A Novel Orthotopic Patient-Derived Xenograft Model of Radiation-Induced Glioma Following Medulloblastoma

 , , , , , , , , ,
, , , , , , , , ,
Simple Summary
Abstract
1. Introduction
2. Results
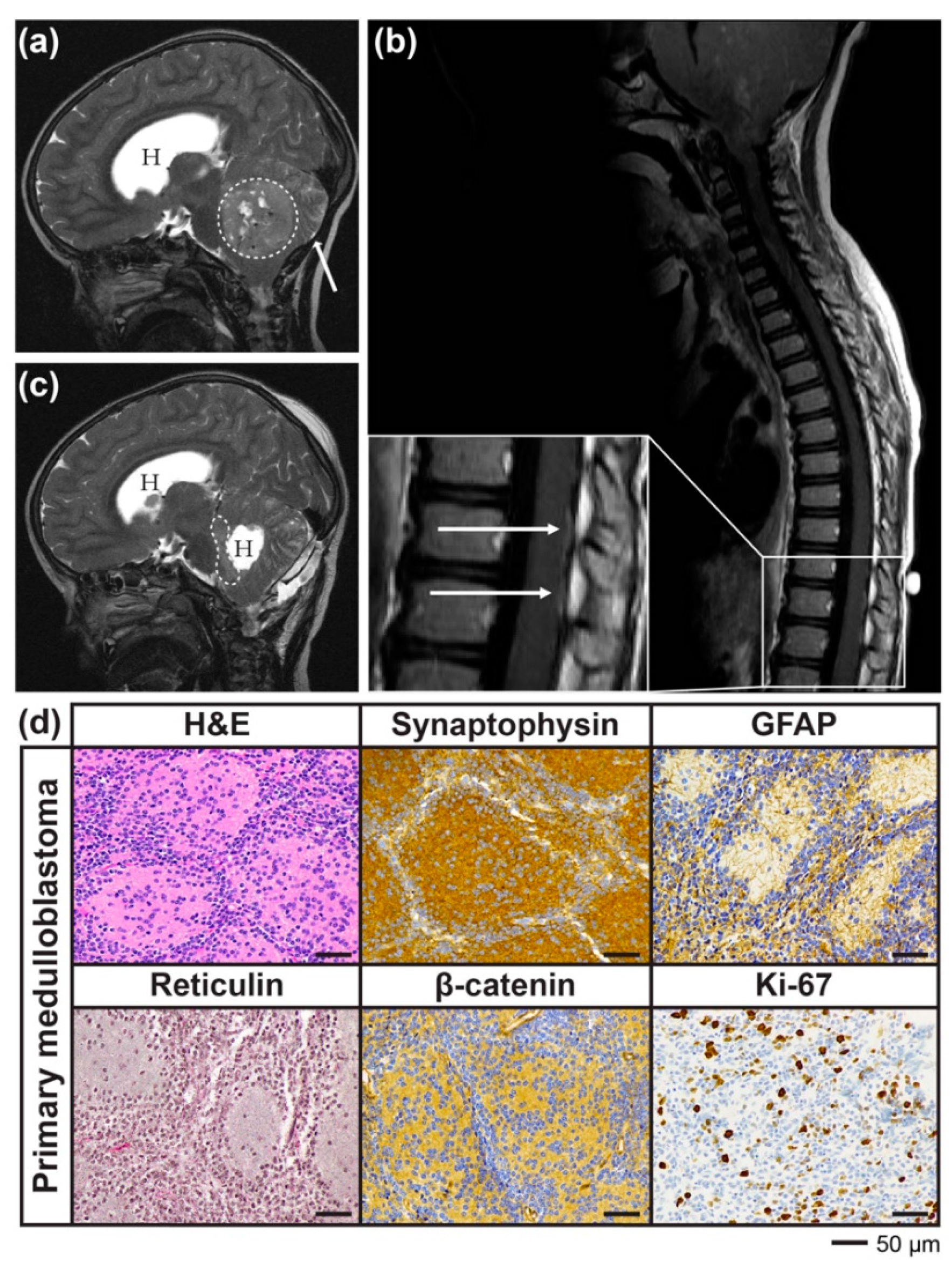
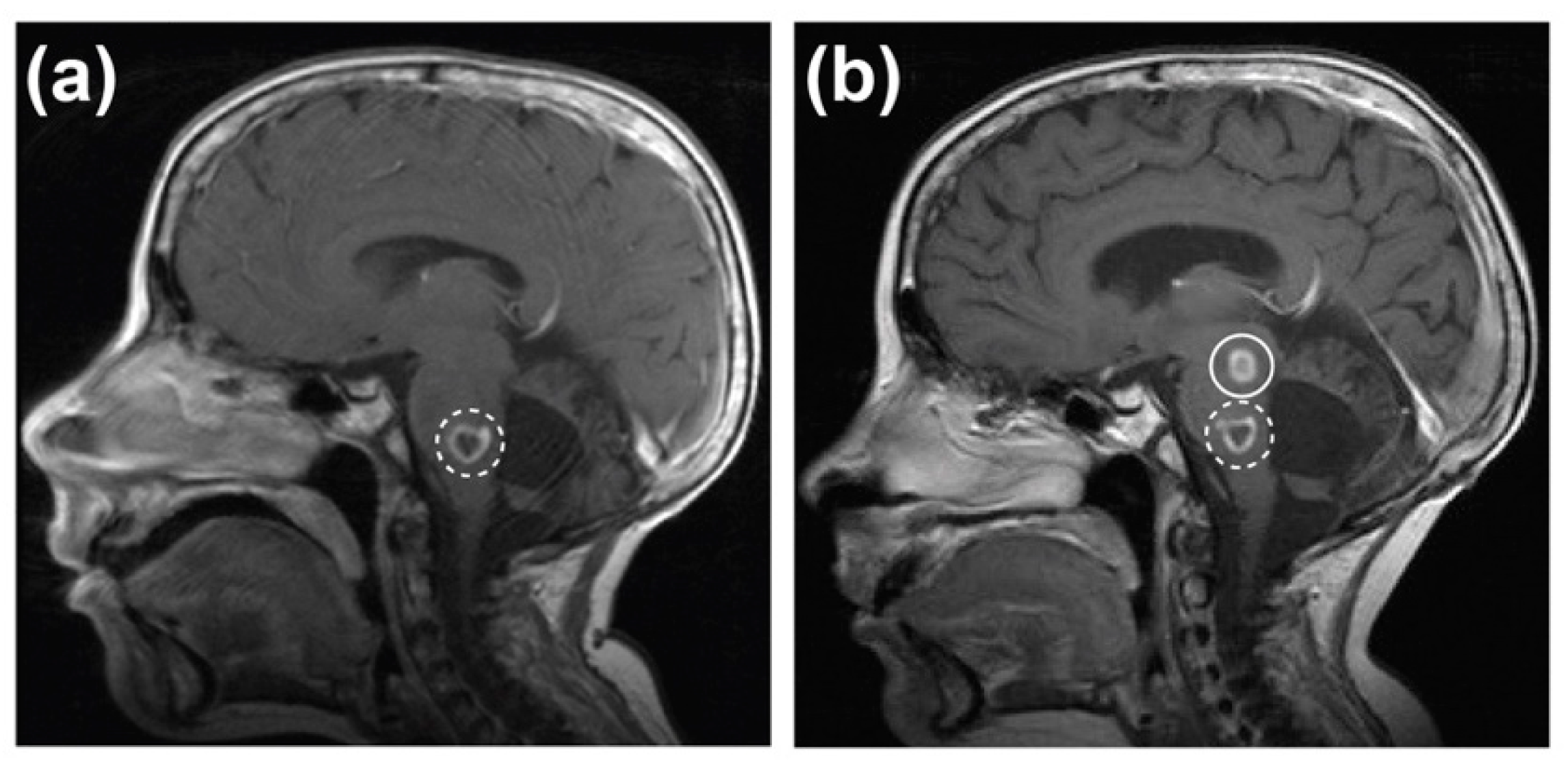
2.1. Case Report
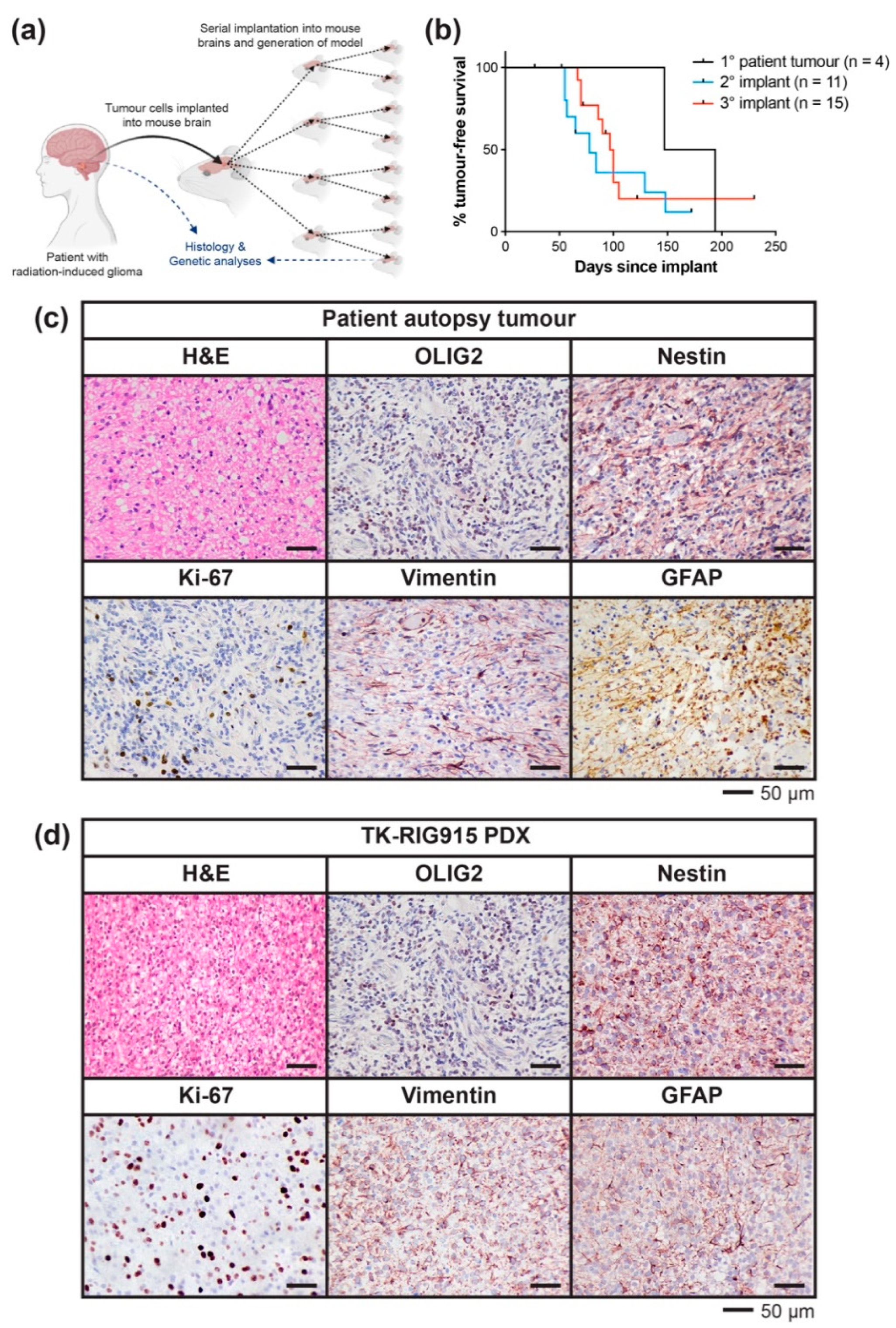
2.2. Development of a PDX Model of RIG Following Medulloblastoma that Histologically Recapitulates the Patient Tumour
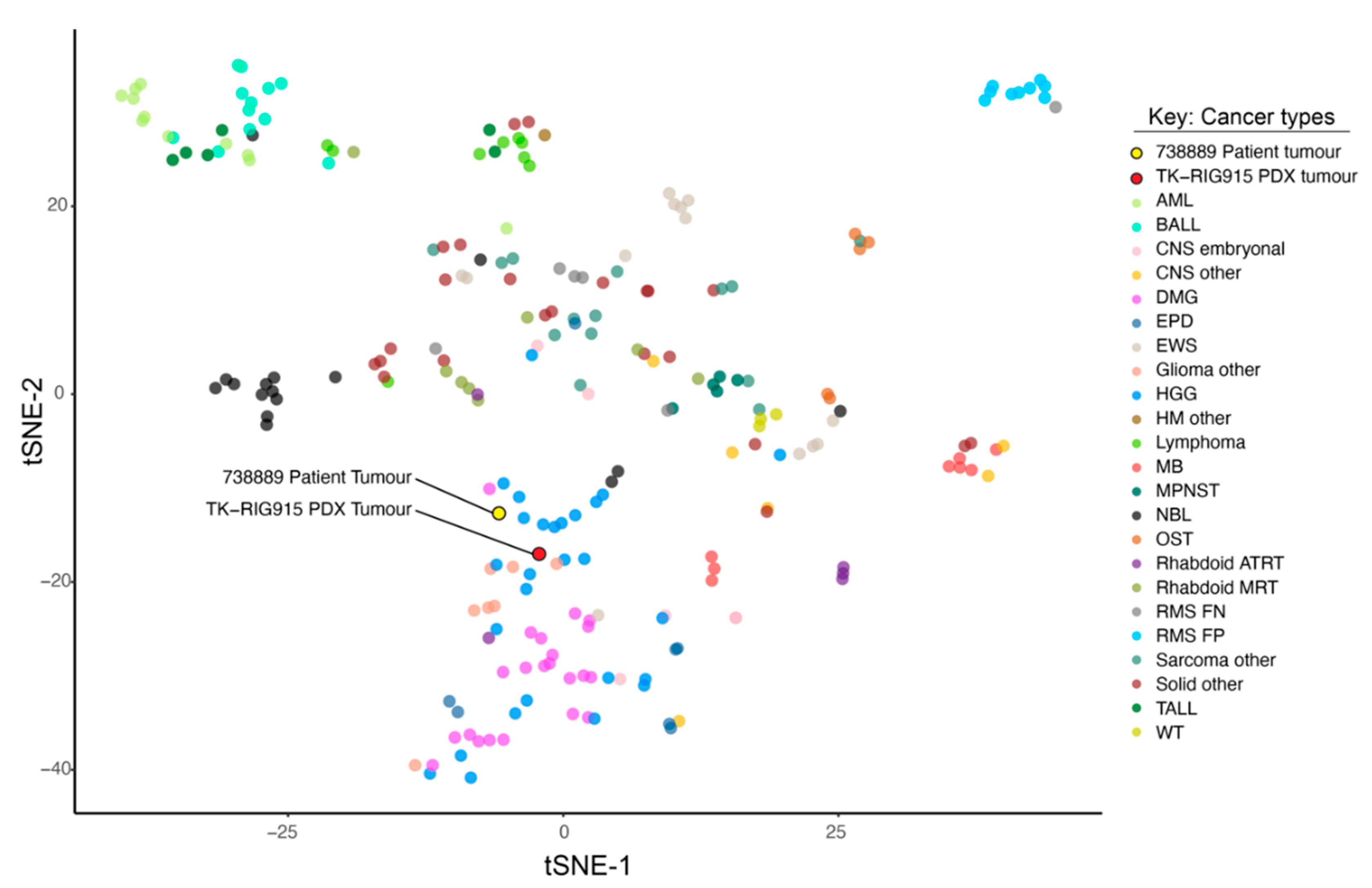
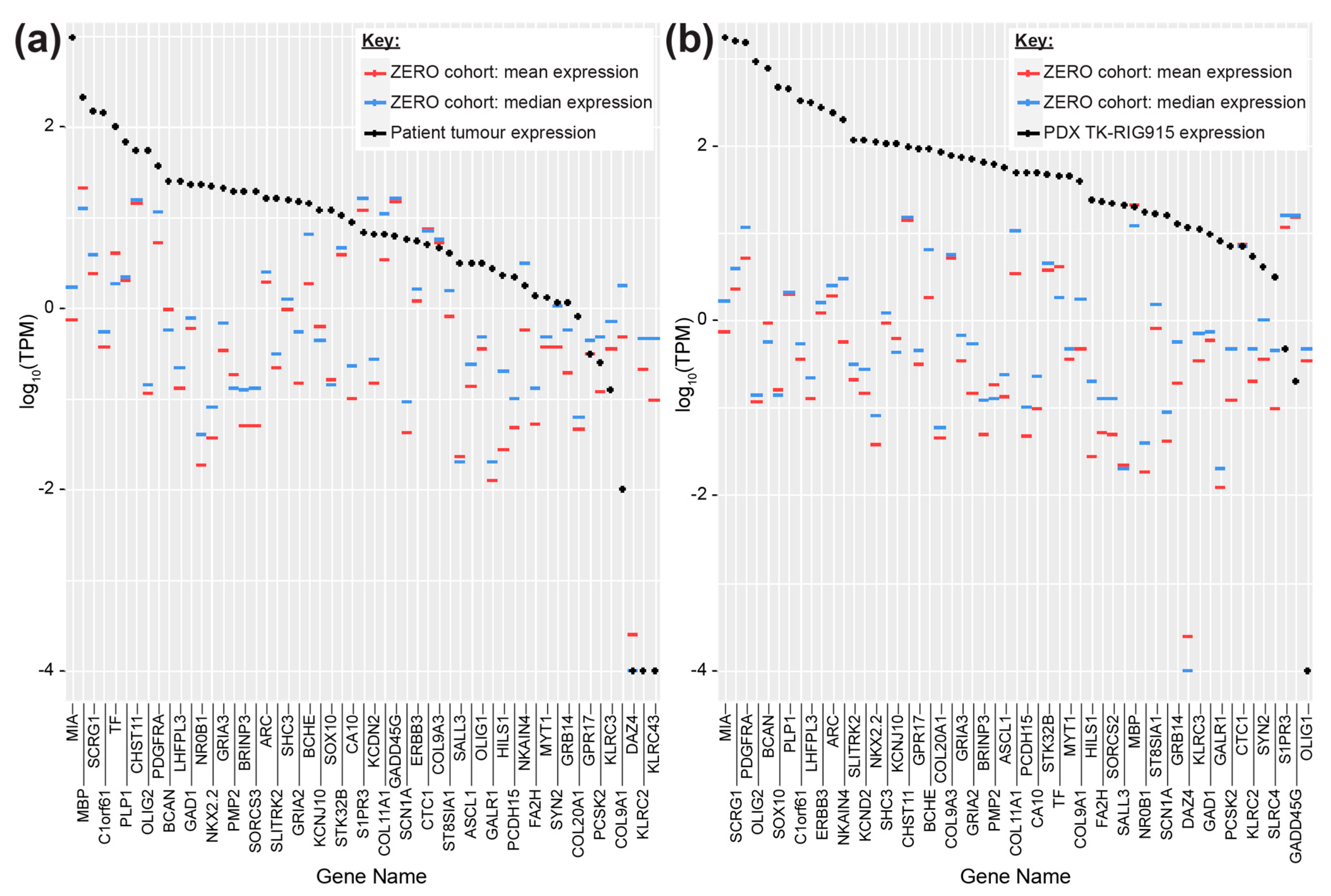
2.3. TK-RIG915 is Molecularly Distinct from Primary DMG and Matches a RIG Expression Profile
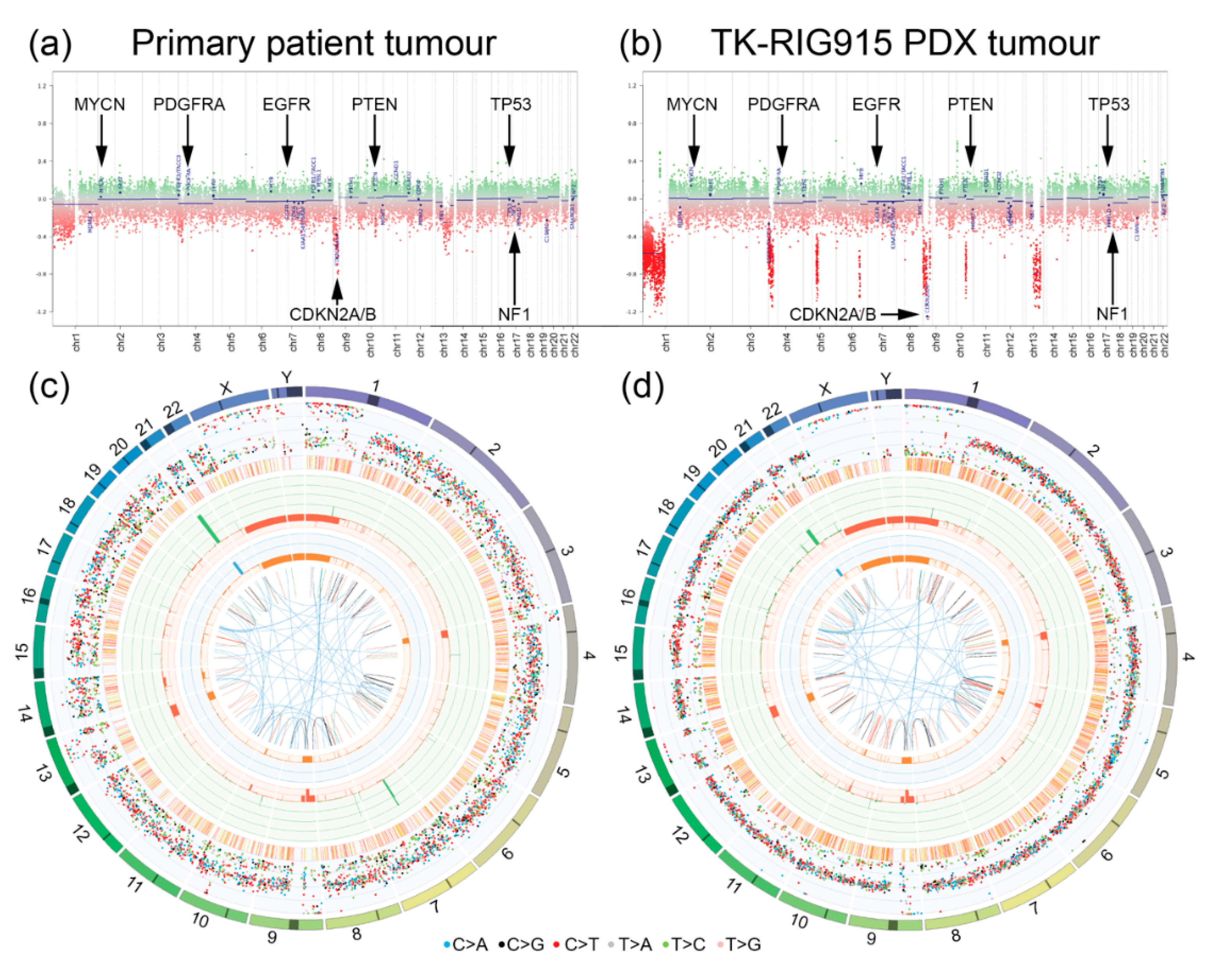
