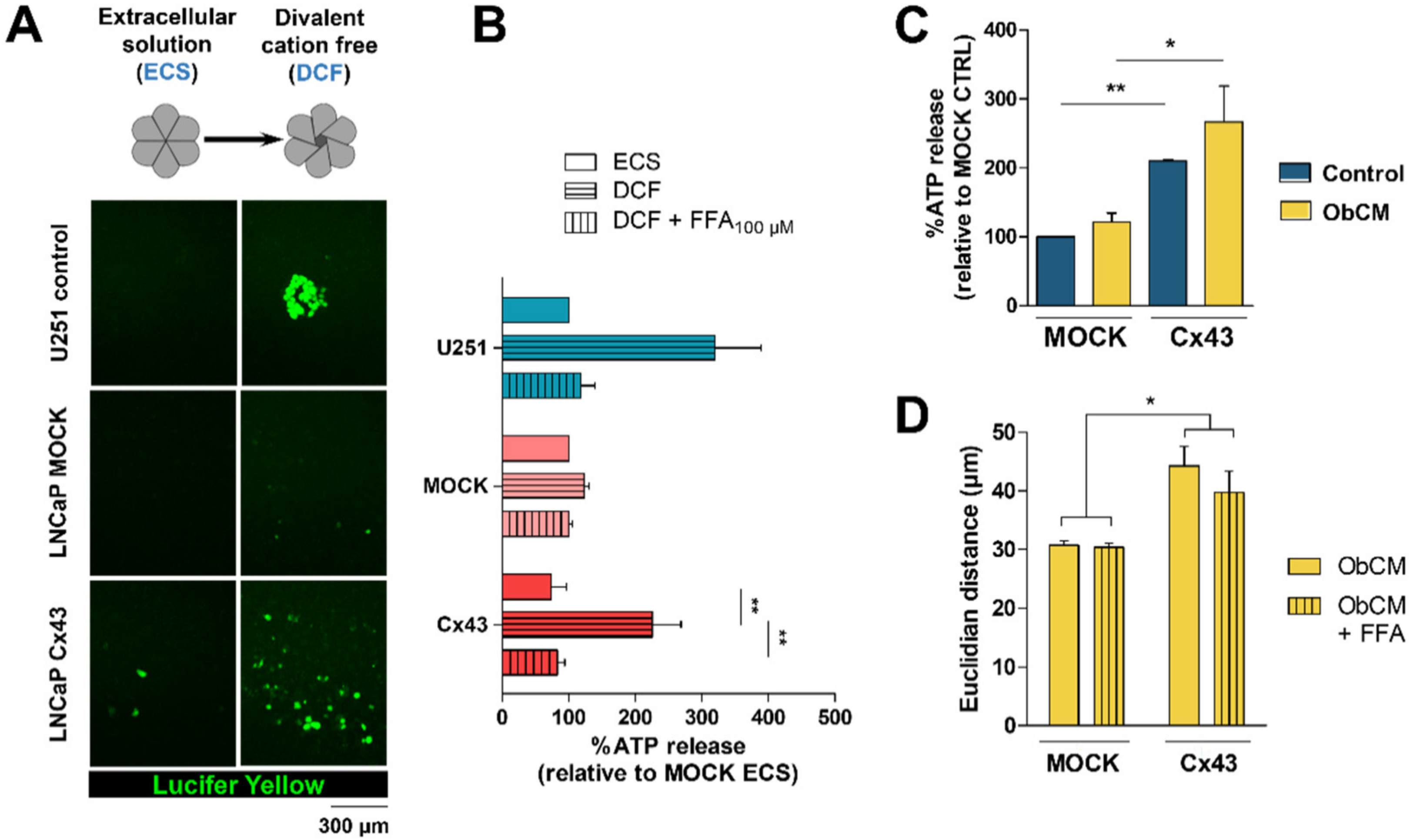
2.3. Membrane Localization of Cx43 Is Required for the Promigratory Effect of Bone-Conditioned Medium but Gap Junctional Intercellular Communication or Hemichannel Permeability Are Not Involved
2.4. Carboxy-Terminal (CT) Domain of Cx43 Is Essential for the Promigratory Effect of Bone-Conditioned Medium
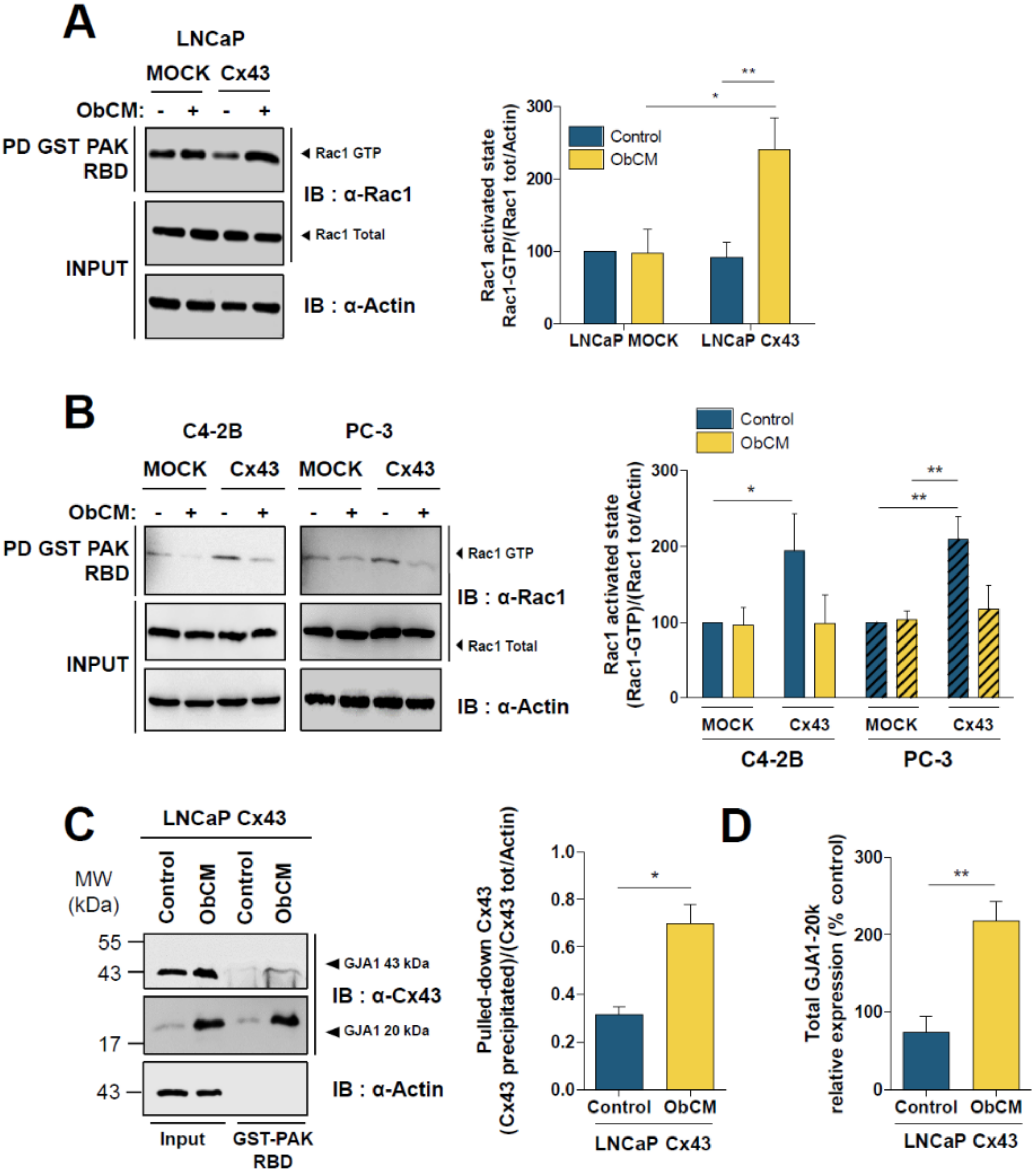
2.5. Cx43 Interacts with Cortactin and Activates the Rac1/PAK1 Pathway under Bone-Conditioned Medium Stimulation
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Plasmid Constructs
4.3. Cell Culture
4.4. Immunohistochemistry on Patient Samples
4.5. Western Blot
4.6. Immunocytofluorescence
4.7. Cell Surface Biotinylation
4.8. Integral Membrane Protein Extraction
4.9. Single Cell Migration by Time-Lapse Videomicroscopy
4.10. Scanning Electron Microscopy (SEM)
4.11. Lucifer Yellow Uptake
4.12. ATP Release
4.13. Immunoprecipitation Assay
4.14. GTP-Bound Rac1 Pull-Down Assay
4.15. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Logothetis, C.J.; Lin, S.-H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 2005, 5, 21–28. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R. Junctional intercellular communication and the control of growth. Biochim. Biophys. Acta 1979, 560, 1–65. [Google Scholar] [CrossRef]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Cronier, L.; Crespin, S.; Strale, P.-O.; Defamie, N.; Mesnil, M. Gap junctions and cancer: New functions for an old story. Antioxid. Redox Signal. 2009, 11, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef]
- Sáez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Czyż, J.; Szpak, K.; Madeja, Z. The role of connexins in prostate cancer promotion and progression. Nat. Rev. Urol. 2012, 9, 274–282. [Google Scholar] [CrossRef]
- Benko, G.; Spajić, B.; Demirović, A.; Stimac, G.; Lin, B.K.S.; Tomas, D. Prognostic value of connexin43 expression in patients with clinically localized prostate cancer. Prostate Cancer Prostatic Dis. 2011, 14, 90–95. [Google Scholar] [CrossRef]
- Govindarajan, R.; Zhao, S.; Song, X.-H.; Guo, R.-J.; Wheelock, M.; Johnson, K.R.; Mehta, P.P. Impaired trafficking of connexins in androgen-independent human prostate cancer cell lines and its mitigation by alpha-catenin. J. Biol. Chem. 2002, 277, 50087–50097. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Hitomi, M.; Bar-Shain, N.; Dalimov, Z.; Ellis, L.; Velpula, K.K.; Fraizer, G.C.; Gourdie, R.G.; Lathia, J.D. Connexin 43 expression is associated with increased malignancy in prostate cancer cell lines and functions to promote migration. Oncotarget 2015, 6, 11640–11651. [Google Scholar] [CrossRef] [PubMed]
