VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. DNA Damage Induces Phosphorylation of Tip60 and Acetylation of H4K16
2.2. Kinase-Active VRK1 Rescues H4K16 Acetylation Induced by DNA Damage
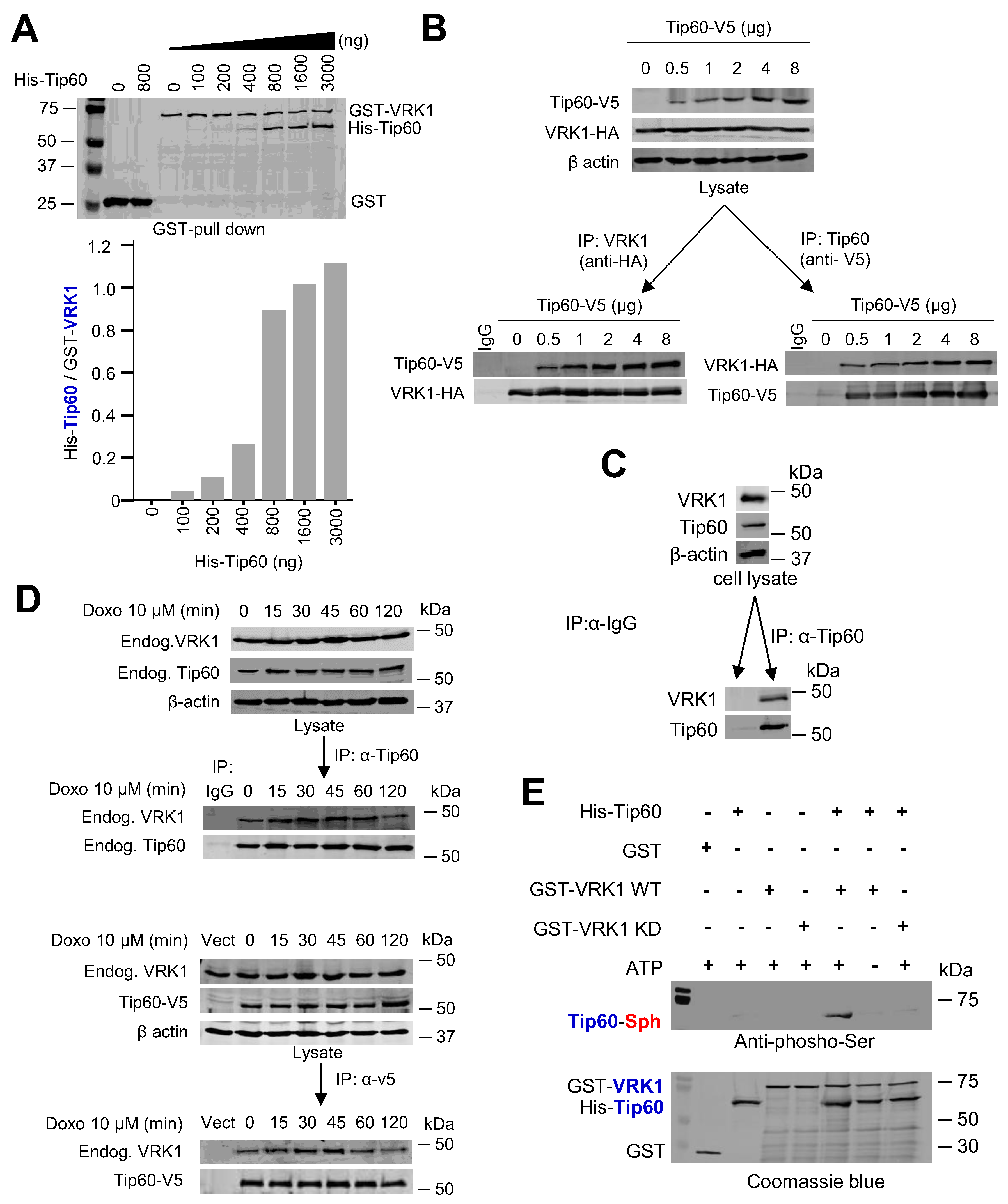
2.3. VRK1 Directly Interacts with and Phosphorylates Tip60
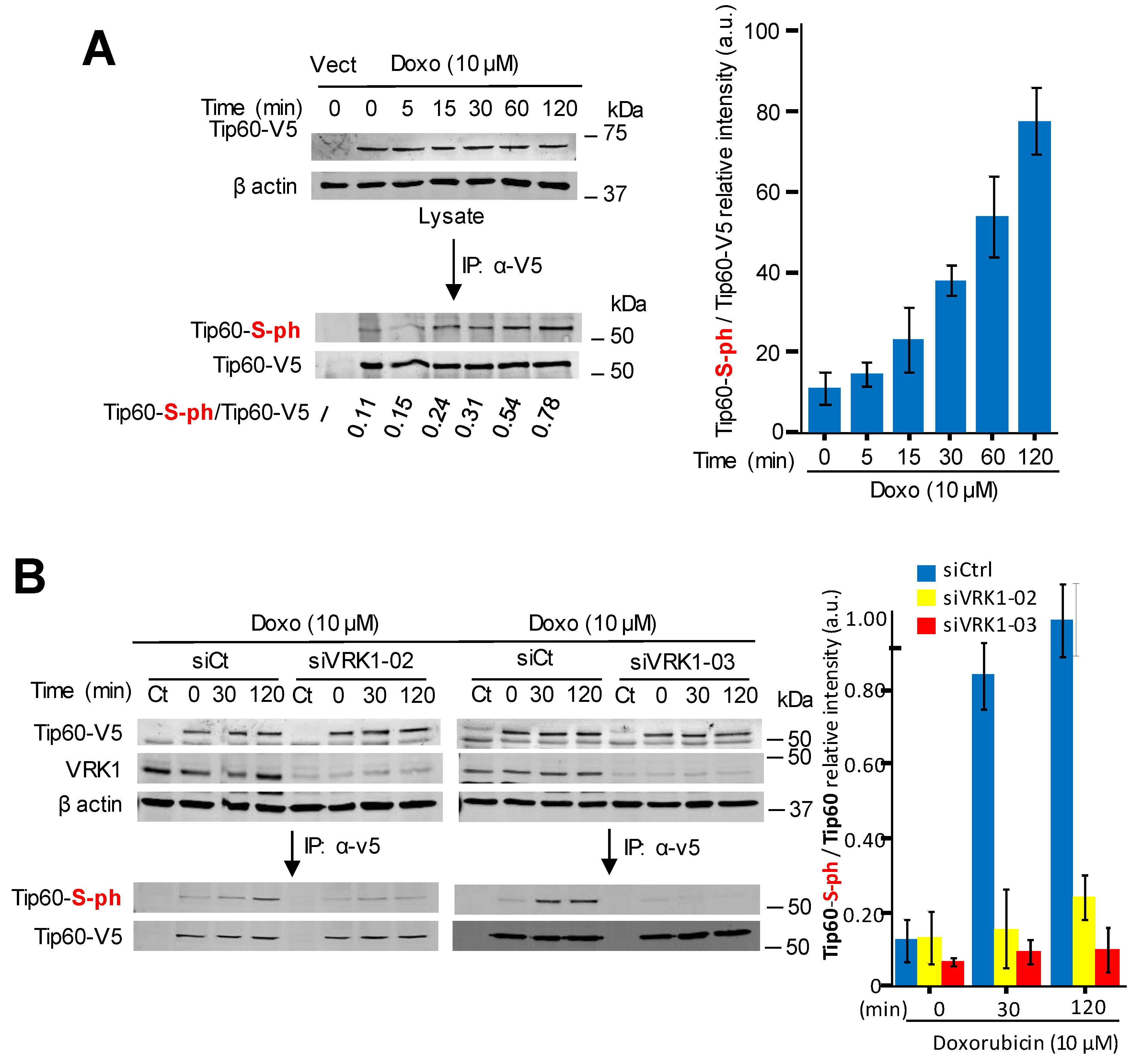
2.4. Kinase-Active VRK1 Mediates the Phosphorylation of Tip60 Induced by DNA-Damage
2.5. VRK1 Depletion Prevents the Accumulation in Chromatin of Phospho-Tip60 Induced by DNA Damage
2.6. The Phosphorylation of Tip60 by VRK1 is Induced by Doxorubicin in ATM Null Cells
2.7. ATM Acetylation by Tip60 Is Insensitive to ATM Kinase Inhibitors but Is Lost by VRK1 Depletion or Tip60 Inhibition
2.8. The Kinase Activity of VRK1 Is Necessary to Rescue ATM Acetylation and Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Plasmids
4.3. Cell Lines, Culture and Transfections
4.4. VRK1 Depletion by siRNA
4.5. Antibodies
4.6. Immunoblots
4.7. Immunoprecipitations
4.8. In Vitro VRK1-Tip60 Protein Interaction and Kinase Assay
4.9. Immunofluorescence and Confocal Microscopy
4.10. Nucleoplasm-Chromatin Fractionation
4.11. Statistical Analysis
4.12. TUNEL Assay
4.13. Database Submission
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, E.; Shilatifard, A. The chromatin signaling pathway: Diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 2010, 40, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell. Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.H.; McGarvey, K.M.; Baylin, S.B. The cancer epigenome–components and functional correlates. Genes Dev. 2006, 20, 3215–3231. [Google Scholar] [CrossRef] [PubMed]
- Beekman, R.; Chapaprieta, V.; Russinol, N.; Vilarrasa-Blasi, R.; Verdaguer-Dot, N.; Martens, J.H.A.; Duran-Ferrer, M.; Kulis, M.; Serra, F.; Javierre, B.M.; et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef]
- Xhemalce, B.; Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 2010, 24, 647–652. [Google Scholar] [CrossRef]
- Murata, K.; Kouzarides, T.; Bannister, A.J.; Gurdon, J.B. Histone H3 lysine 4 methylation is associated with the transcriptional reprogramming efficiency of somatic nuclei by oocytes. Epigenet. Chromatin 2010, 3, 4. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Cantarero, L.; Moura, D.S.; Salzano, M.; Monsalve, D.M.; Campillo-Marcos, I.; Martín-Doncel, E.; Lazo, P.A. VRK1 (vaccinia-related kinase 1). In Encyclopedia of Signaling Molecules, 2nd ed.; Choi, S., Ed.; Springer Science: Berlin, Germany, 2017. [Google Scholar] [CrossRef]