2.4. TK-RIG915 Genetically Recapitulates the Original Patient Tumour and Does Not Harbour a Number of Key Glioma-Associated Genetic Alterations
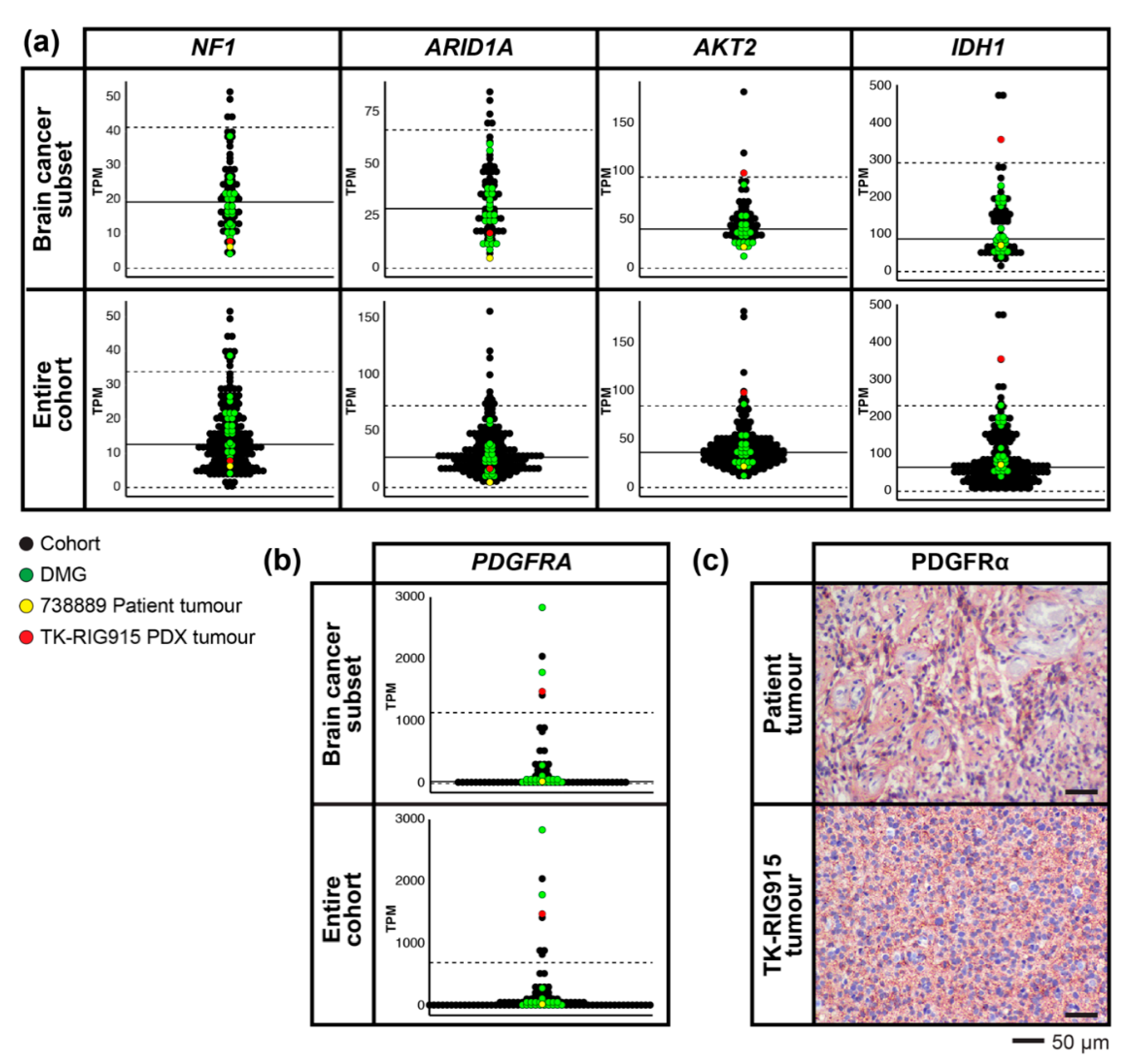
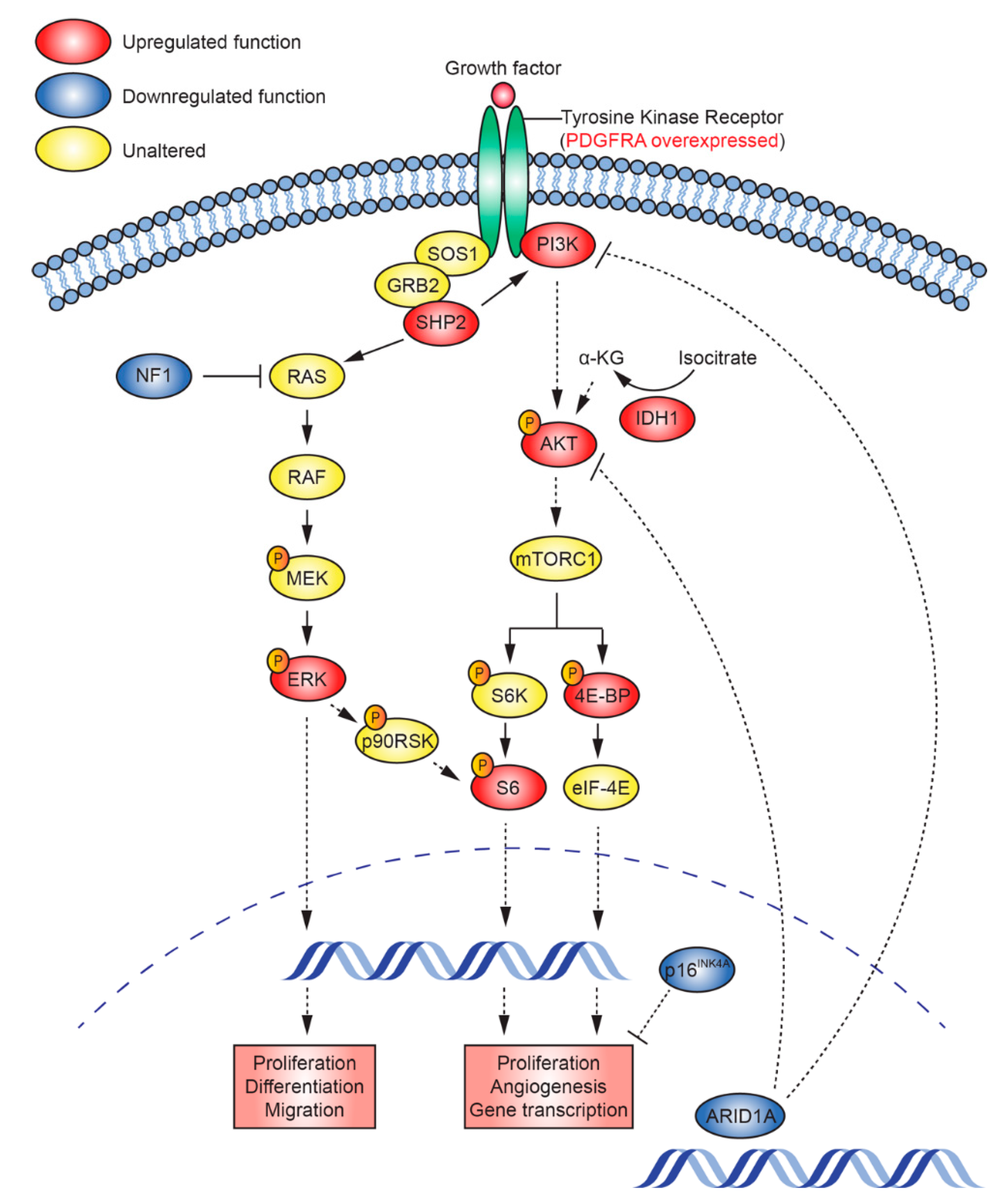
2.5. Mutations that Activate PI3K/AKT and Ras/Raf/MEK/ERK Signalling Are Present in TK-RIG915
2.6. RNA Expression Analysis Supports Over-Activation of PI3K/AKT/mTOR and Ras/Raf/MEK/ERK Pathways in TK-RIG915
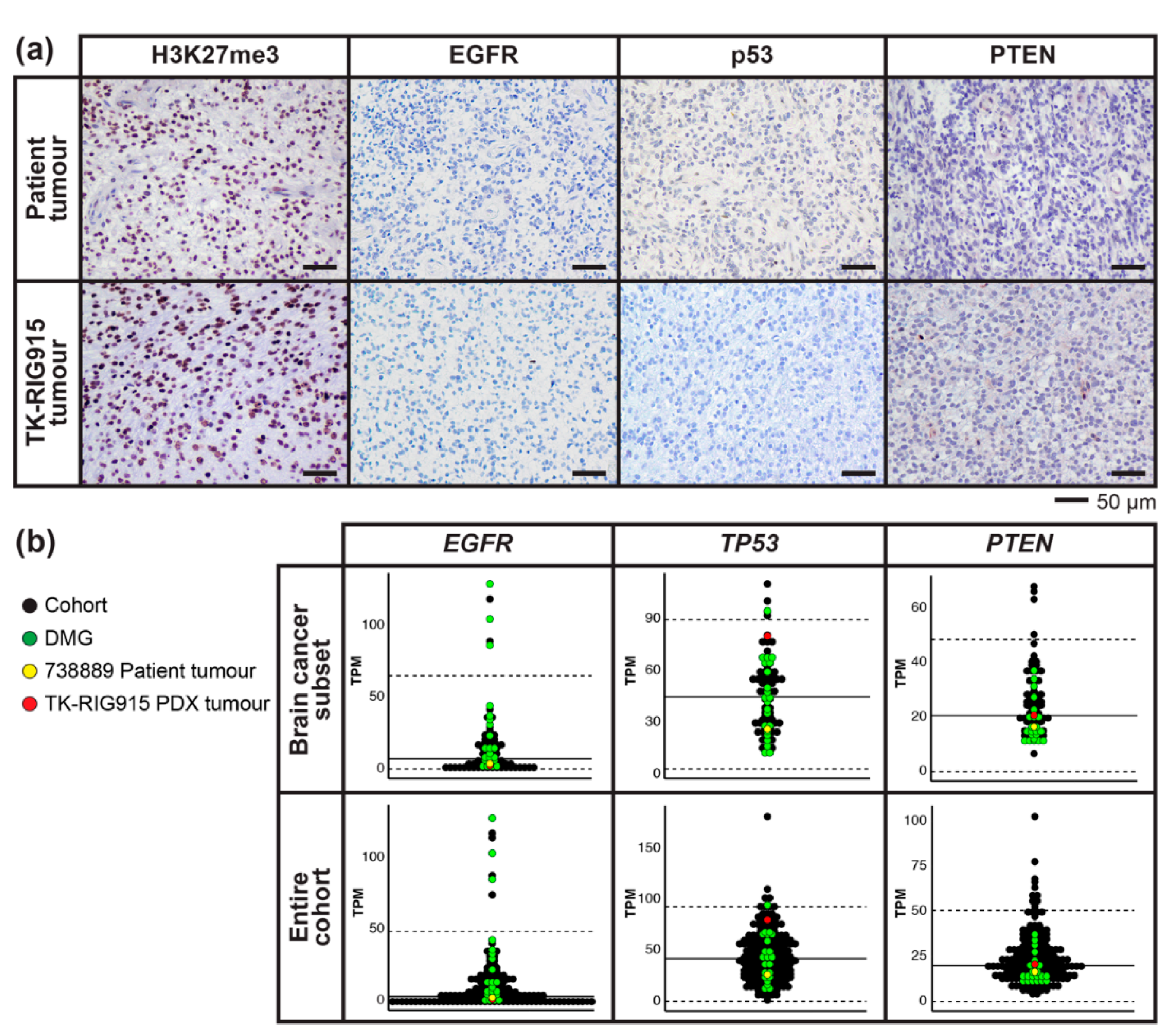
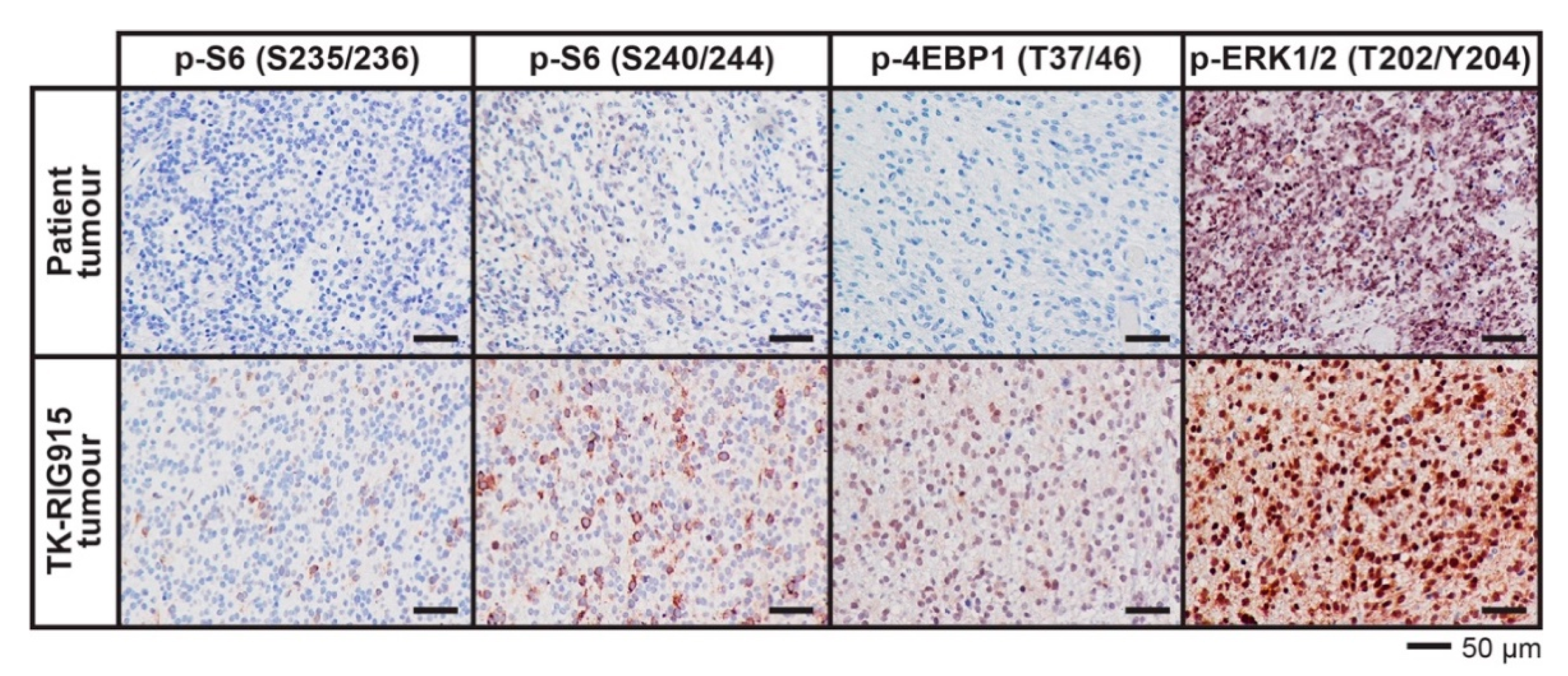
2.7. IHC Confirms Activated PI3K/AKT/mTOR and Ras/Raf/MEK/ERK Signalling in TK-RIG915
3. Discussion
4. Materials and Methods
4.1. Human Samples
