FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer

 , , , ,
, , , , 
Simple Summary
Abstract
1. Introduction
2. Results
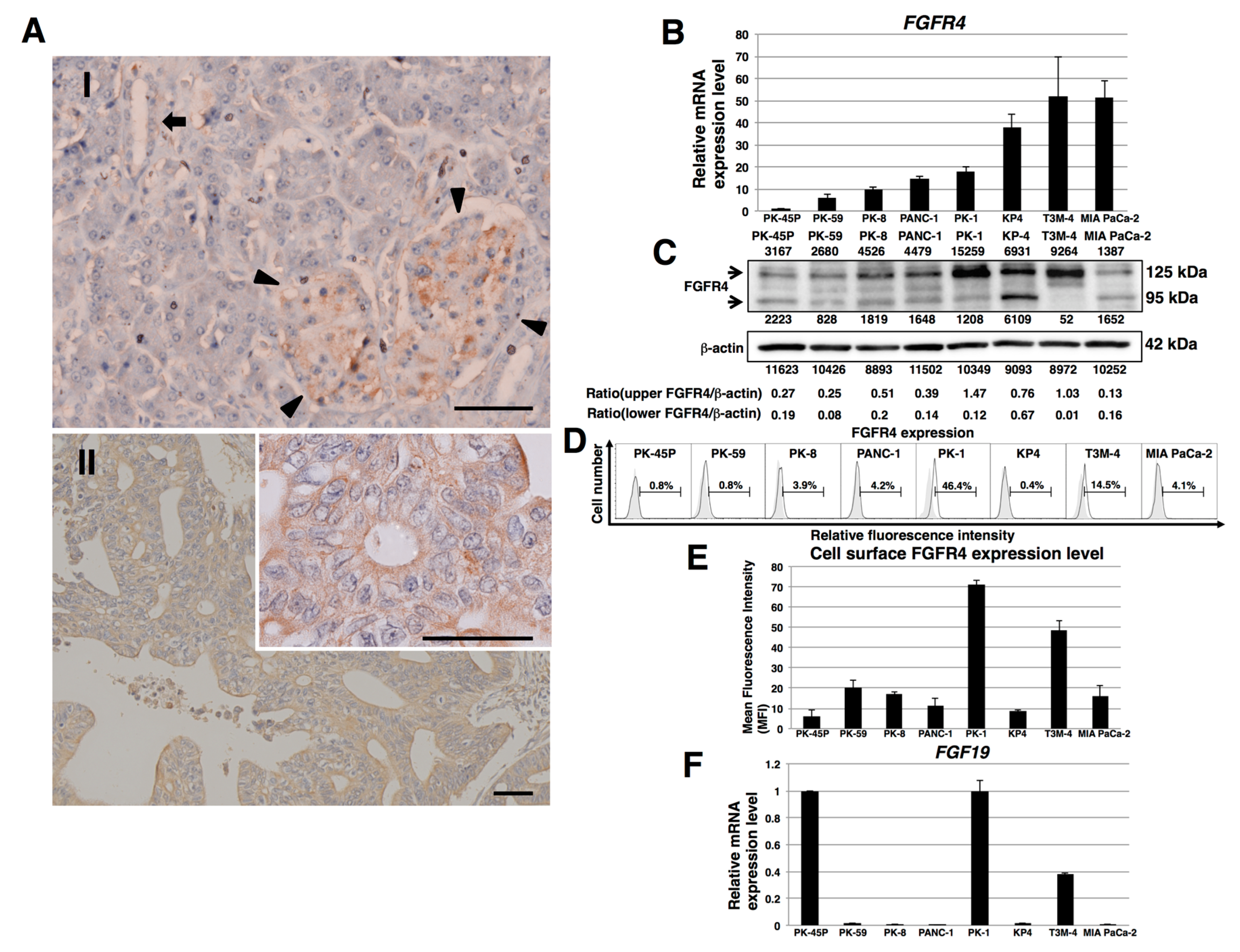
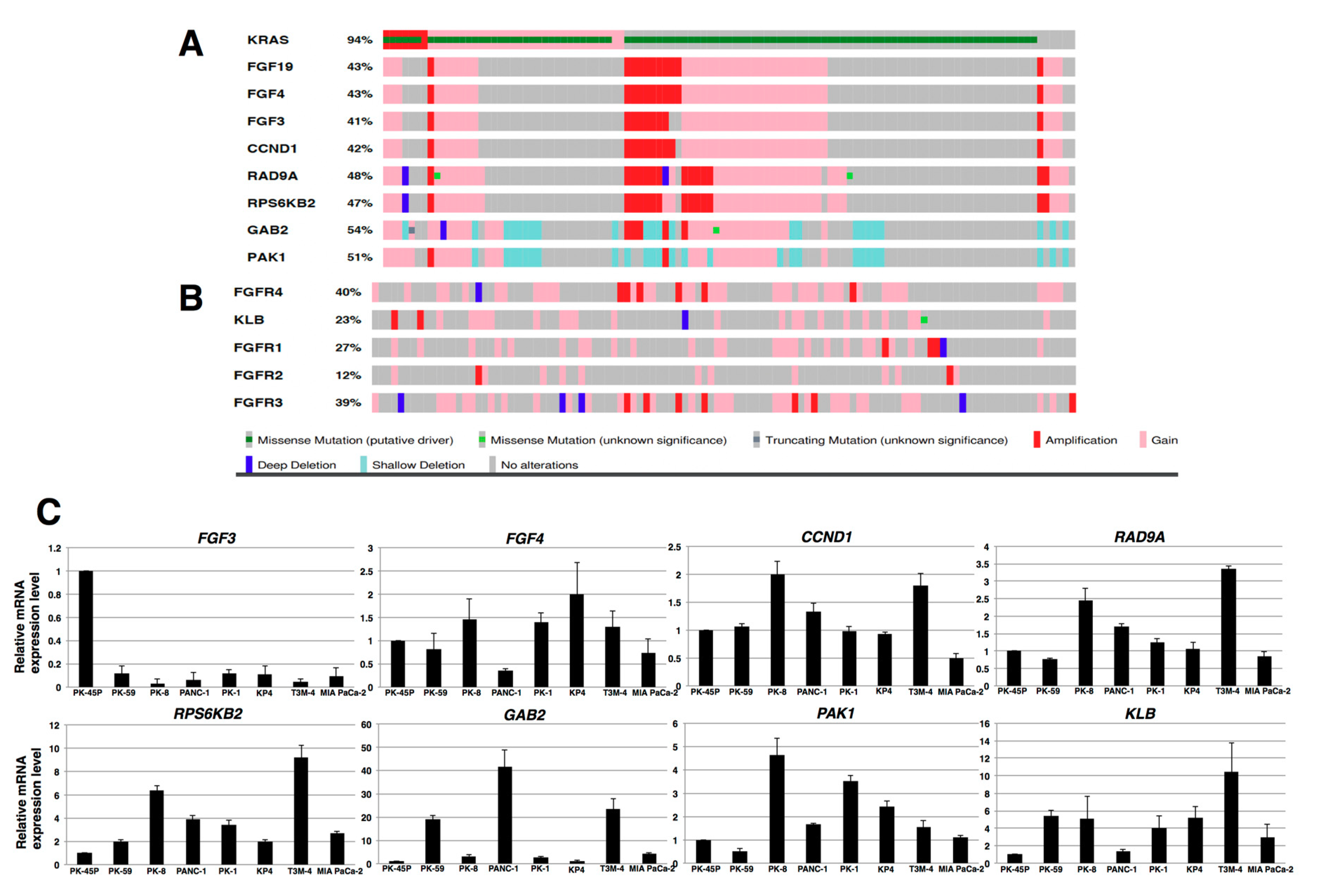
2.1. Expression of FGFR4 in Human PDAC Tissues and PDAC Cell Lines
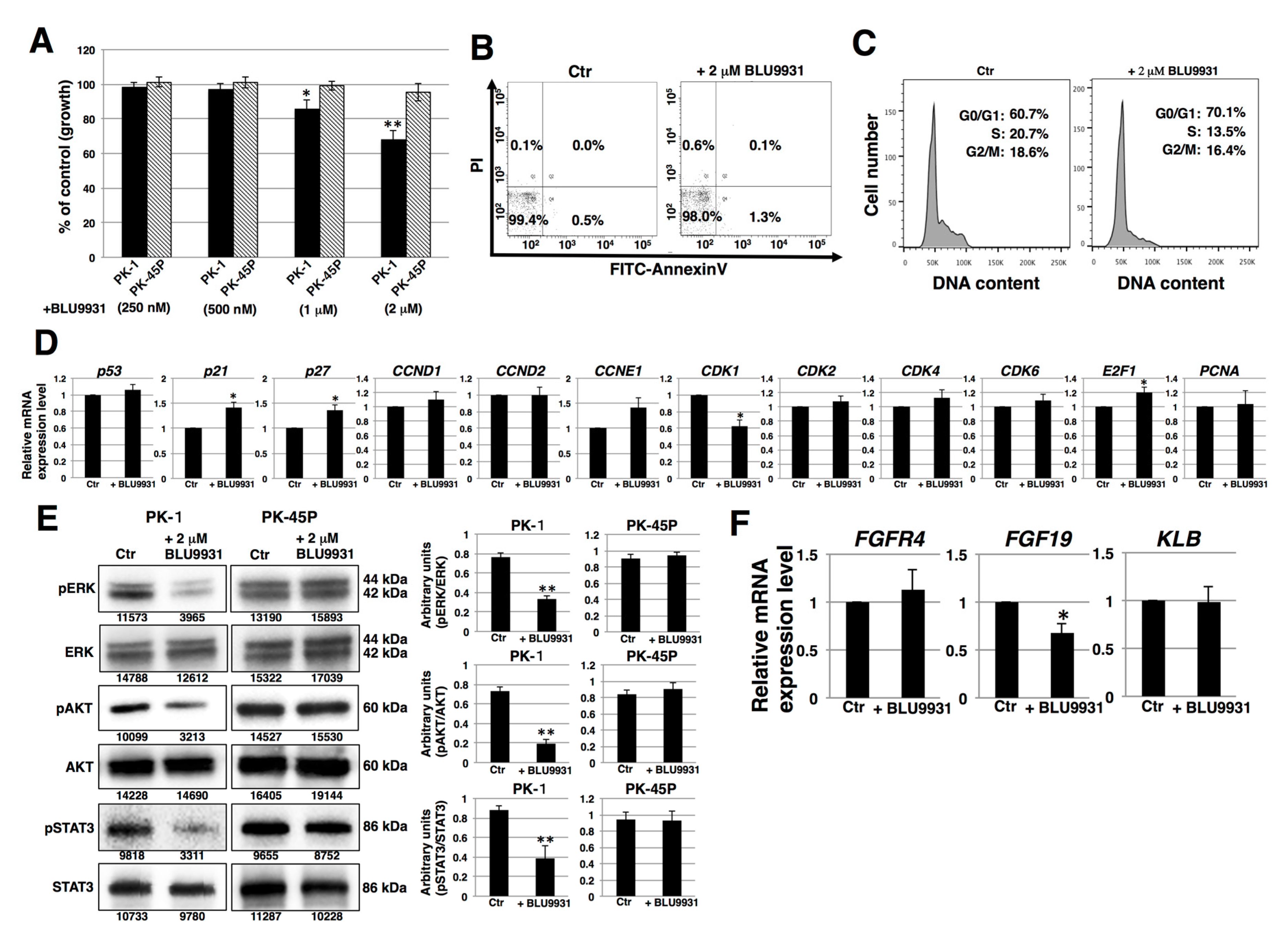
2.2. Effects of BLU9931 on Growth and Cell Cycle of PDAC Cells
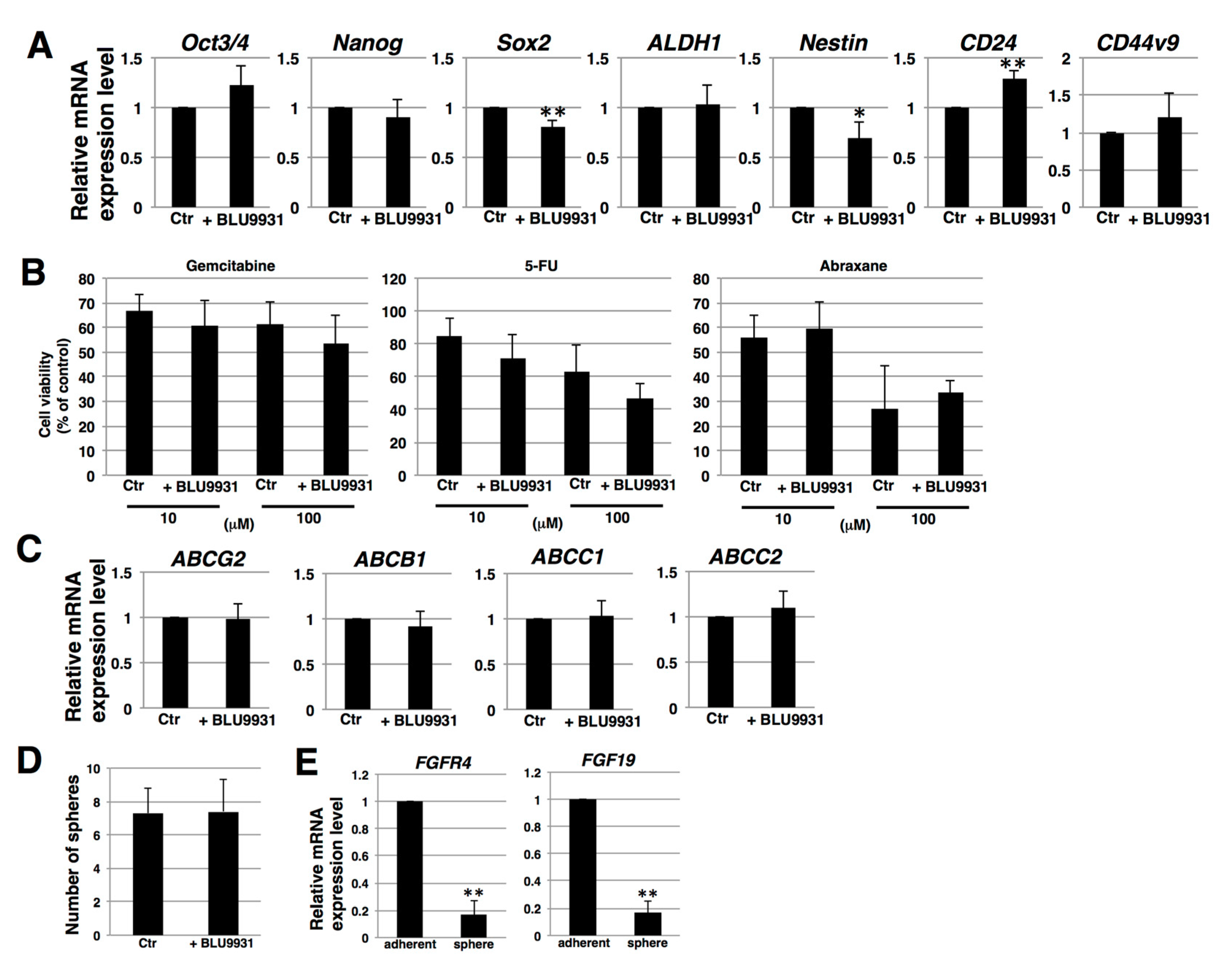
2.3. Effects of BLU9931 on Stemness Features of PDAC Cells
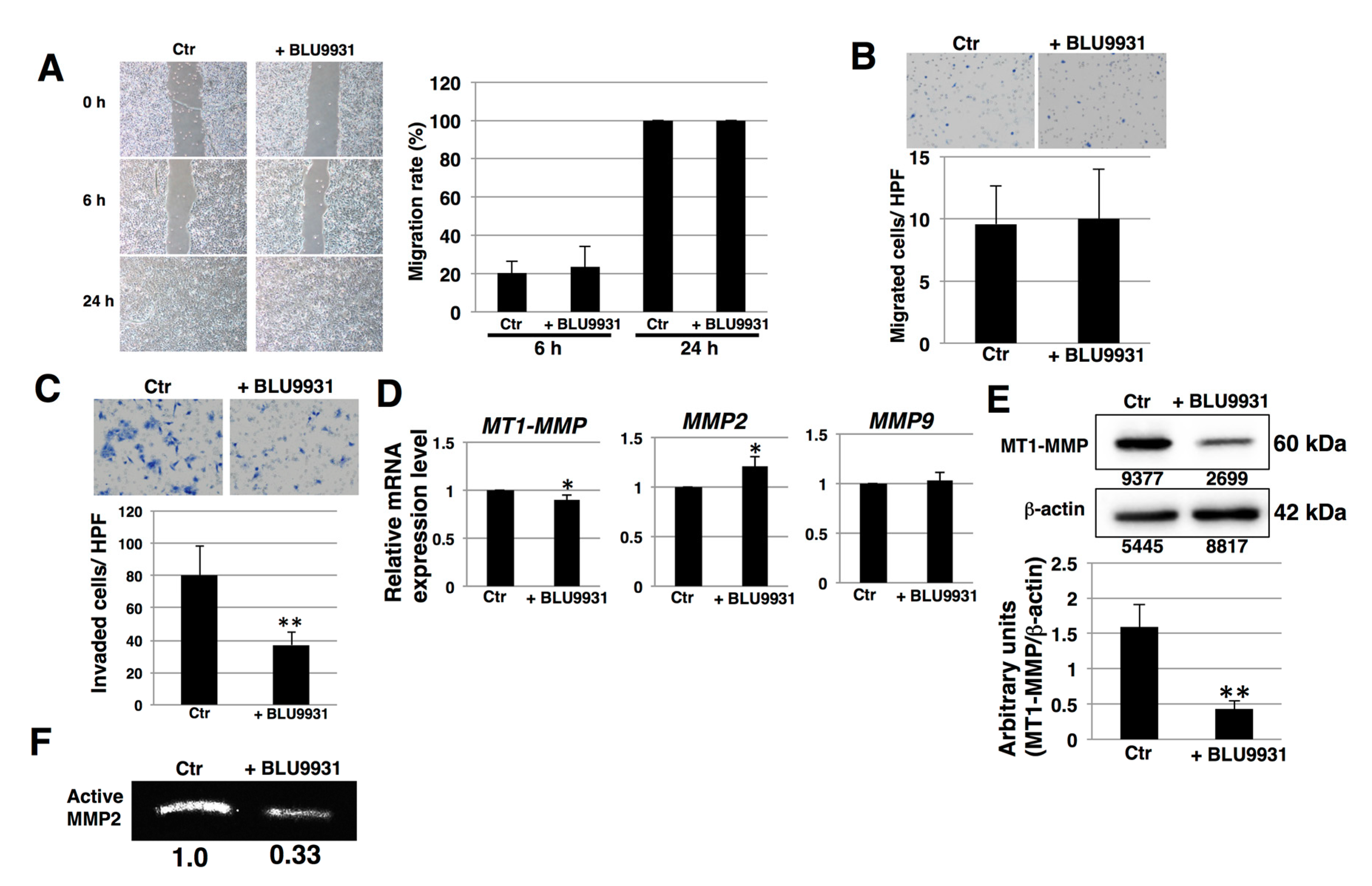
2.4. Effects of BLU9931 on Migration and Invasion of PDAC Cells
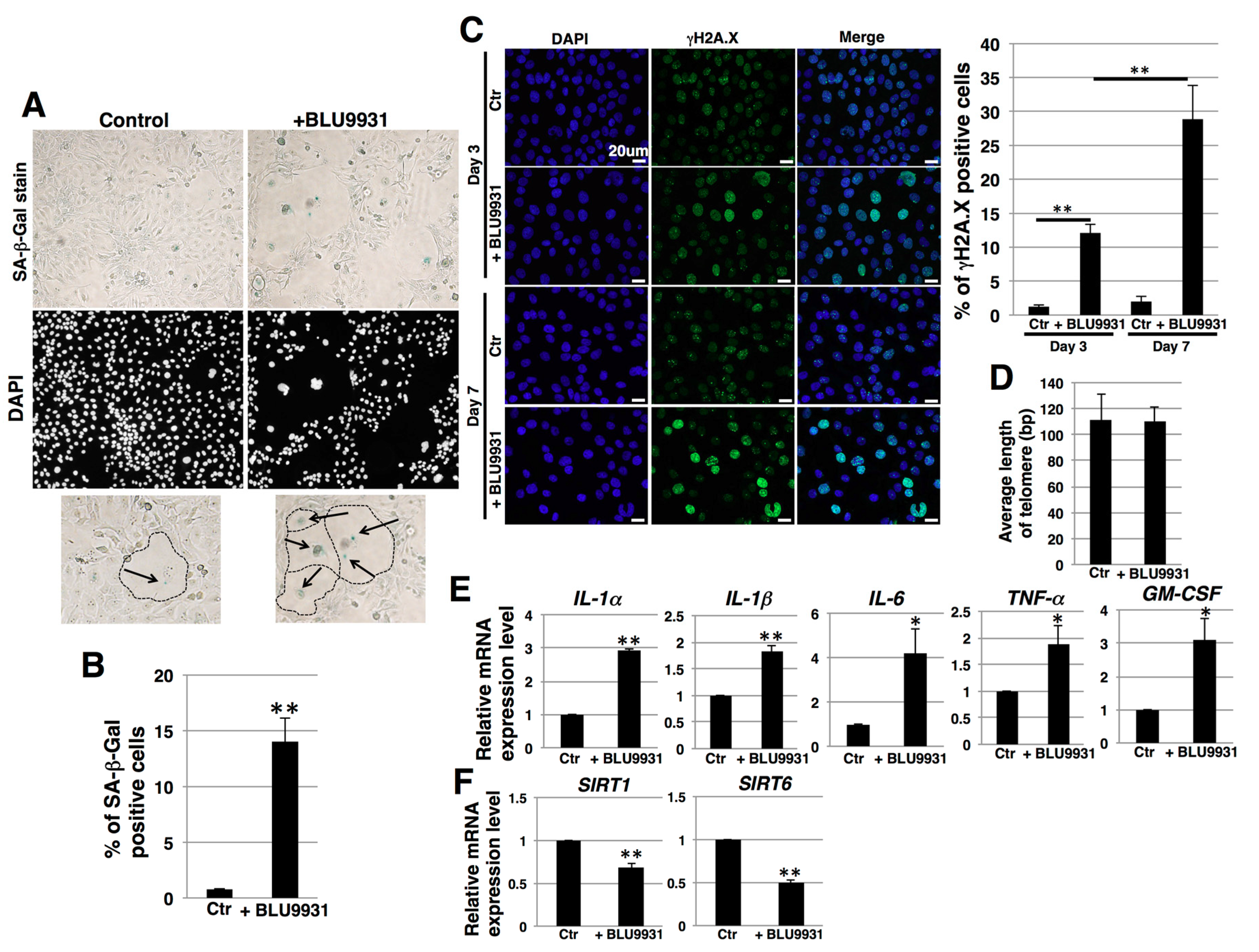
2.5. Induction of Senescence in PDAC Cells by Long-Term Treatment with BLU9931
2.6. Effects of Senolytic Drugs on BLU9931-Induced Senescent PDAC Cells
3. Discussion
4. Materials and Methods
4.1. Immunohistochemistry
4.2. Cell Culture
4.3. Genotyping
4.4. FACS Analysis
4.5. Real-Time qPCR
4.6. Cell Proliferation Assay
4.7. Apoptosis Assay
4.8. Cell Cycle Assay
4.9. Immunoblotting
4.10. Sphere-Forming Assay
4.11. Anti-Drug Resistance Assay
4.12. Wound Healing/Cell Scratch Assay
4.13. Boyden Chamber Assay
4.14. Invasion Assay
4.15. MMP Gelatin Zymography
4.16. SA-β-Gal Assay
4.17. Telomere Length Assay
4.18. Immunostaining
4.19. Senolytic Drug Sensitivity Assay
4.20. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2019, 70, 7–30. [Google Scholar] [CrossRef]
- Korc, M.; Jeon, C.Y.; Edderkaoui, M.; Pandol, S.J.; Petrov, M.S. Consortium for the study of chronic pancreatitis, diabetes, and pancreatic cancer (CPDPC). Tobacco and alcohol as risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Ahrendt, S.A.; Pitt, H.A. Surgical management of pancreatic cancer. Oncology 2002, 16, 725–734. [Google Scholar] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis Primers. 2016, 21, 16022. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [PubMed]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, C.; Yates, E.A.; Jiang, C.; Wilkinson, M.C.; Fernig, D.G. Heparin binding preference and structures in the fibroblast growth factor family parallel their evolutionary diversification. Open Biol. 2016, 3, 150275. [Google Scholar] [CrossRef]
- Fernandes-Freitas, I.; Owen, B.M. Metabolic roles of endocrine fibroblast growth factors. Curr. Opin. Pharmacol. 2015, 25, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Partanen, J.; Makela, T.P.; Eerola, E.; Korhonen, J.; Hirvonen, H.; Claesson-Welsh, L.; Alitalo, K. FGFR-4, a novel acidic fibroblast growth factor receptor with a distinct expression pattern. EMBO J. 1991, 10, 1347–1354. [Google Scholar] [CrossRef]
