The In Vitro and In Vivo Anticancer Properties of Chalcone Flavokawain B through Induction of ROS-Mediated Apoptotic and Autophagic Cell Death in Human Melanoma Cells



 ,
,  , and
, and
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
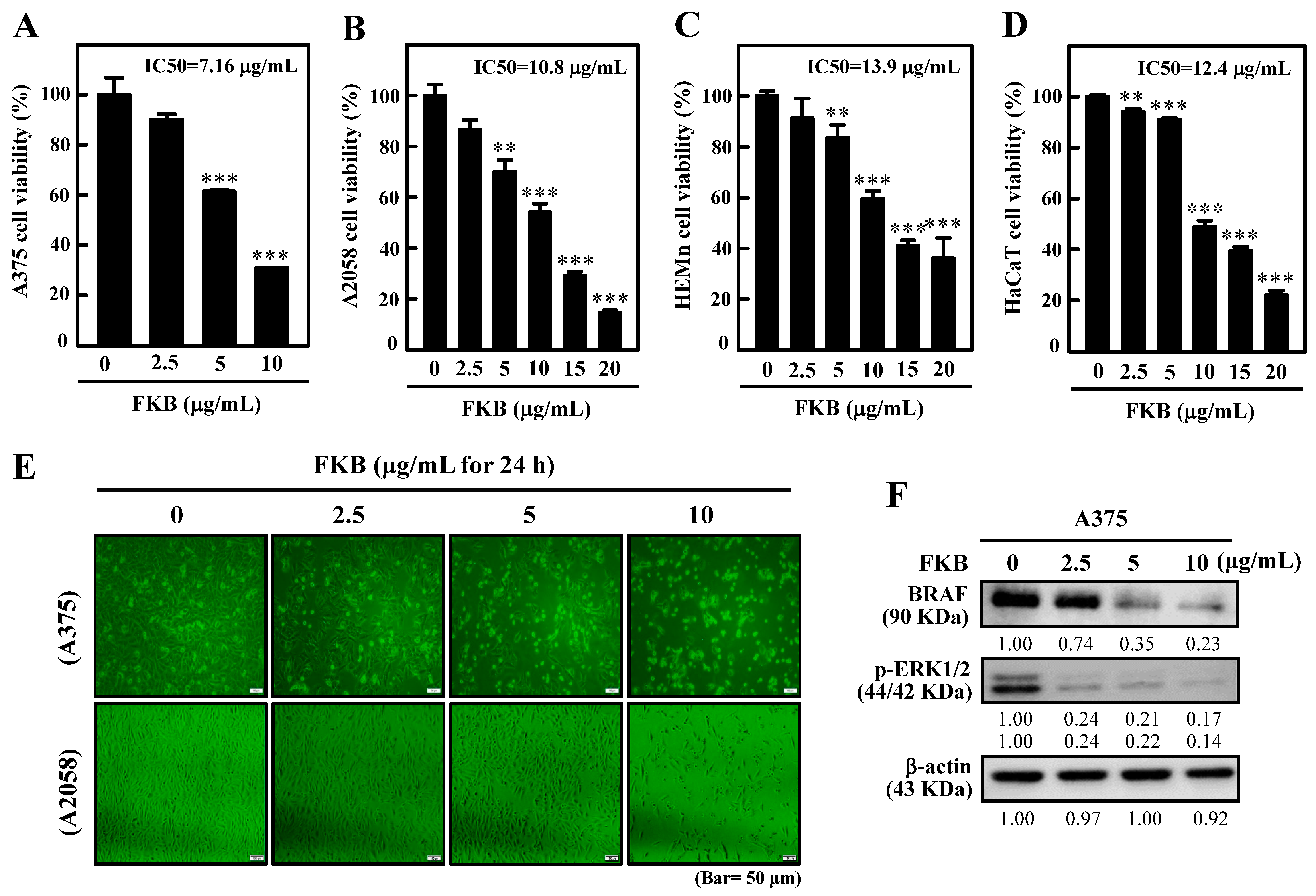
2.1. FKB Suppressed Human Melanoma Cell Growth
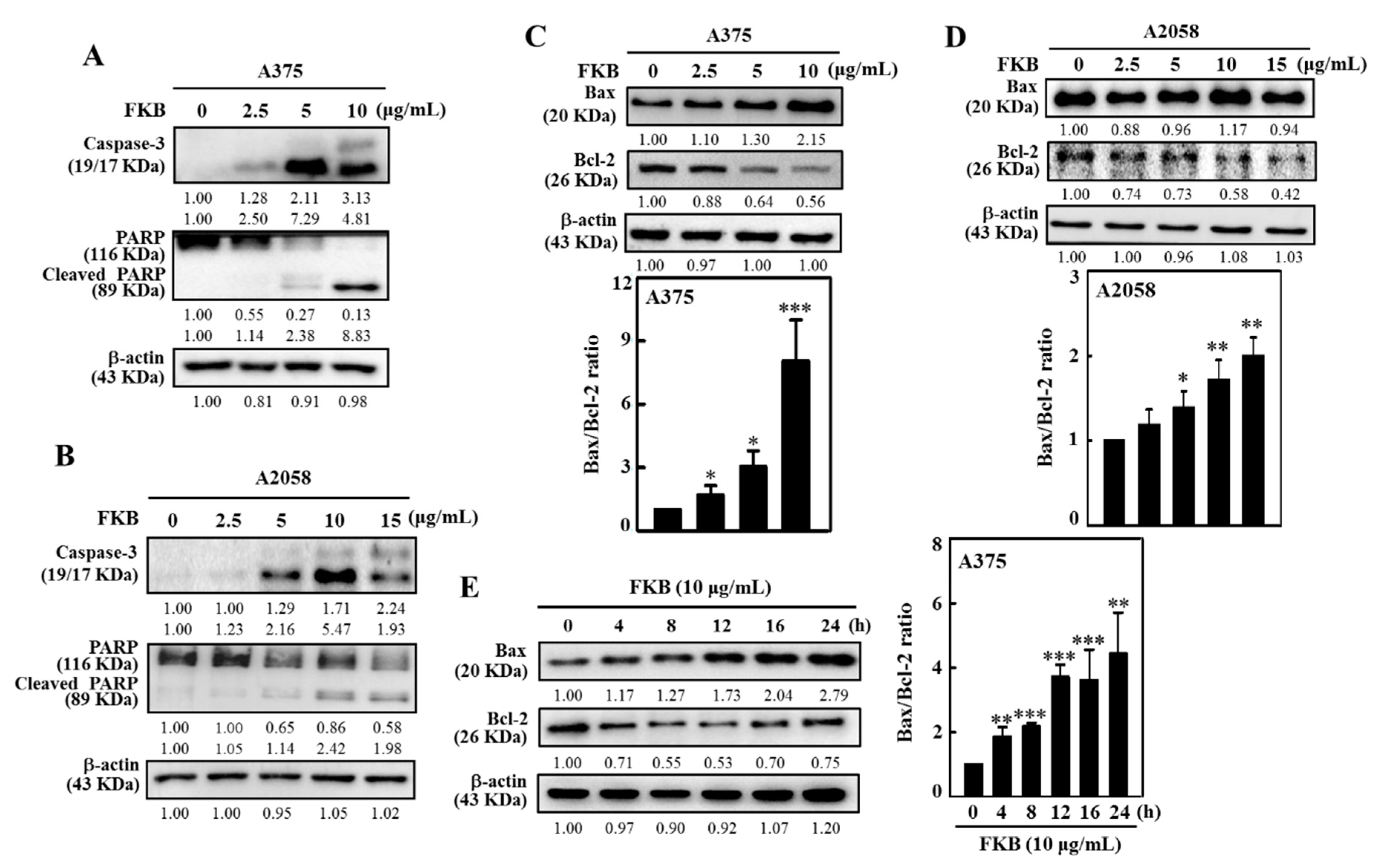
2.2. FKB Induced Apoptosis in Human Melanoma Cells
2.3. FKB Induced Early and Late Apoptotic Cell Death in Melanoma Cells
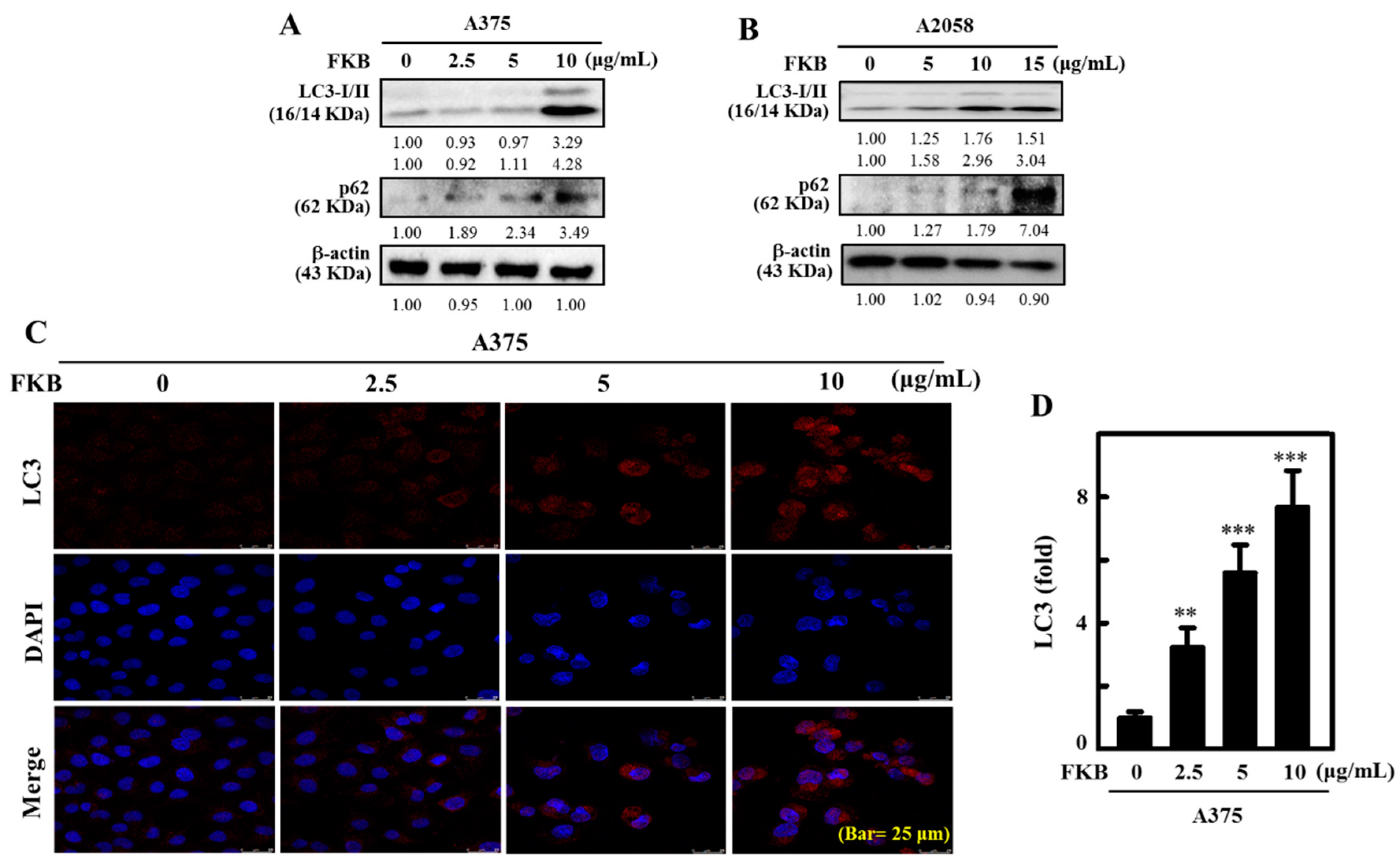
2.4. FKB Increased LC3-II Accumulation and Activated Autophagy in Melanoma Cells
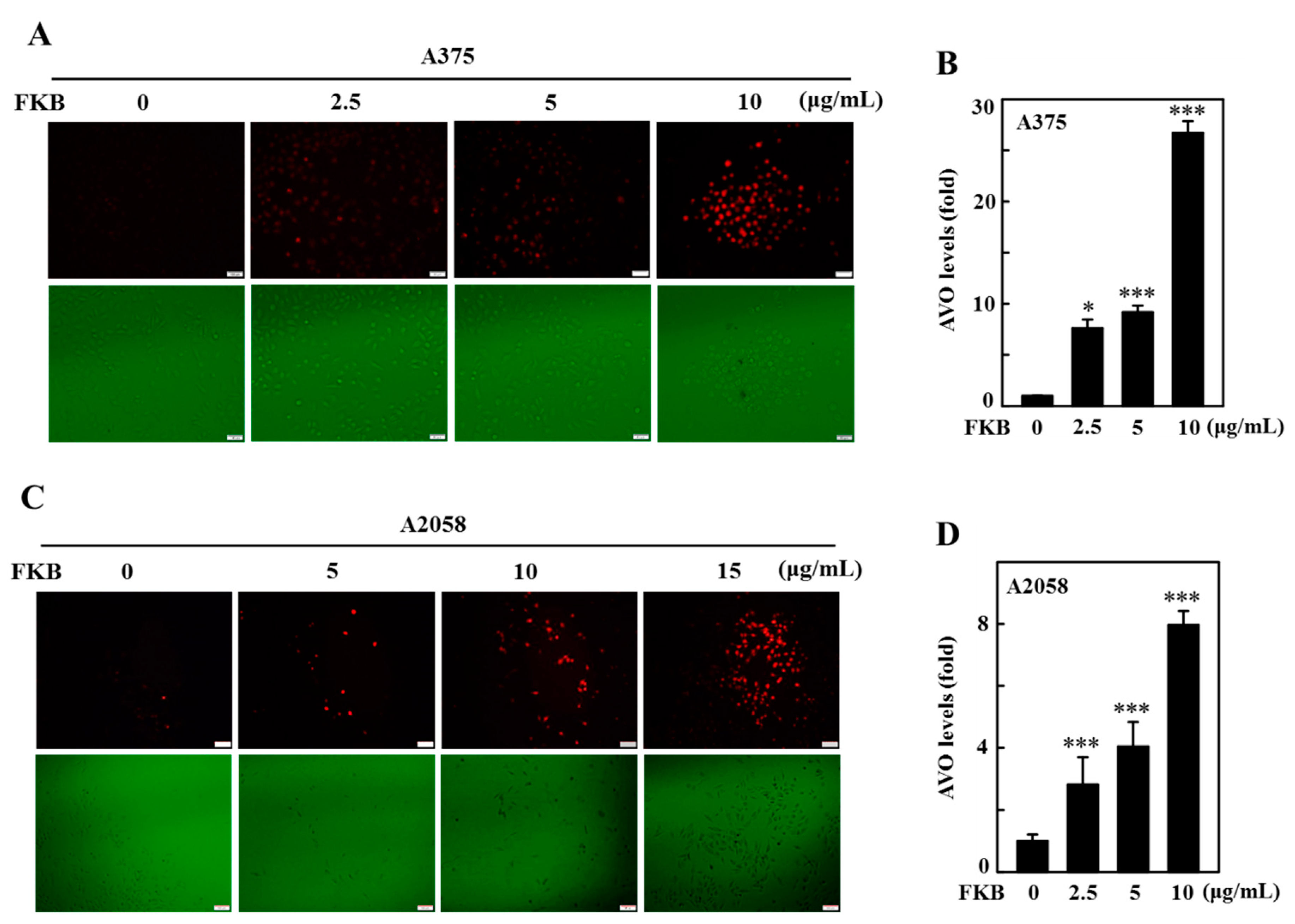
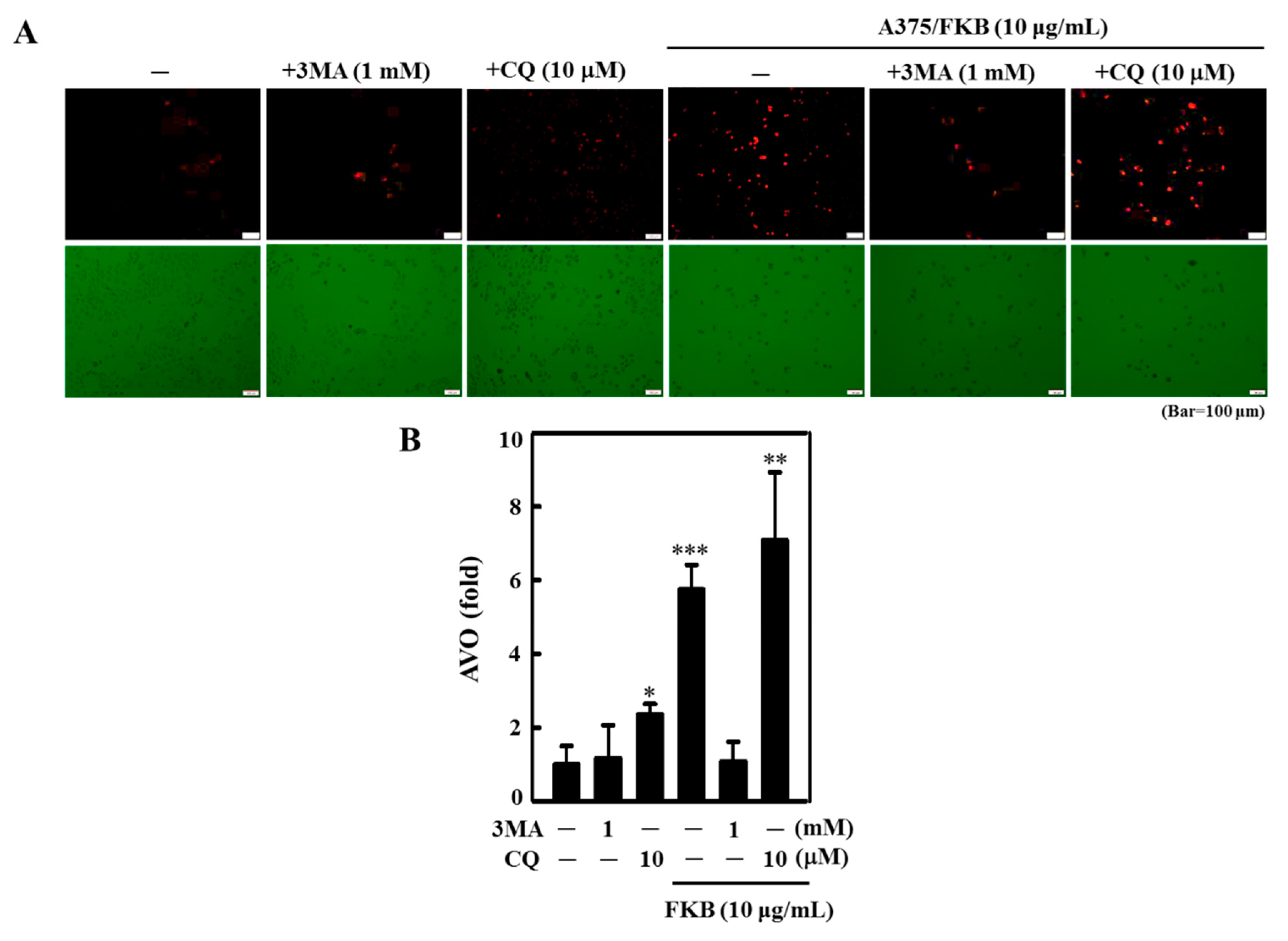
2.5. FKB Enhanced AVO Formation in A375 and A2058 Cells
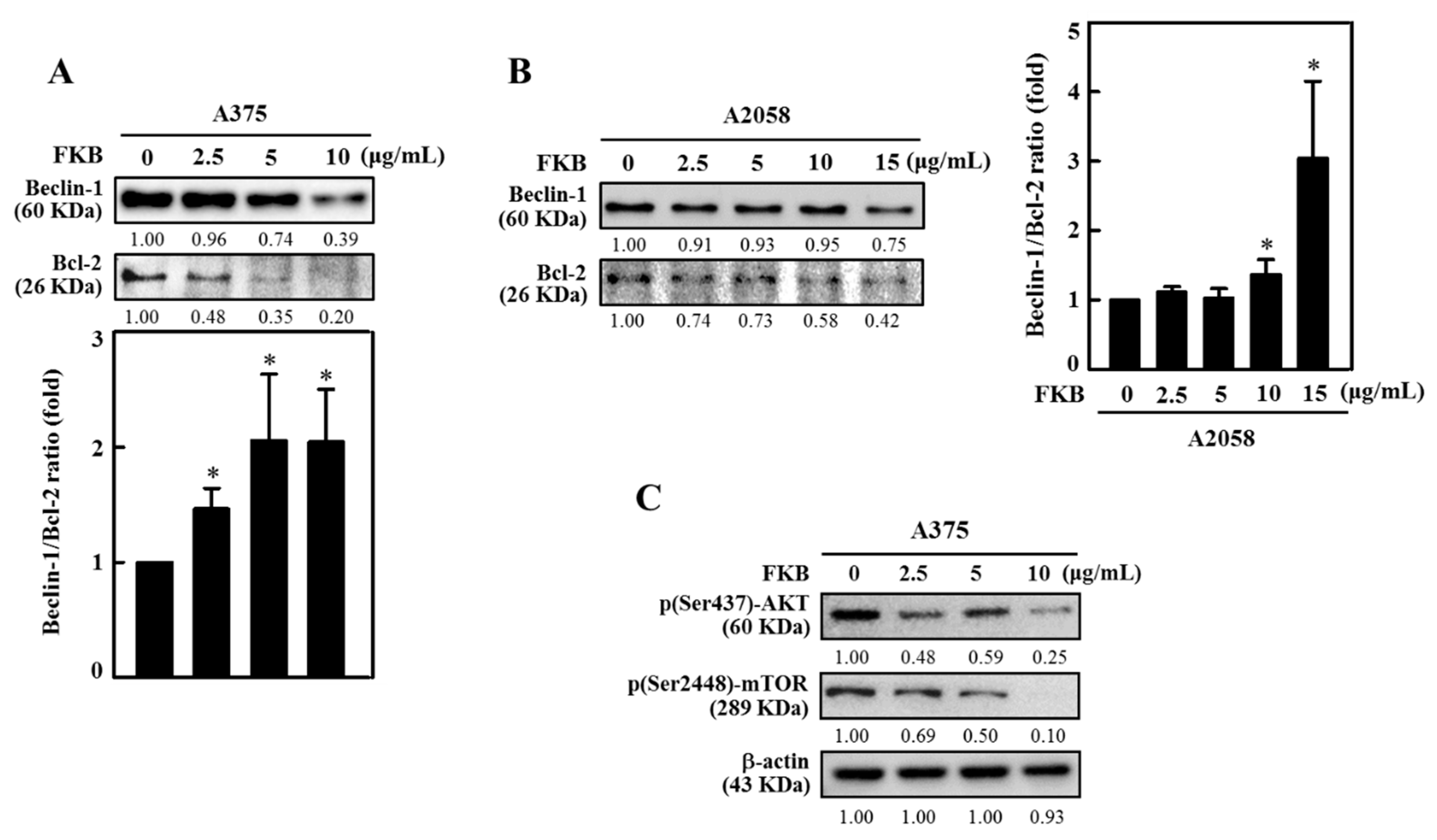
2.6. FKB Dysregulated Beclin-1 and Bcl-2 Ratio and Reduced AKT Phosphorylation and mTOR Expressions Leading to Autophagy in Human Melanoma Cells
2.7. FKB Accelerated Autophagy in A375 Cells as a Death Mechanism
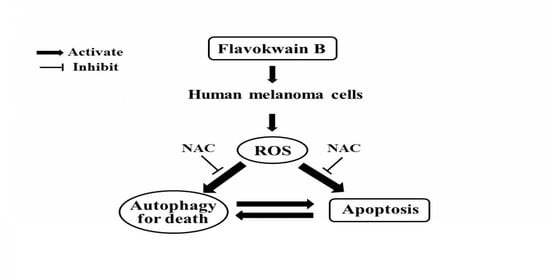
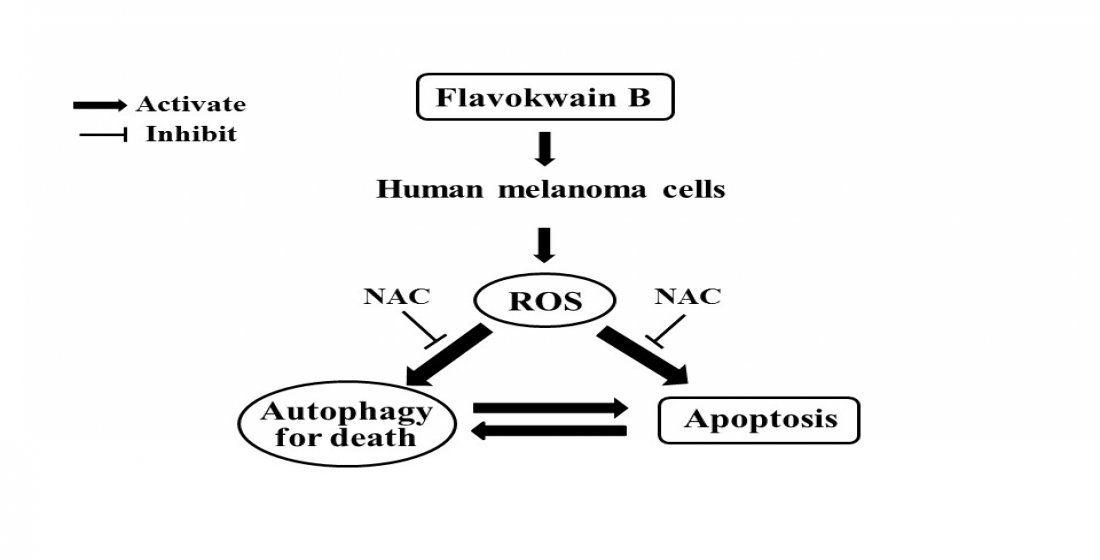
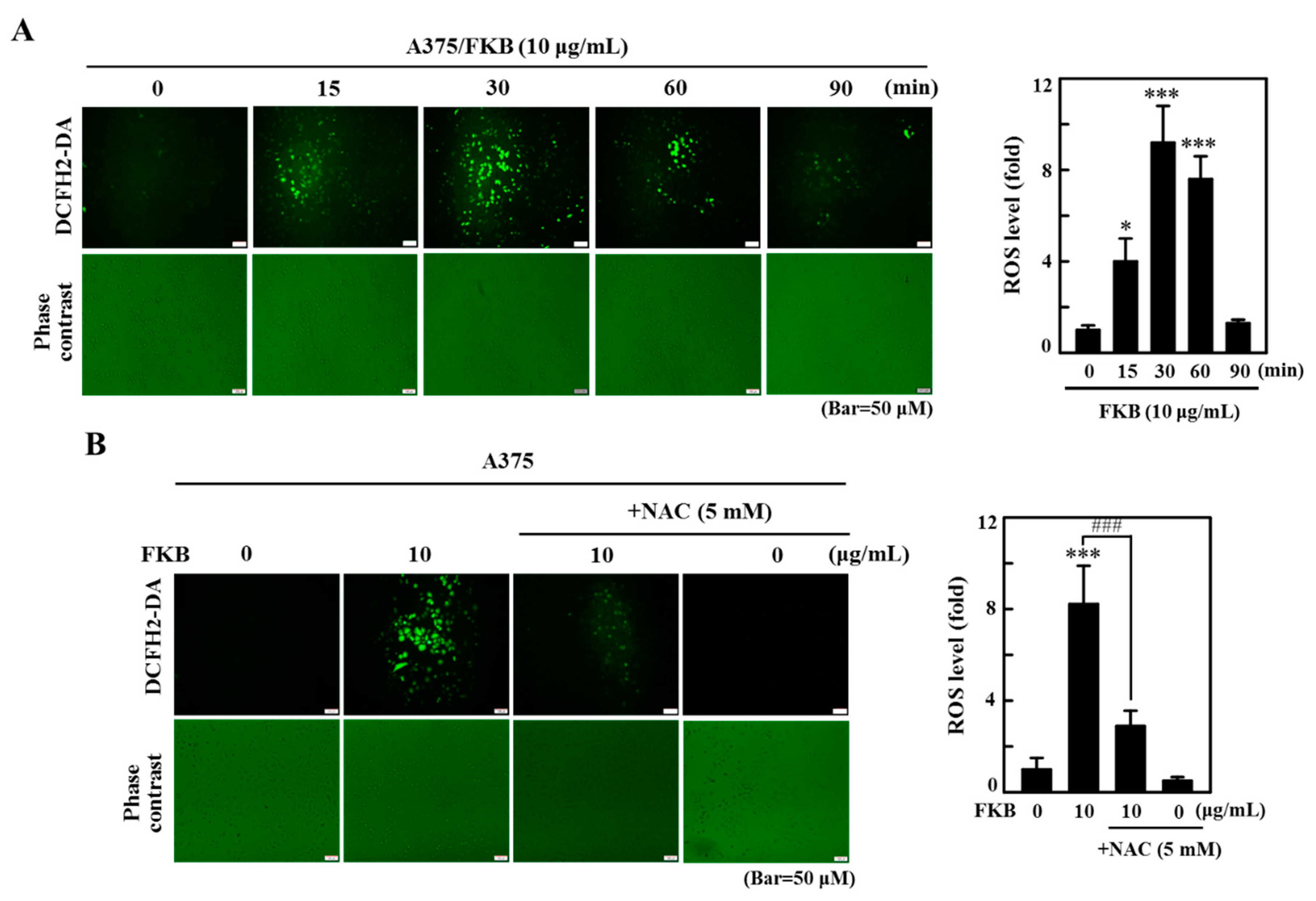
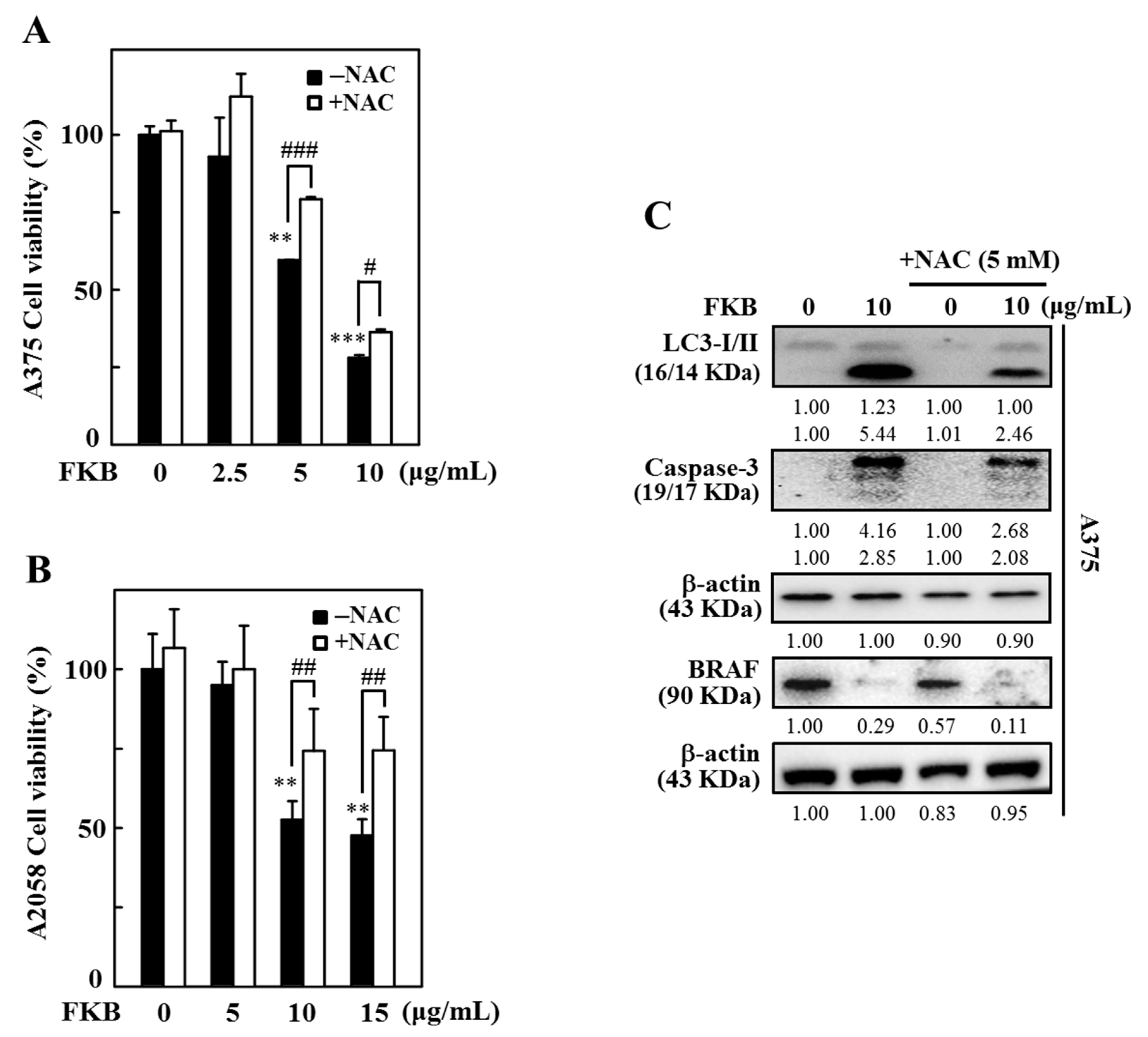
2.8. FKB Induced ROS Triggered Cell Death of Melanoma Cells
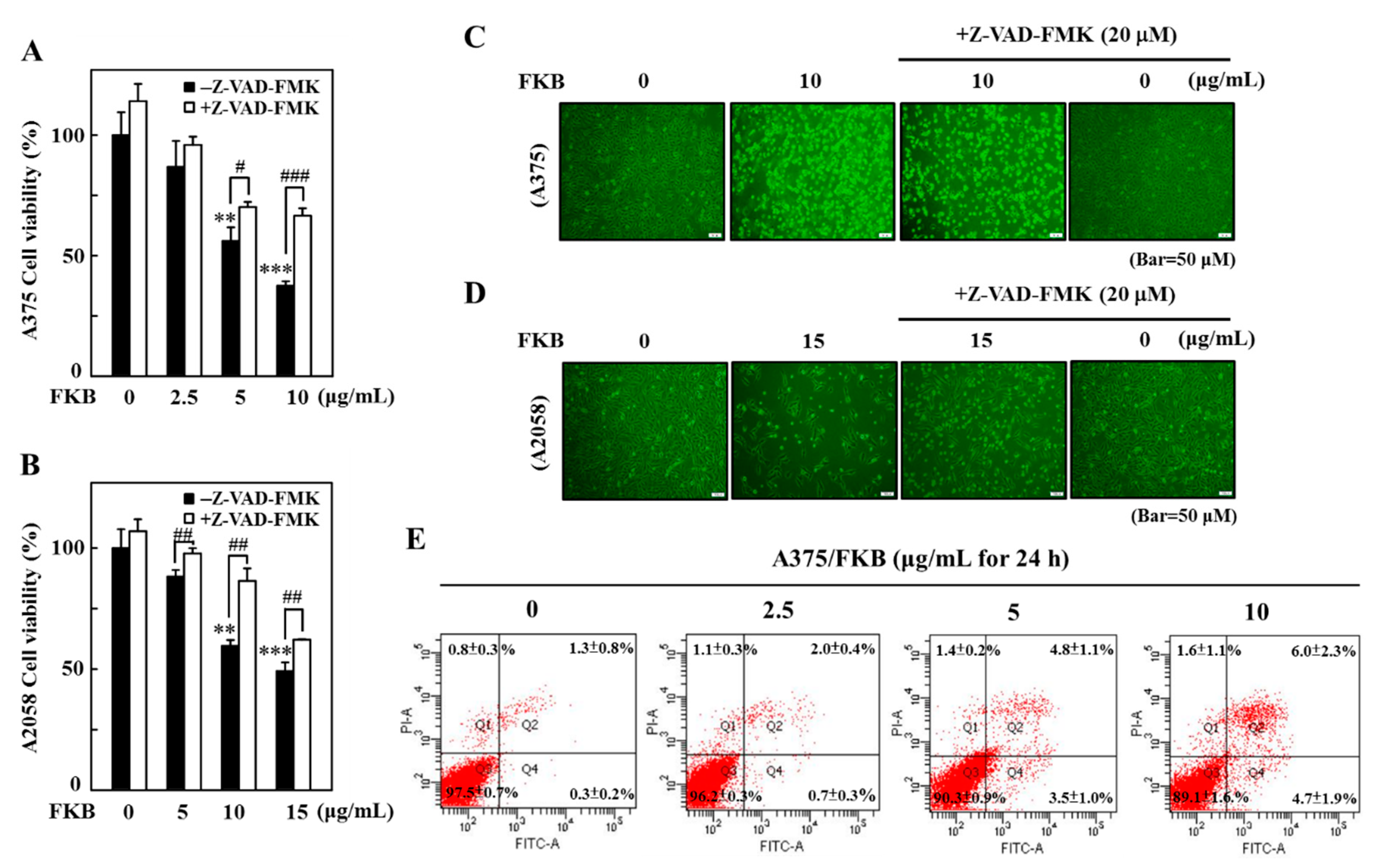
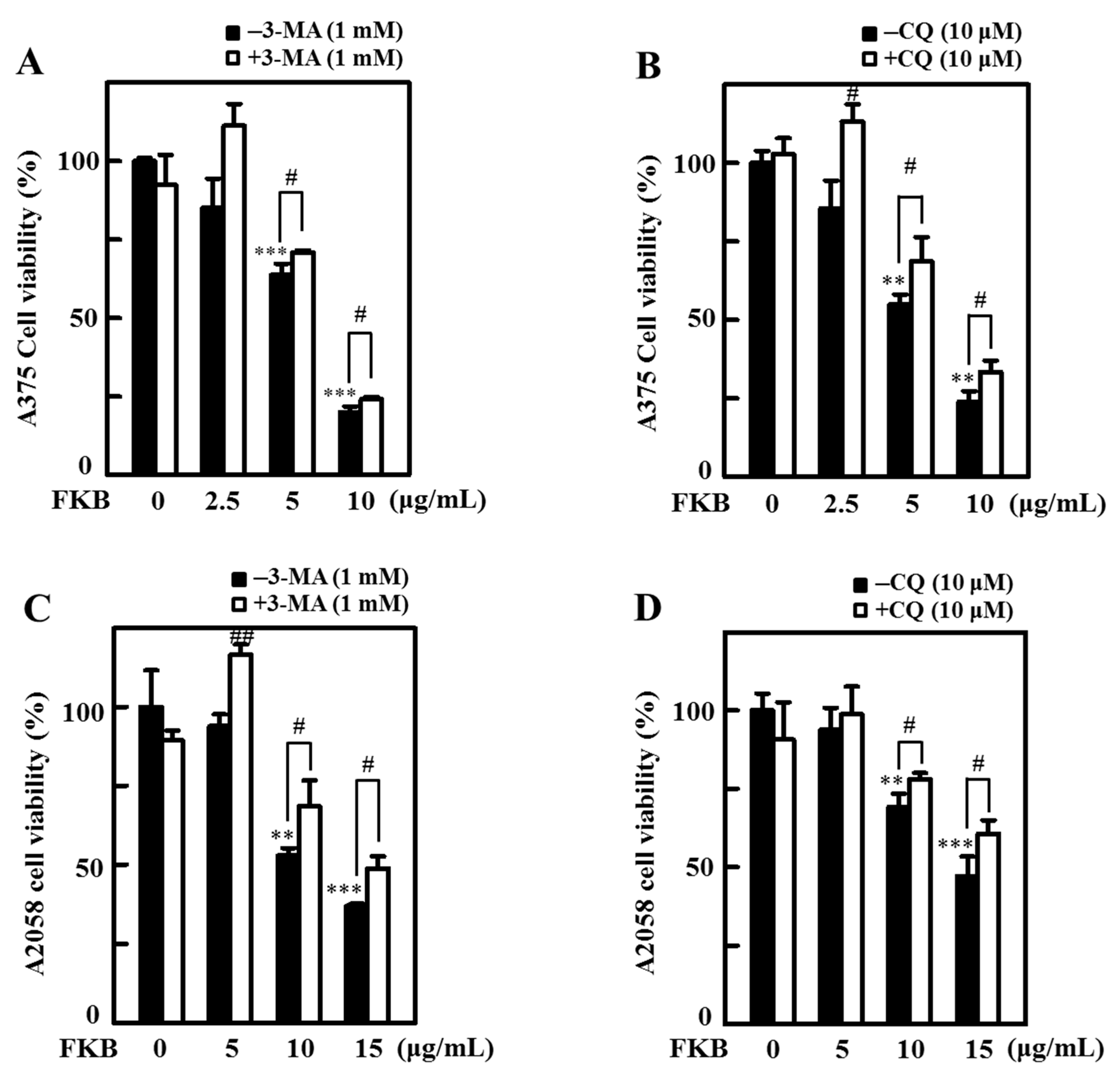
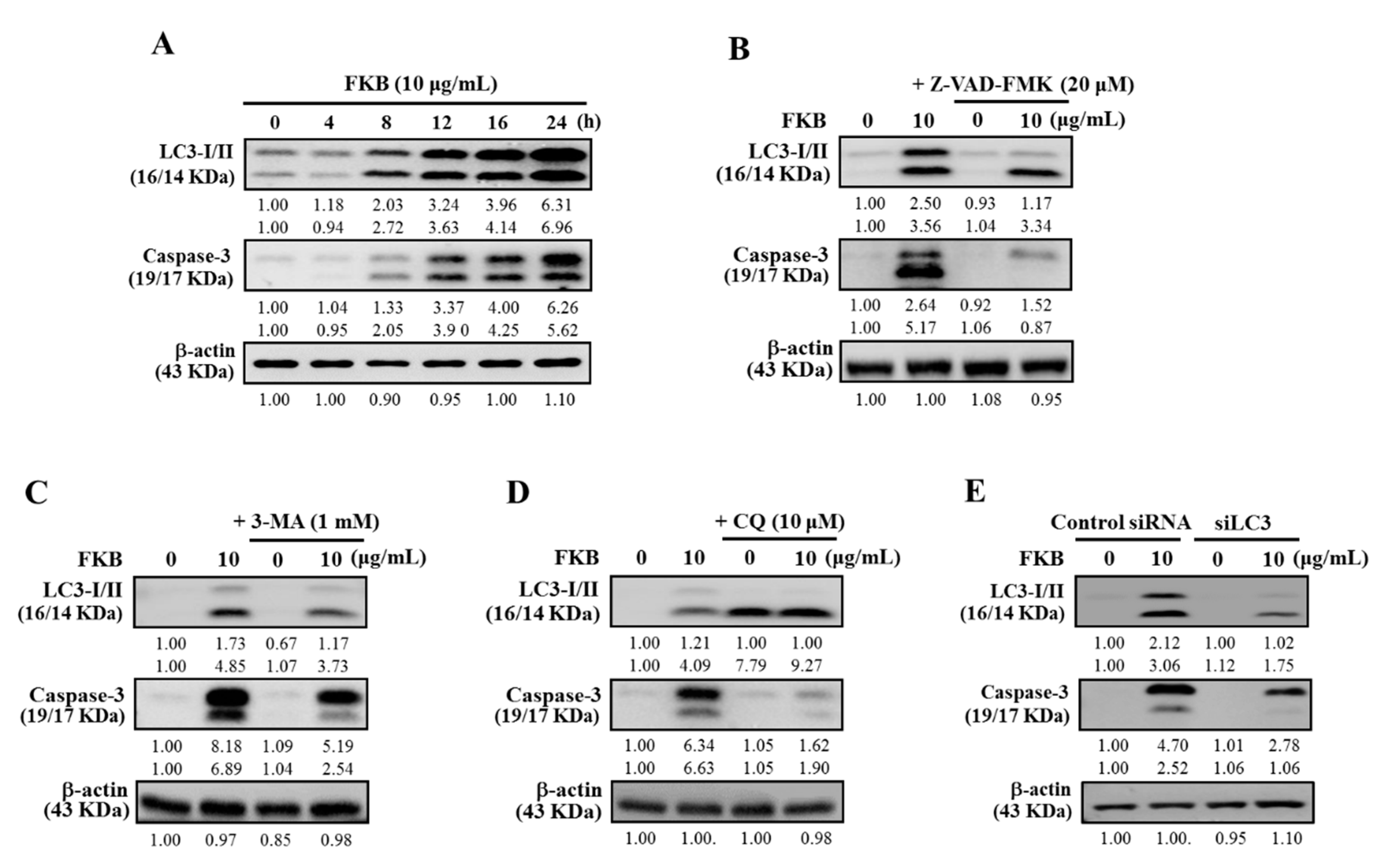
2.9. Inhibition of FKB-Induced Apoptosis Suppressed Autophagy
2.10. Attenuation of Apoptosis Due to the Inhibition of FKB-Induced Autophagy
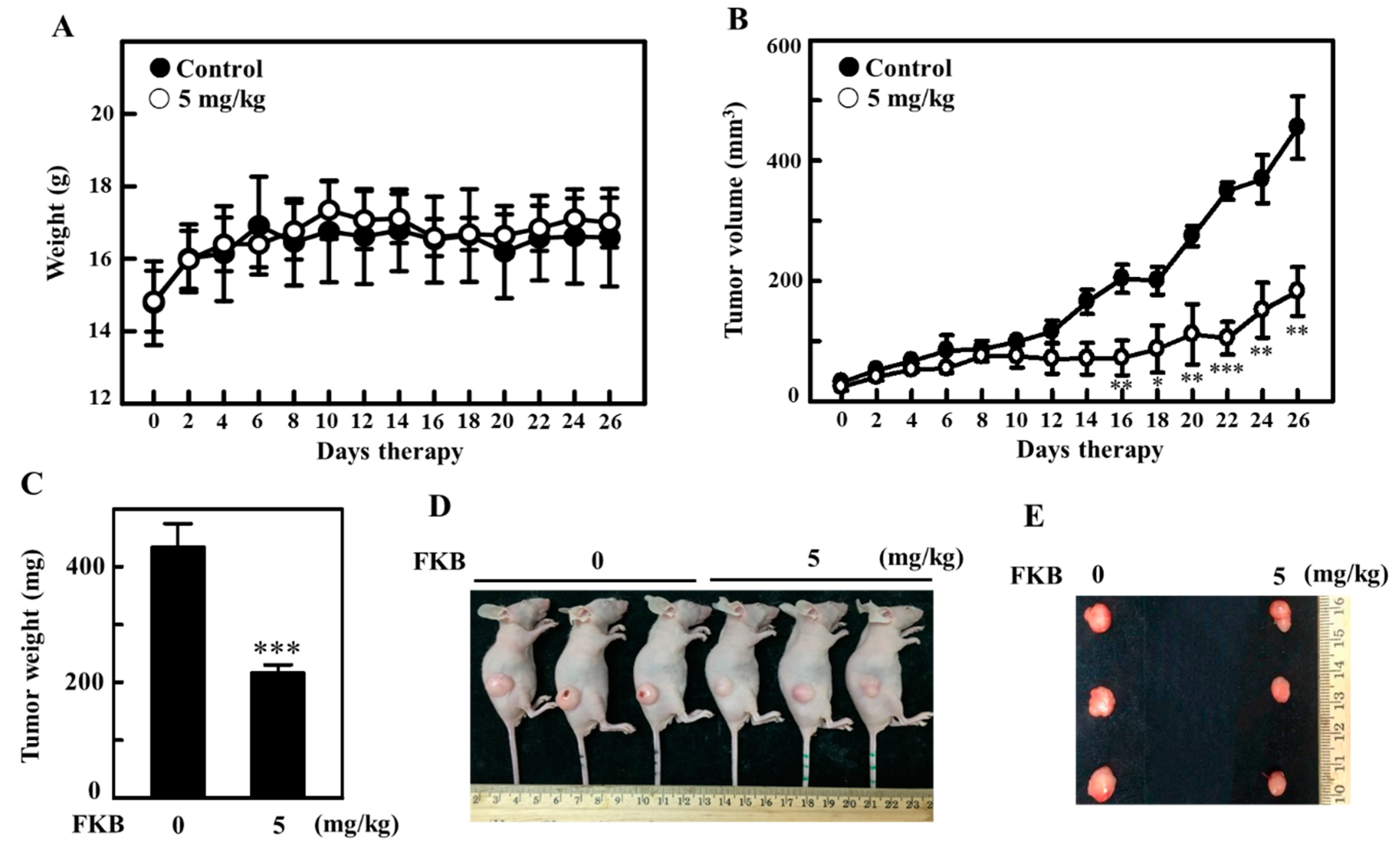
2.11. FKB Treatment Inhibited the Tumor Growth in A375 Xenografted Athymic Nude Mice In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. FKB Treatment
4.3. Cell Culture
4.4. Cell Viability Assay (MTT) and Determination of IC50 Value of FKB