4.2. Implantation of Patient Autopsy Tumour Cells and In Vivo Serial Transplantation
4.3. Histochemical Staining
4.4. DNA and RNA Extraction
4.5. Short Tandem Repeat Analysis
4.6. Methylation Array
4.7. Whole Genome Sequencing
4.8. RNA Sequencing, Clustering Analysis and Expression Profiling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, C.; Baker, S.J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat. Rev. Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Giannini, C.; Capper, D.; Paulus, W.; Figarella-Branger, D.; Lopes, M.B.; Batchelor, T.T.; Cairncross, J.G.; van den Bent, M.; Wick, W.; et al. cIMPACT-NOW update 2: Diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 2018, 135, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Patay, Z.; Howarth, R.; Pai Panandiker, A.S.; Onar-Thomas, A.; Gajjar, A.; Broniscer, A. Clinico-radiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J. Neurooncol. 2013, 114, 339–344. [Google Scholar] [CrossRef]
- Donson, A.M.; Erwin, N.S.; Kleinschmidt-DeMasters, B.K.; Madden, J.R.; Addo-Yobo, S.O.; Foreman, N.K. Unique molecular characteristics of radiation-induced glioblastoma. J. Neuropathol. Exp. Neurol. 2007, 66, 740–749. [Google Scholar] [CrossRef]
- Gits, H.C.; Anderson, M.; Stallard, S.; Pratt, D.; Zon, B.; Howell, C.; Kumar-Sinha, C.; Vats, P.; Kasaian, K.; Polan, D.; et