- Belov, A.A.; Mohammadi, M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol. 2013, 5, a015958. [Google Scholar] [CrossRef] [PubMed]
- Korc, M.; Friesel, R. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cao, R. Hedlund EM. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J. Mol. Med. 2008, 86, 785–789. [Google Scholar] [CrossRef]
- Fons, P.; Gueguen-Dorbes, G.; Herault, J.P.; Geronimi, F.; Tuyaret, J.; Frédérique, D.; Schaeffer, P.; Volle-Challier, C.; Herbert, J.M.; Bono, F. Tumor vasculature is regulated by FGF/FGFR signaling-mediated angiogenesis and bone marrow-derived cell recruitment: This mechanism is inhibited by SSR128129E, the first allosteric antagonist of FGFRs. J. Cell Physiol. 2015, 230, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Matsuda, Y.; Yamamoto, T.; Uchida, E.; Korc, M.; Naito, Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am. J. Pathol. 2012, 180, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Hagio, M.; Seya, T.; Ishiwata, T. Fibroblast growth factor receptor 2 IIIc as a therapeutic target for colorectal cancer cells. Mol. Cancer Ther. 2012, 11, 2010–2020. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Ishiwata, T.; Uchida, E.; Nakazawa, N.; Korc, M.; Naito, Z.; Tajiri, T. Enhanced expression of keratinocyte growth factor and its receptor correlates with venous invasion in pancreatic cancer. Am. J. Pathol. 2007, 170, 1964–1974. [Google Scholar] [CrossRef]
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 2015, 26, 22–29. [Google Scholar] [CrossRef]
- Degirolamo, C.; Sabbà, C.; Moschetta, A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2016, 15, 51–69. [Google Scholar] [CrossRef]
- Li, C.S.; Zhang, S.X.; Liu, H.J.; Shi, Y.L.; Li, L.P.; Guo, X.B.; Zhang, Z.H. Fibroblast growth factor receptor 4 as a potential prognostic and therapeutic marker in colorectal cancer. Biomarkers 2014, 19, 81–85. [Google Scholar] [CrossRef]