- Aihara, H.; Nakagawa, T.; Mizusaki, H.; Yoneda, M.; Kato, M.; Doiguchi, M.; Imamura, Y.; Higashi, M.; Ikura, T.; Hayashi, T.; et al. Histone H2A T120 Phosphorylation Promotes Oncogenic Transformation via Upregulation of Cyclin D1. Mol. Cell 2016, 64, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M.; et al. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef]
- Santos, C.R.; Rodriguez-Pinilla, M.; Vega, F.M.; Rodriguez-Peralto, J.L.; Blanco, S.; Sevilla, A.; Valbuena, A.; Hernandez, T.; van Wijnen, A.J.; Li, F.; et al. VRK1 signaling pathway in the context of the proliferation phenotype in head and neck squamous cell carcinoma. Mol. Cancer Res. 2006, 4, 177–185. [Google Scholar] [CrossRef]
- Valbuena, A.; Lopez-Sanchez, I.; Lazo, P.A. Human VRK1 is an early response gene and its loss causes a block in cell cycle progression. PLoS ONE 2008, 3, e1642. [Google Scholar] [CrossRef]
- Moura, D.S.; Fernández, I.F.; Marín-Royo, G.; López-Sánchez, I.; Martín-Doncel, E.; Vega, F.M.; Lazo, P.A. Oncogenic Sox2 regulates and cooperates with VRK1 in cell cycle progression and differentiation. Sci. Rep. 2016, 6, 28532. [Google Scholar] [CrossRef]
- Sanz-Garcia, M.; Monsalve, D.M.; Sevilla, A.; Lazo, P.A. Vaccinia-related Kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. J. Biol. Chem. 2012, 287, 23757–23768. [Google Scholar] [CrossRef]
- Monsalve, D.M.; Campillo-Marcos, I.; Salzano, M.; Sanz-Garcia, M.; Cantarero, L.; Lazo, P.A. VRK1 phosphorylates and protects NBS1 from ubiquitination and proteasomal degradation in response to DNA damage. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 760–769. [Google Scholar] [CrossRef]
- Salzano, M.; Sanz-Garcia, M.; Monsalve, D.M.; Moura, D.S.; Lazo, P.A. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 2015, 10, 373–383. [Google Scholar] [CrossRef]
- Campillo-Marcos, I.; Lazo, P.A. Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: A therapeutic target? Cell Mol. Life Sci. 2018, 75, 2375–2388. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Borges, S.; Lazo, P.A. The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene 2000, 19, 3656–3664. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Sevilla, A.; Lazo, P.A. p53 Stabilization and accumulation induced by human vaccinia-related kinase 1. Mol. Cell. Biol. 2004, 24, 10366–10380. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, A.; Vega, F.M.; Blanco, S.; Lazo, P.A. p53 downregulates its activating vaccinia-related kinase 1, forming a new autoregulatory loop. Mol. Cell. Biol. 2006, 26, 4782–4793. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, A.; Santos, C.R.; Barcia, R.; Vega, F.M.; Lazo, P.A. c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK). Oncogene 2004, 23, 8950–8958. [Google Scholar] [CrossRef]