- Lamiche, C.; Clarhaut, J.; Strale, P.-O.; Crespin, S.; Pedretti, N.; Bernard, F.-X.; Naus, C.C.; Chen, V.C.; Foster, L.J.; Defamie, N.; et al. The gap junction protein Cx43 is involved in the bone-targeted metastatic behaviour of human prostate cancer cells. Clin. Exp. Metastasis 2012, 29, 111–122. [Google Scholar] [CrossRef]
- Boucher, J.; Monvoisin, A.; Vix, J.; Mesnil, M.; Thuringer, D.; Debiais, F.; Cronier, L. Connexins, important players in the dissemination of prostate cancer cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Ryszawy, D.; Sarna, M.; Rak, M.; Szpak, K.; Kędracka-Krok, S.; Michalik, M.; Siedlar, M.; Zuba-Surma, E.; Burda, K.; Korohoda, W.; et al. Functional links between Snail-1 and Cx43 account for the recruitment of Cx43-positive cells into the invasive front of prostate cancer. Carcinogenesis 2014, 35, 1920–1930. [Google Scholar] [CrossRef]
- Thuringer, D.; Boucher, J.; Jego, G.; Pernet, N.; Cronier, L.; Hammann, A.; Solary, E.; Garrido, C. Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget 2016, 7, 73925–73934. [Google Scholar] [CrossRef]
- Piwowarczyk, K.; Paw, M.; Ryszawy, D.; Rutkowska-Zapała, M.; Madeja, Z.; Siedlar, M.; Czyż, J. Connexin43(high) prostate cancer cells induce endothelial connexin43 up-regulation through the activation of intercellular ERK1/2-dependent signaling axis. Eur. J. Cell Biol. 2017, 96, 337–346. [Google Scholar] [CrossRef]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-independent influence of connexin 43 on cell migration. Biochim. Biophys. Acta 2012, 1818, 1993–2001. [Google Scholar] [CrossRef]
- Kotini, M.; Mayor, R. Connexins in migration during development and cancer. Dev. Biol. 2015, 401, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018, 34, 823–839.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Chen, H.-J.; Chen, S.-H.; Xue, X.-Y.; Chen, H.; Zheng, Q.-S.; Wei, Y.; Li, X.-D.; Huang, J.-B.; Cai, H.; et al. Reduced Connexin 43 expression is associated with tumor malignant behaviors and biochemical recurrence-free survival of prostate cancer. Oncotarget 2016, 7, 67476–67484. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.; Werber, J.; Davia, M.O.; Edelman, M.; Tanaka, K.E.; Melman, A.; Christ, G.J.; Geliebter, J. Reduced connexin 43 expression in high grade, human prostatic adenocarcinoma cells. Biochem. Biophys. Res. Commun. 1996, 227, 64–69. [Google Scholar] [CrossRef]
- Cai, C.; Wang, H.; He, H.H.; Chen, S.; He, L.; Ma, F.; Mucci, L.; Wang, Q.; Fiore, C.; Sowalsky, A.G.; et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Investig. 2013, 123, 1109–1122. [Google Scholar] [CrossRef]