- Ye, Y.W.; Hu, S.; Shi, Y.Q.; Zhang, X.F.; Zhou, Y.; Zhao, C.L.; Wang, G.J.; Wen, J.G.; Zong, H. Combination of the FGFR4 inhibitor PD173074 and 5-fluorouracil reduces proliferation and promotes apoptosis in gastric cancer. Oncol. Rep. 2013, 30, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xie, B.; Zhu, Q.; Xia, Q.; Jiang, S.; Cao, R.; Shi, L.; Qi, D.; Li, X.; Cai, L. FGFR4 and TGF-beta1 expression in hepatocellular carcinoma: Correlation with clinicopathological features and prognosis. Int. J. Med. Sci. 2013, 10, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Motoda, N.; Matsuda, Y.; Onda, M.; Ishiwata, T.; Uchida, E.; Naito, Z. Overexpression of fibroblast growth factor receptor 4 in high-grade pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Int. J. Oncol. 2011, 38, 133–143. [Google Scholar] [PubMed]
- Turkington, R.C.; Longley, D.B.; Allen, W.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.; Blayney, J.K.; Salto-Tellez, M.; Van Schaeybroeck, S.; Johnston, P.G. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef]
- Zaid, T.M.; Yeung, T.L.; Thompson, M.S.; Leung, C.S.; Harding, T.; Co, N.N.; Schmandt, R.S.; Kwan, S.Y.; Rodriguez-Aguay, C.; Lopez-Berestein, G.; et al. Identification of FGFR4 as a potential therapeutic target for advanced-stage, high-grade serous ovarian cancer. Clin. Cancer Res. 2013, 19, 809–820. [Google Scholar] [CrossRef]
- Fang, H.M.; Tian, G.; Zhou, L.J.; Zhou, H.Y.; Fang, Y.Z. FGFR4 genetic polymorphisms determine the chemotherapy response of Chinese patients with non-small cell lung cancer. Acta Pharmacol. Sin. 2013, 34, 549–554. [Google Scholar] [CrossRef]
- Morimoto, Y.; Ozaki, T.; Ouchida, M.; Umehara, N.; Ohata, N.; Yoshida, A.; Shimizu, K.; Inoue, H. Single nucleotide polymorphism in fibroblast growth factor receptor 4 at codon 388 is associated with prognosis in high-grade soft tissue sarcoma. Cancer 2003, 98, 2245–2250. [Google Scholar] [CrossRef]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Müller, S.; Gärtner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002, 62, 840–847. [Google Scholar]
- Serra, S.; Zheng, L.; Hassan, M.; Phan, A.T.; Woodhouse, L.J.; Yao, J.C.; Ezzat, S.; Asa, S.L. The FGFR4-G388R single-nucleotide polymorphism alters pancreatic neuroendocrine tumor progression and response to mTOR inhibition therapy. Cancer Res. 2012, 72, 5683–5691. [Google Scholar] [CrossRef]
- Zhang, F.; Yu, L.; Lin, X.; Cheng, P.; He, L.; Li, X.; Lu, X.; Tan, Y.; Yang, H.; Cai, L.; et al. Minireview: Roles of fibroblast growth factors 19 and 21 in metabolic regulation and chronic diseases. Mol. Endocrinol. 2015, 29, 1400–1413. [Google Scholar] [CrossRef]