- Sevilla, A.; Santos, C.R.; Vega, F.M.; Lazo, P.A. Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on Thr-73 and Ser-62 and cooperates with JNK. J. Biol. Chem. 2004, 279, 27458–27465. [Google Scholar] [CrossRef]
- Kang, T.H.; Park, D.Y.; Kim, W.; Kim, K.T. VRK1 phosphorylates CREB and mediates CCND1 expression. J. Cell. Sci. 2008, 121, 3035–3041. [Google Scholar] [CrossRef]
- Jamin, A.; Wiebe, M.S. Barrier to Autointegration Factor (BANF1): Interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 2015, 34, 61–68. [Google Scholar] [CrossRef]
- Wiebe, M.S.; Jamin, A. The Barrier to Autointegration Factor: Interlocking Antiviral Defense with Genome Maintenance. J. Virol. 2016, 90, 3806–3809. [Google Scholar] [CrossRef]
- Samwer, M.; Schneider, M.W.G.; Hoefler, R.; Schmalhorst, P.S.; Jude, J.G.; Zuber, J.; Gerlich, D.W. DNA Cross-Bridging Shapes a Single Nucleus from a Set of Mitotic Chromosomes. Cell 2017, 170, 956–972.e923. [Google Scholar] [CrossRef]
- Kang, T.H.; Park, D.Y.; Choi, Y.H.; Kim, K.J.; Yoon, H.S.; Kim, K.T. Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol. Cell. Biol. 2007, 27, 8533–8546. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Cedeira, M.; Barcia-Sanjurjo, I.; Sanz-Garcia, M.; Barcia, R.; Lazo, P.A. Differential Inhibitor Sensitivity between Human Kinases VRK1 and VRK2. PLoS ONE 2011, 6, e23235. [Google Scholar] [CrossRef] [PubMed]
- Moura, D.S.; Campillo-Marcos, I.; Vazquez-Cedeira, M.; Lazo, P.A. VRK1 and AURKB form a complex that cross inhibit their kinase activity and the phosphorylation of histone H3 in the progression of mitosis. Cell. Mol. Life Sci. 2018, 76, 2591–2611. [Google Scholar] [CrossRef]
- Aihara, H.; Nakagawa, T.; Yasui, K.; Ohta, T.; Hirose, S.; Dhomae, N.; Takio, K.; Kaneko, M.; Takeshima, Y.; Muramatsu, M.; et al. Nucleosomal histone kinase-1 phosphorylates H2A Thr 119 during mitosis in the early Drosophila embryo. Genes Dev. 2004, 18, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Marcos, I.; Lazo, P.A. Olaparib and ionizing radiation trigger a cooperative DNA-damage repair response that is impaired by depletion of the VRK1 chromatin kinase. J. Exp. Clin. Cancer Res. 2019, 38, 203. [Google Scholar] [CrossRef]
- Choi, Y.H.; Lim, J.K.; Jeong, M.W.; Kim, K.T. HnRNP A1 phosphorylated by VRK1 stimulates telomerase and its binding to telomeric DNA sequence. Nucleic Acids Res. 2012, 40, 8499–8518. [Google Scholar] [CrossRef]