4.5. Protein Isolation and Western Blot
4.6. Detection of Apoptotic Cells by Annexin V and PI Staining
4.7. Detection and Quantification of AVOs with Acridine Orange Staining
4.8. LC3 Immunofluorescence
4.9. siRNA Targeting LC3 Transfection
4.10. Intracellular ROS Measurement
4.11. Animal Care and Maintenance
4.12. Tumor Cell Inoculation
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Riker, A.I.; Zea, N.; Trinh, T. The epidemiology, prevention, and detection of melanoma. Ochsner J. 2010, 10, 56–65. [Google Scholar] [PubMed]
- Elwood, J.M.; Gallagher, R.P. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. Int. J. Cancer 1998, 78, 276–280. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Azoury, S.C.; Lange, J.R. Epidemiology, risk factors, prevention, and early detection of melanoma. Surg. Clin. N. Am. 2014, 94, 945–962. [Google Scholar] [CrossRef]
- Chi, Z.; Li, S.; Sheng, X.; Si, L.; Cui, C.; Han, M.; Guo, J. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: A study of 522 consecutive cases. BMC Cancer 2011, 11, 85. [Google Scholar] [CrossRef]
- Guaman-Ortiz, L.M.; Orellana, M.I.; Ratovitski, E.A. Natural compounds as modulators of non-apoptotic cell death in cancer cells. Curr. Genom. 2017, 18, 132–155. [Google Scholar] [CrossRef]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Sartorius, U.; Schmitz, I.; Krammer, P.H. Molecular mechanisms of death-receptor-mediated apoptosis. ChemBioChem 2001, 2, 20–29. [Google Scholar] [CrossRef]