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2018, 85, 108–116. [Google Scholar] [CrossRef]
- Salat-Canela, C.; Sesé, M.; Peula, C.; y Cajal, S.R.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. CCS 2014, 12, 31. [Google Scholar] [CrossRef]
- Chepied, A.; Daoud-Omar, Z.; Meunier-Balandre, A.-C.; Laird, D.W.; Mesnil, M.; Defamie, N. Involvement of the Gap Junction Protein, Connexin43, in the Formation and Function of Invadopodia in the Human U251 Glioblastoma Cell Line. Cells 2020, 9, 117. [Google Scholar] [CrossRef]
- Aquilina, J.W.; Lipsky, J.J.; Bostwick, D.G. Androgen deprivation as a strategy for prostate cancer chemoprevention. J. Natl. Cancer Inst. 1997, 89, 689–696. [Google Scholar] [CrossRef]
- Chen, R.; Luan, Y.; Liu, Z.; Song, W.; Wu, L.; Li, M.; Yang, J.; Liu, X.; Wang, T.; Liu, J.; et al. AR Pathway Is Involved in the Regulation of CX43 in Prostate Cancer. BioMed Res. Int. 2015, 2015, 514234. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Bellahcène, A.; Bachelier, R.; Detry, C.; Lidereau, R.; Clézardin, P.; Castronovo, V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 135–148. [Google Scholar] [CrossRef]
- Tate, A.W.; Lung, T.; Radhakrishnan, A.; Lim, S.D.; Lin, X.; Edlund, M. Changes in gap junctional connexin isoforms during prostate cancer progression. Prostate 2006, 66, 19–31. [Google Scholar] [CrossRef]
- Lin, J.H.C.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [CrossRef]
- Bates, D.C.; Sin, W.C.; Aftab, Q.; Naus, C.C. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 2007, 55, 1554–1564. [Google Scholar] [CrossRef]
- Khalil, A.A.; Ilina, O.; Vasaturo, A.; Venhuizen, J.-H.; Vullings, M.; Venhuizen, V.; Bilos, A.; Figdor, C.G.; Span, P.N.; Friedl, P. Collective invasion induced by an autocrine purinergic loop through connexin-43 hemichannels. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; Xu, X.; Park, H.; Wei, C.-J.; Chang, S.; Chatterjee, B.; Lo, C. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS ONE 2011, 6, e26379. [Google Scholar] [CrossRef] [PubMed]
- Matsuuchi, L.; Naus, C.C. Gap junction proteins on the move: Connexins, the cytoskeleton and migration. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 94–108. [Google Scholar] [CrossRef]
- Sinha, G.; Ferrer, A.I.; Moore, C.A.; Naaldijk, Y.; Rameshwar, P. Gap Junctions and Breast Cancer Dormancy. Trends Cancer 2020, 6, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Francis, R.; Wei, C.J.; Linask, K.L.; Lo, C.W. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Dev. Camb. Engl. 2006, 133, 3629–3639. [Google Scholar] [CrossRef]