- Raja, A.; Park, I.; Haq, F.; Ahn, S.M. FGF19-FGFR4 signaling in hepatocellular carcinoma. Cells 2019, 8, 536. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Tong, N.; Chen, S.Q.; Hua, L.X.; Wang, Z.J.; Zhang, Z.D.; Chen, M. FGFR4 Gly388Arg polymorphism contributes to prostate cancer development and progression: A meta-analysis of 2618 cases and 2305 controls. BMC Cancer 2011, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Du, F.; Dang, Y.; Li, X.; Qian, M.; Feng, W.; Qiao, C.; Fan, D.; Nie, Y.; Wu, K.; et al. Fibroblast growth factor 19-mediated up-regulation of SYR-related high-mobility group box 18 promotes hepatocellular carcinoma metastasis by transactivating fibroblast growth factor receptor 4 and fms-related tyrosine kinase 4. Hepatology 2020, 5, 1712–1731. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lang, L.; Zhao, X.; Shay, C.; Shull, A.Y.; Teng, Y. FGF19 amplification reveals an oncogenic dependency upon autocrine FGF19/FGFR4 signaling in head and neck squamous cell carcinoma. Oncogene 2019, 38, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Hage, M.; Miduturu, C.; Sheets, M.; Rubin, N.; Weng, W.; Stransky, N.; Bifulco, N.; Kim, J.L.; Hodous, B.; Brooijmans, N.; et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015, 5, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nomura, S.; Hosoi, A.; Nagaoka, K.; Iino, T.; Yasuda, T.; Saito, T.; Matsushita, H.; Uchida, E.; Seto, Y.; et al. Established gastric cancer cell lines transplantable into C57BL/6 mice show fibroblast growth factor receptor 4 promotion of tumor growth. Cancer Sci. 2018, 109, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef]
- Karlsson, E.; Waltersson, M.A.; Bostner, J.; Pérez-Tenorio, G.; Olsson, B.; Hallbeck, A.-L.; Stål, O. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer 2011, 50, 775–787. [Google Scholar] [CrossRef]
- Holzmann, K.; Kohlhammer, H.; Schwaenen, C.; Wessendorf, S.; Kestler, H.A.; Schwoerer, A.; Rau, B.; Radlwimmer, B.; Döhner, H.; Lichter, P.; et al. Genomic DNA-chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res. 2004, 64, 4428–4433. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 4, R41. [Google Scholar] [CrossRef]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 50, 20007–20012. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744–6755. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Hasegawa, F.; Michishita, M.; Sasaki, N.; Ishikawa, N.; Takubo, K.; Matsuda, Y.; Arai, T.; Aida, J. Electron microscopic analysis of different cell types in human pancreatic cancer spheres. Oncol. Lett. 2018, 15, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Toyoda, M.; Hasegawa, F.; Fujiwara, M.; Gomi, F.; Ishiwata, T. Fetal bovine serum enlarges the size of human pancreatic cancer spheres accompanied by an increase in the expression of cancer stem cell markers. Biochem. Biophys. Res. Commun. 2019, 514, 112–117. [Google Scholar] [CrossRef]
- Shichi, Y.; Sasaki, N.; Michishita, M.; Hasegawa, F.; Matsuda, Y.; Arai, T.; Gomi, F.; Aida, J.; Takubo, K.; Toyoda, M.; et al. Enhanced morphological and functional differences of pancreatic cancer with epithelial or mesenchymal characteristics in 3D culture. Sci. Rep. 2019, 9, 10871. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44-46, 207–223. [Google Scholar] [CrossRef]
- Zervos, E.E.; Shafii, A.E.; Haq, M.; Rosemurgy, A.S. Matrix metalloproteinase inhibition suppresses MMP-2 activity and activation of PANC-1 cells in vitro. J. Surg. Res. 1999, 84, 162–167. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro life time of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xu, F.; Dominguez, N.P.; Xiong, Y.; Xiong, Z.; Peng, H.; Shay, C.; Teng, Y. FGFR4 provides the conduit to facilitate FGF19 signaling in breast cancer progression. Mol. Carcinog. 2018, 57, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Tiong, K.H.; Tan, B.S.; Choo, H.L.; Chung, F.F.; Hii, L.W.; Tan, S.H.; Khor, N.T.; Wong, S.F.; See, S.J.; Tan, Y.F.; et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget 2016, 7, 57633–57650. [Google Scholar] [CrossRef]
- Lang, L.; Teng, Y. Fibroblast growth factor receptor 4 targeting in cancer: New insights into mechanisms and therapeutic strategies. Cells 2019, 8, 31. [Google Scholar] [CrossRef]
- Yang, C.; Jin, C.; Li, X.; Wang, F.; McKeehan, W.L.; Luo, Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS ONE 2012, 7, e33870. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR landscape in cancer: Analysis of 4853 tumors by next-generation sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef]