- Bauer, C.; Hees, C.; Sterzik, A.; Bauernfeind, F.; Mak’Anyengo, R.; Duewell, P.; Lehr, H.A.; Noessner, E.; Wank, R.; Trauzold, A.; et al. Proapoptotic and antiapoptotic proteins of the Bcl-2 family regulate sensitivity of pancreatic cancer cells toward gemcitabine and T-cell-mediated cytotoxicity. J. Immunother. 2015, 38, 116–126. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd ed.). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, Y.; Zhong, J.; Bi, Y.; Liu, Y.; Ren, Z.; Li, X.; Jia, J.; Yu, M.; Yu, X. Pristimerin induces apoptosis and autophagy via activation of ROS/ASK1/JNK pathway in human breast cancer in vitro and in vivo. Cell Death Discov. 2019, 5, 125. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Roca-Agujetas, V.; de Dios, C.; Leston, L.; Mari, M.; Morales, A.; Colell, A. Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid. Med. Cell Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Sun, X.; Wang, W.; Chen, J.; Cai, X.; Yang, J.; Yang, Y.; Yan, H.; Cheng, X.; Ye, J.; Lu, W.; et al. The natural diterpenoid isoforretin a inhibits thioredoxin-1 and triggers potent ros-mediated antitumor effects. Cancer Res. 2017, 77, 926–936. [Google Scholar] [CrossRef]
- Li-Weber, M. New therapeutic aspects of flavones: The anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat. Rev. 2009, 35, 57–68. [Google Scholar] [CrossRef]
- Steiner, G.G. The correlation between cancer incidence and kava consumption. Hawaii Med. J. 2000, 59, 420–422. [Google Scholar]
- Abu, N.; Ho, W.Y.; Yeap, S.K.; Akhtar, M.N.; Abdullah, M.P.; Omar, A.R.; Alitheen, N.B. The flavokawains: Uprising medicinal chalcones. Cancer Cell Int. 2013, 13, 102. [Google Scholar] [CrossRef]
- Lebot, V.; Do, T.K.; Legendre, L. Detection of flavokavins (A, B, C) in cultivars of kava (Piper methysticum) using high performance thin layer chromatography (HPTLC). Food Chem. 2014, 151, 554–560. [Google Scholar] [CrossRef]