al. Medulloblastoma therapy generates risk of a poorly-prognostic H3 wild-type subgroup of diffuse intrinsic pontine glioma: A report from the International DIPG Registry. Acta Neuropathol. Commun. 2018, 6, 67. [Google Scholar] [CrossRef]
- Paulino, A.C.; Mai, W.Y.; Chintagumpala, M.; Taher, A.; Teh, B.S. Radiation-induced malignant gliomas: Is there a role for reirradiation? Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 1381–1387. [Google Scholar] [CrossRef]
- Lee, C.Y.; Chen, Y.W.; Lee, Y.Y.; Chang, F.C.; Chen, H.H.; Lin, S.C.; Ho, D.M.; Huang, M.C.; Yen, S.H.; Wong, T.T.; et al. Irradiation-Induced Secondary Tumors following Pediatric Central Nervous System Tumors: Experiences of a Single Institute in Taiwan (1975–2013). Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 1243–1252. [Google Scholar] [CrossRef]
- Albright, A.L.; Packer, R.J.; Zimmerman, R.; Rorke, L.B.; Boyett, J.; Hammond, G.D. Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brain stem gliomas: A report from the Children’s Cancer Group. Neurosurgery 1993, 33, 1026–1029, discussion 1029–1030. [Google Scholar] [CrossRef] [PubMed]
- NCT02233049; Biological Medicine for Diffuse Intrinsic Pontine Glioma (DIPG) Eradication (BIOMEDE). In ClinicalTrials.gov; National Library of Medicine (US): Bethesda, MD, USA, 2014.