- Vignjevic, D.; Montagnac, G. Reorganisation of the dendritic actin network during cancer cell migration and invasion. Semin. Cancer Biol. 2008, 18, 12–22. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar] [CrossRef]
- Crespin, S.; Bechberger, J.; Mesnil, M.; Naus, C.C.; Sin, W.-C. The carboxy-terminal tail of connexin43 gap junction protein is sufficient to mediate cytoskeleton changes in human glioma cells. J. Cell. Biochem. 2010, 110, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.V.; Caggia, S.; Gambrell-Sanders, D.; Khan, S.A. Differential roles and activation of mammalian target of rapamycin complexes 1 and 2 during cell migration in prostate cancer cells. Prostate 2020, 80, 412–423. [Google Scholar] [CrossRef]
- Giepmans, B.N.G. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef]
- Olk, S.; Zoidl, G.; Dermietzel, R. Connexins, cell motility, and the cytoskeleton. Cell Motil. Cytoskeleton 2009, 66, 1000–1016. [Google Scholar] [CrossRef]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein−Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef]
- Behrens, J.; Kameritsch, P.; Wallner, S.; Pohl, U.; Pogoda, K. The carboxyl tail of Cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur. J. Cell Biol. 2010, 89, 828–838. [Google Scholar] [CrossRef]
- Boucher, J. Involvement of Connexin 43 in Prostate Cancer Progression to Bone. Ph.D. Thesis, STIM Laboratory, University of Poitiers, Poitiers, France, 2018. Unpublished work. [Google Scholar]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.; Shaw, R. The “tail” of Connexin43: An unexpected journey from alternative translation to trafficking. Biochim. Biophys. Acta 2016, 1863, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar] [CrossRef]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Kleber, A.G.; Hong, T.; et al. Auxiliary trafficking subunit GJA1-20k protects connexin-43 from degradation and limits ventricular arrhythmias. J. Clin. Investig. 2020, 130, 4858–4870. [Google Scholar] [CrossRef] [PubMed]
- James, C.C.; Zeitz, M.J.; Calhoun, P.J.; Lamouille, S.; Smyth, J.W. Altered translation initiation of Gja1 limits gap junction formation during epithelial-mesenchymal transition. Mol. Biol. Cell 2018, 29, 797–808. [Google Scholar] [CrossRef]
- Blaszczyk, N.; Masri, B.A.; Mawji, N.R.; Ueda, T.; McAlinden, G.; Duncan, C.P.; Bruchovsky, N.; Schweikert, H.-U.; Schnabel, D.; Jones, E.C.; et al. Osteoblast-derived factors induce androgen-independent proliferation and expression of prostate-specific antigen in human prostate cancer cells. Clin. Cancer Res. 2004, 10, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.A.; Morgado, M.; Warren, C.R.; Hinton, C.V.; Farach-Carson, M.C.; Delk, N.A. p62/SQSTM1 is required for cell survival of apoptosis-resistant bone metastatic prostate cancer cell lines. Prostate 2014, 74, 149–163. [Google Scholar] [CrossRef]