- Teschke, R. Kava hepatotoxicity. A clinical review. Ann. Hepatol. 2010, 9, 251–265. [Google Scholar] [CrossRef]
- Narayanapillai, S.C.; Leitzman, P.; O’Sullivan, M.G.; Xing, C. Flavokawains A and B in kava, not dihydromethysticin, potentiate acetaminophen-induced hepatotoxicity in C57BL/6 mice. Chem. Res. Toxicol. 2014, 27, 1871–1876. [Google Scholar] [CrossRef]
- Carreñ, O.I.; Laurenza, E.; Martelloni, A.; Salas, B.; Simõñes, B.G.; Vergona, P.R. German court repeals the withdrawal of marketing authorisation of kava-containing medicinal products. Trade Perspect. 2014, 13, 2–5. [Google Scholar]
- Ji, T.; Lin, C.; Krill, L.S.; Eskander, R.; Guo, Y.; Zi, X.; Hoang, B.H. Flavokawain B, a kava chalcone, inhibits growth of human osteosarcoma cells through G2/M cell cycle arrest and apoptosis. Mol. Cancer 2013, 12, 55. [Google Scholar] [CrossRef]
- Abu, N.; Mohamed, N.E.; Yeap, S.K.; Lim, K.L.; Akhtar, M.N.; Zulfadli, A.J.; Kee, B.B.; Abdullah, M.P.; Omar, A.R.; Alitheen, N.B. In vivo antitumor and antimetastatic effects of flavokawain B in 4T1 breast cancer cell-challenged mice. Drug Des. Dev. Ther. 2015, 9, 1401–1417. [Google Scholar] [CrossRef]
- Chang, C.T.; Hseu, Y.C.; Thiyagarajan, V.; Lin, K.Y.; Way, T.D.; Korivi, M.; Liao, J.W.; Yang, H.L. Chalcone flavokawain B induces autophagic-cell death via reactive oxygen species-mediated signaling pathways in human gastric carcinoma and suppresses tumor growth in nude mice. Arch. Toxicol. 2017, 91, 3341–3364. [Google Scholar] [CrossRef]
- Hua, R.; Pei, Y.; Gu, H.; Sun, Y.; He, Y. Antitumor effects of flavokawain-B flavonoid in gemcitabine-resistant lung cancer cells are mediated via mitochondrial-mediated apoptosis, ROS production, cell migration and cell invasion inhibition and blocking of PI3K/AKT Signaling pathway. J. Buon. 2020, 25, 262–267. [Google Scholar]