- Shin, J.; Chakraborty, G.; Bharatham, N.; Kang, C.; Tochio, N.; Koshiba, S.; Kigawa, T.; Kim, W.; Kim, K.T.; Yoon, H.S. NMR solution structure of human vaccinia-related kinase 1 (VRK1) reveals the C-terminal tail essential for its structural stability and autocatalytic activity. J. Biol. Chem. 2011, 286, 22131–22138. [Google Scholar] [CrossRef]
- Kim, W.; Chakraborty, G.; Kim, S.; Shin, J.; Park, C.H.; Jeong, M.W.; Bharatham, N.; Yoon, H.S.; Kim, K.T. Macro Histone H2A1.2 (MacroH2A1) Protein Suppresses Mitotic Kinase VRK1 during Interphase. J. Biol. Chem. 2012, 287, 5278–5289. [Google Scholar] [CrossRef]
- Sanz-Garcia, M.; Lopez-Sanchez, I.; Lazo, P.A. Proteomics identification of nuclear Ran GTPase as an inhibitor of human VRK1 and VRK2 (vaccinia-related kinase) activities. Mol. Cell. Proteom. 2008, 7, 2199–2214. [Google Scholar] [CrossRef]
- Robinson, P.J.; An, W.; Routh, A.; Martino, F.; Chapman, L.; Roeder, R.G.; Rhodes, D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J. Mol. Biol. 2008, 381, 816–825. [Google Scholar] [CrossRef]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell. Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Stante, M.; Minopoli, G.; Passaro, F.; Raia, M.; Vecchio, L.D.; Russo, T. Fe65 is required for Tip60-directed histone H4 acetylation at DNA strand breaks. Proc. Natl. Acad. Sci. USA 2009, 106, 5093–5098. [Google Scholar] [CrossRef]
- Krishnan, V.; Chow, M.Z.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef]
- Ikura, M.; Furuya, K.; Matsuda, S.; Matsuda, R.; Shima, H.; Adachi, J.; Matsuda, T.; Shiraki, T.; Ikura, T. Acetylation of Histone H2AX at Lys 5 by the TIP60 Histone Acetyltransferase Complex Is Essential for the Dynamic Binding of NBS1 to Damaged Chromatin. Mol. Cell. Biol. 2015, 35, 4147–4157. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Renaud, E.; Barascu, A.; Rosselli, F. Impaired TIP60-mediated H4K16 acetylation accounts for the aberrant chromatin accumulation of 53BP1 and RAP80 in Fanconi anemia pathway-deficient cells. Nucleic Acids Res. 2016, 44, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef]
- Chen, G.; Cheng, Y.; Tang, Y.; Martinka, M.; Li, G. Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. J. Investig. Dermatol. 2012, 132, 2632–2641. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Noguchi, T.; Nakajima, Y.; Ohmiya, Y.; Shiota, M.; Kidani, A.; Tawara, A.; Kohno, K. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J. Biol. Chem. 2008, 283, 18218–18226. [Google Scholar] [CrossRef]
- Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Chabannon, C.; Charpin, C.; Chaffanet, M.; Jacquemier, J.; Viens, P.; Birnbaum, D.; Bertucci, F. Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res. 2008, 68, 767–776. [Google Scholar] [CrossRef]
- Salzano, M.; Vazquez-Cedeira, M.; Sanz-Garcia, M.; Valbuena, A.; Blanco, S.; Fernandez, I.F.; Lazo, P.A. Vaccinia-related kinase 1 (VRK1) confers resistance to DNA-damaging agents in human breast cancer by affecting DNA damage response. Oncotarget 2014, 5, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef]
- Martin-Doncel, E.; Rojas, A.M.; Cantarero, L.; Lazo, P.A. VRK1 functional insufficiency due to alterations in protein stability or kinase activity of human VRK1 pathogenic variants implicated in neuromotor syndromes. Sci. Rep. 2019, 9, 13381. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef]
- Zhang, T.; Shen, Y.; Chen, Y.; Hsieh, J.T.; Kong, Z. The ATM inhibitor KU55933 sensitizes radioresistant bladder cancer cells with DAB2IP gene defect. Int. J. Radiat. Biol. 2015, 91, 368–378. [Google Scholar] [CrossRef]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar]
- Zhou, B.B.; Chaturvedi, P.; Spring, K.; Scott, S.P.; Johanson, R.A.; Mishra, R.; Mattern, M.R.; Winkler, J.D.; Khanna, K.K. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J. Biol. Chem. 2000, 275, 10342–10348. [Google Scholar] [CrossRef]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef] [PubMed]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 2011, 42, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Brauns-Schubert, P.; Schubert, F.; Wissler, M.; Weiss, M.; Schlicher, L.; Bessler, S.; Safavi, M.; Miething, C.; Borner, C.; Brummer, T.; et al. CDK9-mediated phosphorylation controls the interaction of TIP60 with the transcriptional machinery. EMBO Rep. 2018, 19, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Lemercier, C.; Legube, G.; Caron, C.; Louwagie, M.; Garin, J.; Trouche, D.; Khochbin, S. Tip60 acetyltransferase activity is controlled by phosphorylation. J. Biol. Chem. 2003, 278, 4713–4718. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar] [CrossRef]