- Nagase, H. Cell surface activation of progelatinase A (proMMP-2) and cell migration. Cell Res. 1998, 8, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.T.; Tsao, P.N.; Lin, H.L.; Tung, C.C.; Change, M.C.; Chang, Y.T.; Wong, J.M.; Wei, S.C. Hes1 Increases the invasion ability of colorectal cancer cells via the STAT3-MMP14 pathway. PLoS ONE 2015, 10, e0144322. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.Y.; et al. First-in-Human phase I study of Fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yu, Y.; Li, Z.; Wang, G.; Li, F.; Xia, W.; Lu, S. FGFR1 promotes the stem cell-like phenotype of FGFR1-amplified non-small cell lung cancer cells through the Hedgehog pathway. Oncotarget 2016, 7, 15118–15134. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Bamodu, O.A.; Tsai, W.C.; Chang, Y.M.; Lee, W.H.; Yeh, C.T.; Chao, T.Y. The therapeutic targeting of the FGFR1/Src/NF-κB signaling axis inhibits pancreatic ductal adenocarcinoma stemness and oncogenicity. Clin. Exp. Metastasis. 2018, 35, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Peláez-García, A.; Barderas, R.; Torres, S.; Hernández-Varas, P.; Teixidó, J.; Bonilla, F.; de Herreros, A.G.; Casal, J.I. FGFR4 role in epithelial-mesenchymal transition and its therapeutic value in colorectal cancer. PLoS ONE 2013, 8, e63695. [Google Scholar] [CrossRef]
- Zhao, H.; Lv, F.; Liang, G.; Huang, X.; Wu, G.; Zhang, W.; Yu, L.; Shi, L.; Teng, Y. FGF19 promotes epithelial-mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3β/β- catenin signaling cascade via FGFR4 activation. Oncotarget 2016, 7, 13575–13586. [Google Scholar] [CrossRef]
- Luo, Y.; Ye, S.; Li, X.; Lu, W. Emerging structure-function paradigm of endocrine FGFs in metabolic diseases. Trends Pharmacol. Sci. 2019, 40, 142–153. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, Y.; Liu, M.; Wang, D.; Wang, F.; Bi, Y.; Ji, J.; Li, S.; Liu, Y.; Chen, R.; et al. Oncogenic KRAS reduces expression of FGF21 in acinar cells to promote pancreatic tumorigenesis in mice on a high-fat diet. Gastroenterology 2019, 157, 1413–1428. [Google Scholar] [CrossRef]
- Kobrin, M.S.; Yamanaka, Y.; Friess, H.; Lopez, M.E.; Korc, M. Aberrant expression of type I fibroblast growth factor receptor in human pancreatic adenocarcinomas. Cancer Res. 1993, 53, 4741–4744. [Google Scholar]
- Opalinski, L.; Szczepara, M.; Sokolowska-Wedzina, A.; Zakrzewska, M.; Otlewski, J. The autoinhibitory function of D1 domain of FGFR1 goes beyond the inhibition of ligand binding. Int. J. Biochem. Cell Biol. 2017, 89, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.L.; Narita, M. Short-term gain, long-term pain: The senescence life cycle and cancer. Genes Dev. 2019, 33, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 7686, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Schulte, B.A.; Wang, G.Y. Role of senescence induction in cancer treatment. World J. Clin. Oncol. 