- Hseu, Y.C.; Huang, Y.C.; Thiyagarajan, V.; Mathew, D.C.; Lin, K.Y.; Chen, S.C.; Liu, J.Y.; Hsu, L.S.; Li, M.L.; Yang, H.L. Anticancer activities of chalcone flavokawain B from Alpinia pricei Hayata in human lung adenocarcinoma (A549) cells via induction of reactive oxygen species-mediated apoptotic and autophagic cell death. J. Cell Physiol. 2019, 234, 17514–17526. [Google Scholar] [CrossRef]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Lin, R.W.; Shen, Y.C.; Lin, K.Y.; Liao, J.W.; Thiyagarajan, V.; Yang, H.L. Flavokawain B and doxorubicin work synergistically to impede the propagation of gastric cancer cells via ROS-mediated apoptosis and autophagy pathways. Cancers 2020, 12, 2475. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Tanida, I.; Waguri, S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010, 648, 193–214. [Google Scholar] [CrossRef]
- Kanematsu, S.; Uehara, N.; Miki, H.; Yoshizawa, K.; Kawanaka, A.; Yuri, T.; Tsubura, A. Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells. Anticancer Res. 2010, 30, 3381–3390. [Google Scholar] [PubMed]
- Marquez, R.T.; Xu, L. Bcl-2: Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Kim, J.-H.; Chi, G.Y.; Kim, G.-Y.; Chang, Y.-C.; Moon, S.-K.; Nam, S.-W.; Kim, W.-J.; Yoo, Y.H.; Choi, Y.H. Induction of apoptosis and autophagy by sodium selenite in A549 human lung carcinoma cells through generation of reactive oxygen species. Toxicol. Lett. 2012, 212, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef]