- Millimaggi, D.; Festuccia, C.; Angelucci, A.; D’Ascenzo, S.; Rucci, N.; Flati, S.; Bologna, M.; Teti, A.; Pavan, A.; Dolo, V. Osteoblast-conditioned media stimulate membrane vesicle shedding in prostate cancer cells. Int. J. Oncol. 2006, 28, 909–914. [Google Scholar] [CrossRef]
- Festuccia, C.; Bologna, M.; Gravina, G.L.; Guerra, F.; Angelucci, A.; Villanova, I.; Millimaggi, D.; Teti, A. Osteoblast conditioned media contain TGF-beta1 and modulate the migration of prostate tumor cells and their interactions with extracellular matrix components. Int. J. Cancer 1999, 81, 395–403. [Google Scholar] [CrossRef]
- Chang, A.-C.; Chen, P.-C.; Lin, Y.-F.; Su, C.-M.; Liu, J.-F.; Lin, T.-H.; Chuang, S.-M.; Tang, C.-H. Osteoblast-secreted WISP-1 promotes adherence of prostate cancer cells to bone via the VCAM-1/integrin α4β1 system. Cancer Lett. 2018, 426, 47–56. [Google Scholar] [CrossRef]
- Yu-Lee, L.-Y.; Yu, G.; Lee, Y.-C.; Lin, S.-C.; Pan, J.; Pan, T.; Yu, K.-J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFβRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Jacob, K.; Webber, M.; Benayahu, D.; Kleinman, H.K. Osteonectin promotes prostate cancer cell migration and invasion: A possible mechanism for metastasis to bone. Cancer Res. 1999, 59, 4453–4457. [Google Scholar]
- Karlsson, T.; Sundar, R.; Widmark, A.; Landström, M.; Persson, E. Osteoblast-derived factors promote metastatic potential in human prostate cancer cells, in part via non-canonical transforming growth factor β (TGFβ) signaling. Prostate 2018, 78, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Qu, X.; Fan, Q.; Wang, L.; Tang, T.; Hao, Y.; Dai, K. Regulation of prostate cancer cell migration toward bone marrow stromal cell-conditioned medium by Wnt5a signaling. Mol. Med. Rep. 2013, 8, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Urata, S.; Izumi, K.; Hiratsuka, K.; Maolake, A.; Natsagdorj, A.; Shigehara, K.; Iwamoto, H.; Kadomoto, S.; Makino, T.; Naito, R.; et al. C-C motif ligand 5 promotes migration of prostate cancer cells in the prostate cancer bone metastasis microenvironment. Cancer Sci. 2018, 109, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Choi, C.H.; Li, Q.K.; Rahmatpanah, F.B.; Chen, X.; Kim, S.R.; Veltri, R.; Chia, D.; Zhang, Z.; Mercola, D.; et al. Overexpression of periostin in stroma positively associated with aggressive prostate cancer. PLoS ONE 2015, 10, e0121502. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Role of microenvironmental periostin in pancreatic cancer progression. Oncotarget 2017, 8, 89552–89565. [Google Scholar] [CrossRef]