2018, 9, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-step senescence-focused cancer therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Ovadya, Y.; Krizhanovsky, V. Strategies targeting cellular senescence. J. Clin. Investig. 2018, 128, 1247–1254. [Google Scholar] [CrossRef]
- Tang, S.M.; Deng, X.T.; Zhou, J.; Li, Q.P.; Ge, X.X.; Miao, L. Pharmacological basis and new insights of quercetin action in respect to its anti-cancer effects. Biomed. Pharmacother. 2020, 121, 109604. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Nagai, K.; Matsushita, T.; Matsuzaki, T.; Takayama, K.; Matsumoto, T.; Kuroda, R.; Kurosaka, M. Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes. Osteoarthr. Cartil. 2015, 23, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, H.; Xu, C.; Wang, P.; Chen, J.; Wang, P.; Sun, Z.; Su, Y.; Wang, Z.; Han, L.; et al. SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitin-proteasome degradation. Aging 2016, 8, 2308–2323. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, P.; Rahnasto-Rilla, M.; Mellini, P.; Jarho, E.; Lahtela-Kakkonen, M.; Kokkola, T. Studying SIRT6 regulation using H3K56 based substrate and small molecules. Eur. J. Pharm Sci. 2014, 63, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer. 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Di Magliano, M.P.; Welling, T.H.; et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Sugiyama, N.; Varjosalo, M.; Meller, P.; Lohi, J.; Hyytiäinen, M.; Kilpinen, S.; Kallioniemi, O.; Ingvarsen, S.; Engelholm, L.H.; Taipale, J. Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 2010, 70, 7851–7861. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef]
- Hong, C.S.; Sun, E.G.; Choi, J.N.; Kim, D.; Kim, J.; Ryu, K.; Shim, H.; Hwang, J.; Bae, W.; Kim, H.; et al. Fibroblast growth factor receptor 4 increases epidermal growth factor receptor (EGFR) signaling by inducing amphiregulin expression and attenuates response to EGFR inhibitors in colon cancer. Cancer Sci. 2020. [Google Scholar] [CrossRef]