- Leiter, U.; Garbe, C. Epidemiology of melanoma and nonmelanoma skin cancer—The role of sunlight. Adv. Exp. Med. Biol. 2008, 624, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Cho, H.J.; Gowrisankar, Y.V.; Thiyagarajan, V.; Chen, X.Z.; Lin, K.Y.; Huang, H.C.; Yang, H.L. Kalantuboside B induced apoptosis and cytoprotective autophagy in human melanoma A2058cells: An in vitro and in vivo study. Free Radic. Biol. Med. 2019, 143, 397–411. [Google Scholar] [CrossRef]
- Lin, E.; Lin, W.H.; Wang, S.Y.; Chen, C.S.; Liao, J.W.; Chang, H.W.; Chen, S.C.; Lin, K.Y.; Wang, L.; Yang, H.L.; et al. Flavokawain B inhibits growth of human squamous carcinoma cells: Involvement of apoptosis and cell cycle dysregulation in vitro and in vivo. J. Nutr. Biochem. 2012, 23, 368–378. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Oliveira, E.A.; Faiao-Flores, F.; Alicea-Rebecca, G.; Weeraratna, A.T.; Smalley, K.S.M.; Barros, S.B.M.; Maria-Engler, S.S. ER stress promotes antitumor effects in BRAFi/MEKi resistant human melanoma induced by natural compound 4-nerolidylcathecol (4-NC). Pharm. Res. 2019, 141, 63–72. [Google Scholar] [CrossRef]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef]
- Tseng, T.-H.; Chien, M.-H.; Lin, W.-L.; Wen, Y.-C.; Chow, J.-M.; Chen, C.-K.; Kuo, T.-C.; Lee, W.-J. Inhibition of MDA-MB-231 breast cancer cell proliferation and tumor growth by apigenin through induction of G2/M arrest and histone H3 acetylation-mediated p21WAF1/CIP1 expression. Environ. Toxicol. 2016, 32, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, C.; Bao, J.; Jia, X.; Liang, Y.; Wang, X.; Chen, M.; Su, H.; Li, P.; Wan, J.-B.; et al. Synergistic chemopreventive effects of curcumin and berberine on human breast cancer cells through induction of apoptosis and autophagic cell death. Sci. Rep. 2016, 6, 26064. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Lee, M.S.; Wu, C.R.; Cho, H.J.; Lin, K.Y.; Lai, G.H.; Wang, S.Y.; Kuo, Y.H.; Kumar, K.J.; Yang, H.L. The chalcone flavokawain B induces G2/M cell-cycle arrest and apoptosis in human oral carcinoma HSC-3 cells through the intracellular ROS generation and downregulation of the Akt/p38 MAPK signaling pathway. J. Agric. Food Chem. 2012, 60, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Hultqvist, M.; Olsson, L.M.; Gelderman, K.A.; Holmdahl, R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009, 30, 201–208. [Google Scholar] [CrossRef]
- Nakazato, T.; Ito, K.; Ikeda, Y.; Kizaki, M. Green tea component, catechin, induces apoptosis of human malignant B cells via production of reactive oxygen species. Clin. Cancer Res. 2005, 11, 6040–6049. [Google Scholar] [CrossRef]
- Erlank, H.; Elmann, A.; Kohen, R.; Kanner, J. Polyphenols activate Nrf2 in astrocytes via H2O2, semiquinones and quinones. Free Radic. Biol. Med. 2011, 51, 2319–2327. [Google Scholar] [CrossRef]
- Mai, T.T.; Moon, J.; Song, Y.; Viet, P.Q.; Van Phuc, P.; Lee, J.M.; Yi, T.-H.; Cho, M.; Cho, S.K. Ginsenoside F2 induces apoptosis accompanied by protective autophagy in breast cancer stem cells. Cancer Lett. 2012, 321, 144–153. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef] [PubMed]
- Gowrisankar, Y.V.; Clark, M.A. Regulation of angiotensinogen expression by angiotensin II in spontaneously hypertensive rat primary astrocyte cultures. Brain Res. 2016, 1643, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef]
- Kuma, A.; Matsui, M.; Mizushima, N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: Caution in the interpretation of LC3 localization. Autophagy 2007, 3, 323–328. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Ho, Y.G.; Mathew, D.C.; Yen, H.R.; Chen, X.Z.; Yang, H.L. The in vitro and in vivo depigmenting activity of Coenzyme Q10 through the down-regulation of alpha-MSH signaling pathways and induction of Nrf2/ARE-mediated antioxidant genes in UVA-irradiated skin keratinocytes. Biochem. Pharm. 2019, 164, 299–310. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hseu, Y.-C.; Chiang, Y.-C.; Vudhya Gowrisankar, Y.; Lin, K.-Y.; Huang, S.-T.; Shrestha, S.; Chang, G.-R.; Yang, H.-L. The In Vitro and In Vivo Anticancer Properties of Chalcone Flavokawain B through Induction of ROS-Mediated Apoptotic and Autophagic Cell Death in Human Melanoma Cells. Cancers 2020, 12, 2936. https://doi.org/10.3390/cancers12102936
Hseu Y-C, Chiang Y-C, Vudhya Gowrisankar Y, Lin K-Y, Huang S-T, Shrestha S, Chang G-R, Yang H-L. The In Vitro and In Vivo Anticancer Properties of Chalcone Flavokawain B through Induction of ROS-Mediated Apoptotic and Autophagic Cell Death in Human Melanoma Cells. Cancers. 2020; 12(10):2936. https://doi.org/10.3390/cancers12102936
Chicago/Turabian StyleHseu, You-Cheng, Yu-Chi Chiang, Yugandhar Vudhya Gowrisankar, Kai-Yuan Lin, Sheng-Teng Huang, Sirjana Shrestha, Geng-Ruei Chang, and Hsin-Ling Yang. 2020. "The In Vitro and In Vivo Anticancer Properties of Chalcone Flavokawain B through Induction of ROS-Mediated Apoptotic and Autophagic Cell Death in Human Melanoma Cells" Cancers 12, no. 10: 2936. https://doi.org/10.3390/cancers12102936
APA StyleHseu, Y.-C., Chiang, Y.-C., Vudhya Gowrisankar, Y., Lin, K.-Y., Huang, S.-T., Shrestha, S., Chang, G.-R., & Yang, H.-L. (2020). The In Vitro and In Vivo Anticancer Properties of Chalcone Flavokawain B through Induction of ROS-Mediated Apoptotic and Autophagic Cell Death in Human Melanoma Cells. Cancers, 12(10), 2936. https://doi.org/10.3390/cancers12102936