- Williams, J.R.; Young, C.C.; Vitanza, N.A.; McGrath, M.; Feroze, A.H.; Browd, S.R.; Hauptman, J.S. Progress in diffuse intrinsic pontine glioma: Advocating for stereotactic biopsy in the standard of care. Neurosurg. Focus 2020, 48, E4. [Google Scholar] [CrossRef] [PubMed]
- Phi, J.H.; Park, A.K.; Lee, S.; Choi, S.A.; Baek, I.P.; Kim, P.; Kim, E.H.; Park, H.C.; Kim, B.C.; Bhak, J.; et al. Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas. Acta Neuropathol. 2018, 135, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.S.; Xu, K.; Mercer, K.S.; Boop, F.; Klimo, P.; DeCupyere, M.; Grenet, J.; Robinson, S.; Dunphy, P.; Baker, S.J.; et al. Patient-derived orthotopic xenografts of pediatric brain tumors: A St. Jude resource. Acta Neuropathol. 2020, 140, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; James, C.D.; Jedlicka, A.E.; Connolly, D.C.; Chang, E.; Castellani, R.J.; Schmid, M.; Schiller, M.; Carson, D.A.; Burger, P.C. Molecular genetic alterations in radiation-induced astrocytomas. Am. J. Pathol. 1999, 154, 1431–1438. [Google Scholar] [CrossRef]
- Gessi, M.; Maderna, E.; Guzzetti, S.; Cefalo, G.; Massimino, M.; Solero, C.L.; Finocchiaro, G.; Pollo, B. Radiation-induced glioblastoma in a medulloblastoma patient: A case report with molecular features. Neuropathology 2008, 28, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.Y.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Clarke, J.; Oberheim Bush, N.A.; Taylor, J.; Chang, S.; et al. The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol. 2019, 137, 139–150. [Google Scholar] [CrossRef]
- Romeike, B.F.; Kim, Y.J.; Steudel, W.I.; Graf, N. Diffuse high-grade gliomas as second malignant neoplasms after radio-chemotherapy for pediatric malignancies. Childs Nerv. Syst. 2007, 23, 185–193. [Google Scholar] [CrossRef]
- Tada, M.; Sawamura, Y.; Abe, H.; Iggo, R. Homozygous p53 gene mutation in a radiation-induced glioblastoma 10 years after treatment for an intracranial germ cell tumor: Case report. Neurosurgery 1997, 40, 393–396. [Google Scholar] [CrossRef]
- Mascelli, S.; Raso, A.; Biassoni, R.; Severino, M.; Sak, K.; Joost, K.; Milanaccio, C.; Barra, S.; Grillo-Ruggieri, F.; Vanni, I.; et al. Analysis of NADP+-dependent isocitrate dehydrogenase-1/2 gene mutations in pediatric brain tumors: Report of a secondary anaplastic astrocytoma carrying the IDH1 mutation. J. Neurooncol. 2012, 109, 477–484. [Google Scholar] [CrossRef]
- Nakao, T.; Sasagawa, Y.; Nobusawa, S.; Takabatake, Y.; Sabit, H.; Kinoshita, M.; Miyashita, K.; Hayashi, Y.; Yokoo, H.; Nakada, M. Radiation-induced gliomas: A report of four cases and analysis of molecular biomarkers. Brain Tumor Pathol. 2017, 34, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Meel, M.H.; Sewing, A.C.P.; Waranecki, P.; Metselaar, D.S.; Wedekind, L.E.; Koster, J.; van Vuurden, D.G.; Kaspers, G.J.L.; Hulleman, E. Culture methods of diffuse intrinsic pontine glioma cells determine response to targeted therapies. Exp. Cell Res. 2017, 360, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Dobson, T.H.W.; Gopalakrishnan, V. Preclinical Models of Pediatric Brain Tumors-Forging Ahead. Bioengineering (Basel) 2018, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- NCT00003203; Carboplatin and Vincristine Plus Radiation Therapy Followed By Adjuvant Chemotherapy in Treating Young Patients With Newly Diagnosed CNS Embryonal Tumors. In ClinicalTrials.gov; National Library of Medicine (US): Bethesda, MD, USA, 2003.
- NCT01189266; Vorinostat and Radiation Therapy Followed by Maintenance Therapy With Vorinostat in Treating Younger Patients With Newly Diagnosed Diffuse Intrinsic Pontine Glioma. In ClinicalTrials.gov; National Library of Medicine (US): Bethesda, MD, USA, 2010.
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Mayoh, C.; Lau, L.M.S.; Khuong-Quang, D.A.; Pinese, M.; Kumar, A.; Barahona, P.; Wilkie, E.E.; Sullivan, P.; Bowen-James, R.; et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk paediatric cancer. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Cahan, W.G.; Woodard, H.Q.; Higinbotham, N.L.; Stewart, F.W.; Coley, B.L. Sarcoma arising in irradiated bone; report of 11 cases. Cancer 1948, 1, 3–29. [Google Scholar] [CrossRef]
- Singh, G.K.; Yadav, V.; Singh, P.; Bhowmik, K.T. Radiation-Induced Malignancies Making Radiotherapy a “Two-Edged Sword”: A Review of Literature. World J. Oncol. 2017, 8, 1–6. [Google Scholar] [CrossRef]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar; [VCV000013653.4]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000013653.4 (accessed on 15 September 2020).
- Gymnopoulos, M.; Elsliger, M.A.; Vogt, P.K. Rare cancer-specific mutations in PIK3CA show gain of function. Proc. Natl. Acad. Sci. USA 2007, 104, 5569–5574. [Google Scholar] [CrossRef] [PubMed]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, G.; Militello, L.; Steelman, L.S.; Cavallaro, A.; Basile, F.; Nicoletti, F.; Stivala, F.; McCubrey, J.A.; Libra, M. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Hon, W.C.; Berndt, A.; Williams, R.L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31, 3655–3666. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Collins, F.S. The neurofibromatosis type 1 gene and its protein product, neurofibromin. Neuron 1993, 10, 335–343. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar; [VCV000481993.3]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/481993/ (accessed on 15 September 2020).
- National Center for Biotechnology Information. ClinVar; [VCV000545744.6]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/545744/ (accessed on 15 September 2020).
- National Center for Biotechnology Information. ClinVar; [VCV000429015.2]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/429015/ (accessed on 15 September 2020).
- National Center for Biotechnology Information. ClinVar; [VCV000013335.6]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/13335/ (accessed on 15 September 2020).
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef]
- Loh, M.L.; Vattikuti, S.; Schubbert, S.; Reynolds, M.G.; Carlson, E.; Lieuw, K.H.; Cheng, J.W.; Lee, C.M.; Stokoe, D.; Bonifas, J.M.; et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Sonnenblick, A.; Gennissen, A.; Brohee, S.; Hijmans, E.M.; Evers, B.; Fumagalli, D.; Desmedt, C.; Loibl, S.; Denkert, C.; et al. Loss of ARID1A Activates ANXA1, which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin. Cancer Res. 2016, 22, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih Ie, M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef]