- Chailleux, C.; Tyteca, S.; Papin, C.; Boudsocq, F.; Puget, N.; Courilleau, C.; Grigoriev, M.; Canitrot, Y.; Trouche, D. Physical interaction between the histone acetyl transferase Tip60 and the DNA double-strand breaks sensor MRN complex. Biochem. J. 2010, 426, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.Y.; Mizzen, C.A. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J. Mol. Cell. Biol. 2013, 5, 157–165. [Google Scholar] [CrossRef]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, I.; Valbuena, A.; Vazquez-Cedeira, M.; Khadake, J.; Sanz-Garcia, M.; Carrillo-Jimenez, A.; Lazo, P.A. VRK1 interacts with p53 forming a basal complex that is activated by UV-induced DNA damage. FEBS Lett. 2014, 588, 692–700. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Cantarero, L.; Sanz-Garcia, M.; Vinograd-Byk, H.; Renbaum, P.; Levy-Lahad, E.; Lazo, P.A. VRK1 regulates Cajal body dynamics and protects coilin from proteasomal degradation in cell cycle. Sci. Rep. 2015, 5, 10543. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, I.; Sanz-Garcia, M.; Lazo, P.A. Plk3 interacts with and specifically phosphorylates VRK1 in Ser342, a downstream target in a pathway that induces Golgi fragmentation. Mol. Cell. Biol. 2009, 29, 1189–1201. [Google Scholar] [CrossRef]
- Marcos, A.T.; Martin-Doncel, E.; Morejon-Garcia, P.; Marcos-Alcalde, I.; Gomez-Puertas, P.; Segura-Puimedon, M.; Armengol, L.; Navarro-Pando, J.M.; Lazo, P.A. VRK1 (Y213H) homozygous mutant impairs Cajal bodies in a hereditary case of distal motor neuropathy. Ann. Clin. Transl. Neurol. 2020, 7, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, A.; Castro-Obregon, S.; Lazo, P.A. Downregulation of VRK1 by p53 in Response to DNA Damage Is Mediated by the Autophagic Pathway. PLoS ONE 2011, 6, e17320. [Google Scholar] [CrossRef]
- Bremer, M.; Doerge, R.M. Statistics at the Bench: A Step-By Step Handbook for Biologists; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009. [Google Scholar]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-González, R.; Morejón-García, P.; Campillo-Marcos, I.; Salzano, M.; Lazo, P.A. VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers 2020, 12, 2986. https://doi.org/10.3390/cancers12102986
García-González R, Morejón-García P, Campillo-Marcos I, Salzano M, Lazo PA. VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers. 2020; 12(10):2986. https://doi.org/10.3390/cancers12102986
Chicago/Turabian StyleGarcía-González, Raúl, Patricia Morejón-García, Ignacio Campillo-Marcos, Marcella Salzano, and Pedro A. Lazo. 2020. "VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage" Cancers 12, no. 10: 2986. https://doi.org/10.3390/cancers12102986
APA StyleGarcía-González, R., Morejón-García, P., Campillo-Marcos, I., Salzano, M., & Lazo, P. A. (2020). VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers, 12(10), 2986. https://doi.org/10.3390/cancers12102986