- Dutra, R.L.; de Carvalho, M.B.; Dos Santos, M.; Mercante, A.M.; Gazito, D.; de Cicco, R.; Group, G.; Tajara, E.H.; Louro, I.D.; da Silva, A.M. FGFR4 profile as a prognostic marker in squamous cell carcinoma of the mouth and oropharynx. PLoS ONE 2012, 7, e50747. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Itakura, Y.; Toyoda, M. Ganglioside GM1 Contributes to the state of insulin resistance in senescent human arterial endothelial cells. J. Biol. Chem. 2015, 290, 25475–25486. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | FGFR4 Expression | |||
|---|---|---|---|---|
| Low (<2) | High (2~3) | p-Value | ||
| Tumor Grade | 0.100 | |||
| Grade 1 | 15 | 8 | ||
| Grade 2 | 33 | 42 | ||
| Grade 3 | 19 | 12 | ||
| Primary Tumor | <0.001 | |||
| T1 | 2 | 0 | ||
| T2 | 27 | 8 | ||
| T3 | 39 | 53 | ||
| T4 | 1 | 6 | ||
| Regional Lymph Node | 0.083 | |||
| N0 | 62 | 53 | ||
| N1 | 7 | 14 | ||
| Stage | <0.001 | |||
| Stage I | 27 | 2 | ||
| Stage II | 34 | 45 | ||
| Stage III | 7 | 16 | ||
| Stage IV | 1 | 4 | ||
| PDAC Cell Lines | FGFR4 Genotype | FGFR4 Amino Acid (388) |
|---|---|---|
| MIA PaCa-2 | G/G | Gly/Gly |
| PK-8 | G/G | Gly/Gly |
| PK-1 | G/A | Gly/Arg |
| PANC-1 | G/A | Gly/Arg |
| PK-45P | G/A | Gly/Arg |
| PK-59 | A/A | Arg/Arg |
| T3M-4 | A/A | Arg/Arg |
| KP4 | A/A | Arg/Arg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sasaki, N.; Gomi, F.; Yoshimura, H.; Yamamoto, M.; Matsuda, Y.; Michishita, M.; Hatakeyama, H.; Kawano, Y.; Toyoda, M.; Korc, M.; et al. FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer. Cancers 2020, 12, 2976. https://doi.org/10.3390/cancers12102976
Sasaki N, Gomi F, Yoshimura H, Yamamoto M, Matsuda Y, Michishita M, Hatakeyama H, Kawano Y, Toyoda M, Korc M, et al. FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer. Cancers. 2020; 12(10):2976. https://doi.org/10.3390/cancers12102976
Chicago/Turabian StyleSasaki, Norihiko, Fujiya Gomi, Hisashi Yoshimura, Masami Yamamoto, Yoko Matsuda, Masaki Michishita, Hitoshi Hatakeyama, Yoichi Kawano, Masashi Toyoda, Murray Korc, and et al. 2020. "FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer" Cancers 12, no. 10: 2976. https://doi.org/10.3390/cancers12102976
APA StyleSasaki, N., Gomi, F., Yoshimura, H., Yamamoto, M., Matsuda, Y., Michishita, M., Hatakeyama, H., Kawano, Y., Toyoda, M., Korc, M., & Ishiwata, T. (2020). FGFR4 Inhibitor BLU9931 Attenuates Pancreatic Cancer Cell Proliferation and Invasion While Inducing Senescence: Evidence for Senolytic Therapy Potential in Pancreatic Cancer. Cancers, 12(10), 2976. https://doi.org/10.3390/cancers12102976