- Zhu, Z.; He, X.; Johnson, C.; Stoops, J.; Eaker, A.E.; Stoffer, D.S.; Bell, A.; Zarnegar, R.; DeFrances, M.C. PI3K is negatively regulated by PIK3IP1, a novel p110 interacting protein. Biochem. Biophys. Res. Commun. 2007, 358, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gu, F.; She, C.; Guo, H.; Li, W.; Niu, R.; Fu, L.; Zhang, N.; Ma, Y. Reduction of Akt2 inhibits migration and invasion of glioma cells. Int. J. Cancer 2009, 125, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wu, S.; Zhang, J.; Li, M.; Xu, F.; Wang, A.; Lei, Y.; Zhu, G. Wildtype IDH1 affects cell migration by modulating the PI3K/AKT/mTOR pathway in primary glioblastoma cells. Mol. Med. Rep. 2020, 22, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Hayano, A.; Kanayama, T. Radiation-induced gliomas: A comprehensive review and meta-analysis. Neurosurg. Rev. 2018, 41, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Bartels, U.; Bouffet, E.; Becher, O.; Hawkins, C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: Diagnostic and therapeutic implications. Acta Neuropathol. 2014, 128, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Lapin, D.H.; Tsoli, M.; Ziegler, D.S. Genomic Insights into Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2017, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Meel, M.H.; Kaspers, G.J.L.; Hulleman, E. Preclinical therapeutic targets in diffuse midline glioma. Drug Resist. Updat. 2019, 44, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; da Silva, N.S.; et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J. Clin. Oncol. 2011, 29, 3999–4006. [Google Scholar] [CrossRef]
- Addo-Yobo, S.O.; Straessle, J.; Anwar, A.; Donson, A.M.; Kleinschmidt-Demasters, B.K.; Foreman, N.K. Paired overexpression of ErbB3 and Sox10 in pilocytic astrocytoma. J. Neuropathol. Exp. Neurol. 2006, 65, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Colin, C.; Virard, I.; Baeza, N.; Tchoghandjian, A.; Fernandez, C.; Bouvier, C.; Calisti, A.; Tong, S.; Durbec, P.; Figarella-Branger, D. Relevance of combinatorial profiles of intermediate filaments and transcription factors for glioma histogenesis. Neuropathol. Appl. Neurobiol. 2007, 33, 431–439. [Google Scholar] [CrossRef]
- Takei, H.; Yogeswaren, S.T.; Wong, K.K.; Mehta, V.; Chintagumpala, M.; Dauser, R.C.; Lau, C.C.; Adesina, A.M. Expression of oligodendroglial differentiation markers in pilocytic astrocytomas identifies two clinical subsets and shows a significant correlation with proliferation index and progression free survival. J. Neurooncol. 2008, 86, 183–190. [Google Scholar] [CrossRef]
- Bar, E.E.; Lin, A.; Tihan, T.; Burger, P.C.; Eberhart, C.G. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J. Neuropathol. Exp. Neurol. 2008, 67, 878–887. [Google Scholar] [CrossRef]
- Jones, D.T.; Hutter, B.; Jager, N.; Korshunov, A.; Kool, M.; Warnatz, H.J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.; et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Perry, A.; Gutmann, D.H.; O’Neill, B.P.; Leonard, J.; Bryant, S.; Giannini, C. Gliomas in neurofibromatosis type 1: A clinicopathologic study of 100 patients. J. Neuropathol. Exp. Neurol. 2008, 67, 240–249. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino Mdel, C.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Martinho, O.; Longatto-Filho, A.; Lambros, M.B.; Martins, A.; Pinheiro, C.; Silva, A.; Pardal, F.; Amorim, J.; Mackay, A.; Milanezi, F.; et al. Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br. J. Cancer 2009, 101, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, L.; Zhang, A.; Wang, Y.; Yue, X.; You, Y.; Pu, P.; Kang, C. AKT2 expression is associated with glioma malignant progression and required for cell survival and invasion. Oncol. Rep. 2010, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Dogruluk, T.; Tsang, Y.H.; Espitia, M.; Chen, F.; Chen, T.; Chong, Z.; Appadurai, V.; Dogruluk, A.; Eterovic, A.K.; Bonnen, P.E.; et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015, 75, 5341–5354. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Dresser, J.; Wilder-Romans, K.; Parsels, J.D.; Zhao, S.G.; Davis, M.; Zhao, L.; Kachman, M.; Wernisch, S.; Burant, C.F.; et al. Glioblastoma Therapy Can Be Augmented by Targeting IDH1-Mediated NADPH Biosynthesis. Cancer Res. 2017, 77, 960–970. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, H.; Hou, B. Decreased expression of ARID1A is related to the poor prognosis of glioma patients. Int. J. Clin. Exp. Pathol. 2016, 9, 2009–2014. [Google Scholar]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tsuda, H.; Takano, M.; Tamai, S.; Matsubara, O. PIK3CA mutations and loss of ARID1A protein expression are early events in the development of cystic ovarian clear cell adenocarcinoma. Virchows Arch. 2012, 460, 77–87. [Google Scholar] [CrossRef]
- Jongmans, M.C.; van der Burgt, I.; Hoogerbrugge, P.M.; Noordam, K.; Yntema, H.G.; Nillesen, W.M.; Kuiper, R.P.; Ligtenberg, M.J.; van Kessel, A.G.; van Krieken, J.H.; et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. 2011, 19, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zheng, H.; Li, X.; Wang, S.; Meyerson, H.J.; Yang, W.; Neel, B.G.; Qu, C.K. Gain-of-function mutations of Ptpn11 (Shp2) cause aberrant mitosis and increase susceptibility to DNA damage-induced malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, 984–989. [Google Scholar] [CrossRef] [PubMed]
- See, W.L.; Tan, I.L.; Mukherjee, J.; Nicolaides, T.; Pieper, R.O. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012, 72, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; McArthur, G.; Gan, H.; Cher, L. Prolonged disease control with MEK inhibitor in neurofibromatosis type I-associated glioblastoma. J. Clin. Pharm. Ther. 2016, 41, 357–359. [Google Scholar] [CrossRef]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS ONE 2013, 8, e77639. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- NCT03336931; PRecISion Medicine for Children With Cancer (PRISM). In ClinicalTrials.gov; Library of Medicine (US): Bethesda, MD, USA, 2017.
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA, USA, 2014.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Coding Mutation | Impact | Likely Effect | Amino Acid Alteration | Allelic Frequency | |
|---|---|---|---|---|---|---|
| Patient Tumour | TK-RIG915 | |||||
| PIK3CA | c.3140A > T | Missense | Activating | p. His1047Leu | 24% | 43% |
| NF1 | c.3367G > T | Stop-gain | Inactivating | p.Glu1123Ter | 32% | 45% |
| NF1 | c.233dupA | Frameshift | Inactivating | p.Asn78LysfsTer29 | 20% | 62% |
| PTPN11 | c.854T > C | Missense | Gain of function | p. Phe285Ser | 2% | 55% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitehouse, J.P.; Howlett, M.; Hii, H.; Mayoh, C.; Wong, M.; Barahona, P.; Ajuyah, P.; White, C.L.; Buntine, M.K.; Dyke, J.M.; et al. A Novel Orthotopic Patient-Derived Xenograft Model of Radiation-Induced Glioma Following Medulloblastoma. Cancers 2020, 12, 2937. https://doi.org/10.3390/cancers12102937
Whitehouse JP, Howlett M, Hii H, Mayoh C, Wong M, Barahona P, Ajuyah P, White CL, Buntine MK, Dyke JM, et al. A Novel Orthotopic Patient-Derived Xenograft Model of Radiation-Induced Glioma Following Medulloblastoma. Cancers. 2020; 12(10):2937. https://doi.org/10.3390/cancers12102937
Chicago/Turabian StyleWhitehouse, Jacqueline P., Meegan Howlett, Hilary Hii, Chelsea Mayoh, Marie Wong, Paulette Barahona, Pamela Ajuyah, Christine L. White, Molly K. Buntine, Jason M. Dyke, and et al. 2020. "A Novel Orthotopic Patient-Derived Xenograft Model of Radiation-Induced Glioma Following Medulloblastoma" Cancers 12, no. 10: 2937. https://doi.org/10.3390/cancers12102937
APA StyleWhitehouse, J. P., Howlett, M., Hii, H., Mayoh, C., Wong, M., Barahona, P., Ajuyah, P., White, C. L., Buntine, M. K., Dyke, J. M., Lee, S., Valvi, S., Stanley, J., Andradas, C., Carline, B., Kuchibhotla, M., Ekert, P. G., Cowley, M. J., Gottardo, N. G., & Endersby, R. (2020). A Novel Orthotopic Patient-Derived Xenograft Model of Radiation-Induced Glioma Following Medulloblastoma. Cancers, 12(10), 2937. https://doi.org/10.3390/cancers12102937