- Webber, J.; Yeung, V.; Clayton, A. Extracellular vesicles as modulators of the cancer microenvironment. Semin. Cell Dev. Biol. 2015, 40, 27–34. [Google Scholar] [CrossRef]
- Jiang, S.; Mo, C.; Guo, S.; Zhuang, J.; Huang, B.; Mao, X. Human bone marrow mesenchymal stem cells-derived microRNA-205-containing exosomes impede the progression of prostate cancer through suppression of RHPN2. J. Exp. Clin. Cancer Res. CR 2019, 38, 495. [Google Scholar] [CrossRef]
- Tong, D.; Li, T.Y.; Naus, K.E.; Bai, D.; Kidder, G.M. In vivo analysis of undocked connexin43 gap junction hemichannels in ovarian granulosa cells. J. Cell Sci. 2007, 120, 4016–4024. [Google Scholar] [CrossRef]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. Biochim. Biophys. Acta 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boucher, J.; Balandre, A.-C.; Debant, M.; Vix, J.; Harnois, T.; Bourmeyster, N.; Péraudeau, E.; Chépied, A.; Clarhaut, J.; Debiais, F.; et al. Cx43 Present at the Leading Edge Membrane Governs Promigratory Effects of Osteoblast-Conditioned Medium on Human Prostate Cancer Cells in the Context of Bone Metastasis. Cancers 2020, 12, 3013. https://doi.org/10.3390/cancers12103013
Boucher J, Balandre A-C, Debant M, Vix J, Harnois T, Bourmeyster N, Péraudeau E, Chépied A, Clarhaut J, Debiais F, et al. Cx43 Present at the Leading Edge Membrane Governs Promigratory Effects of Osteoblast-Conditioned Medium on Human Prostate Cancer Cells in the Context of Bone Metastasis. Cancers. 2020; 12(10):3013. https://doi.org/10.3390/cancers12103013
Chicago/Turabian StyleBoucher, Jonathan, Annie-Claire Balandre, Marjolaine Debant, Justine Vix, Thomas Harnois, Nicolas Bourmeyster, Elodie Péraudeau, Amandine Chépied, Jonathan Clarhaut, Françoise Debiais, and et al. 2020. "Cx43 Present at the Leading Edge Membrane Governs Promigratory Effects of Osteoblast-Conditioned Medium on Human Prostate Cancer Cells in the Context of Bone Metastasis" Cancers 12, no. 10: 3013. https://doi.org/10.3390/cancers12103013
APA StyleBoucher, J., Balandre, A.-C., Debant, M., Vix, J., Harnois, T., Bourmeyster, N., Péraudeau, E., Chépied, A., Clarhaut, J., Debiais, F., Monvoisin, A., & Cronier, L. (2020). Cx43 Present at the Leading Edge Membrane Governs Promigratory Effects of Osteoblast-Conditioned Medium on Human Prostate Cancer Cells in the Context of Bone Metastasis. Cancers, 12(10), 3013. https://doi.org/10.3390/cancers12103013