Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases



Simple Summary
Abstract
1. Introduction
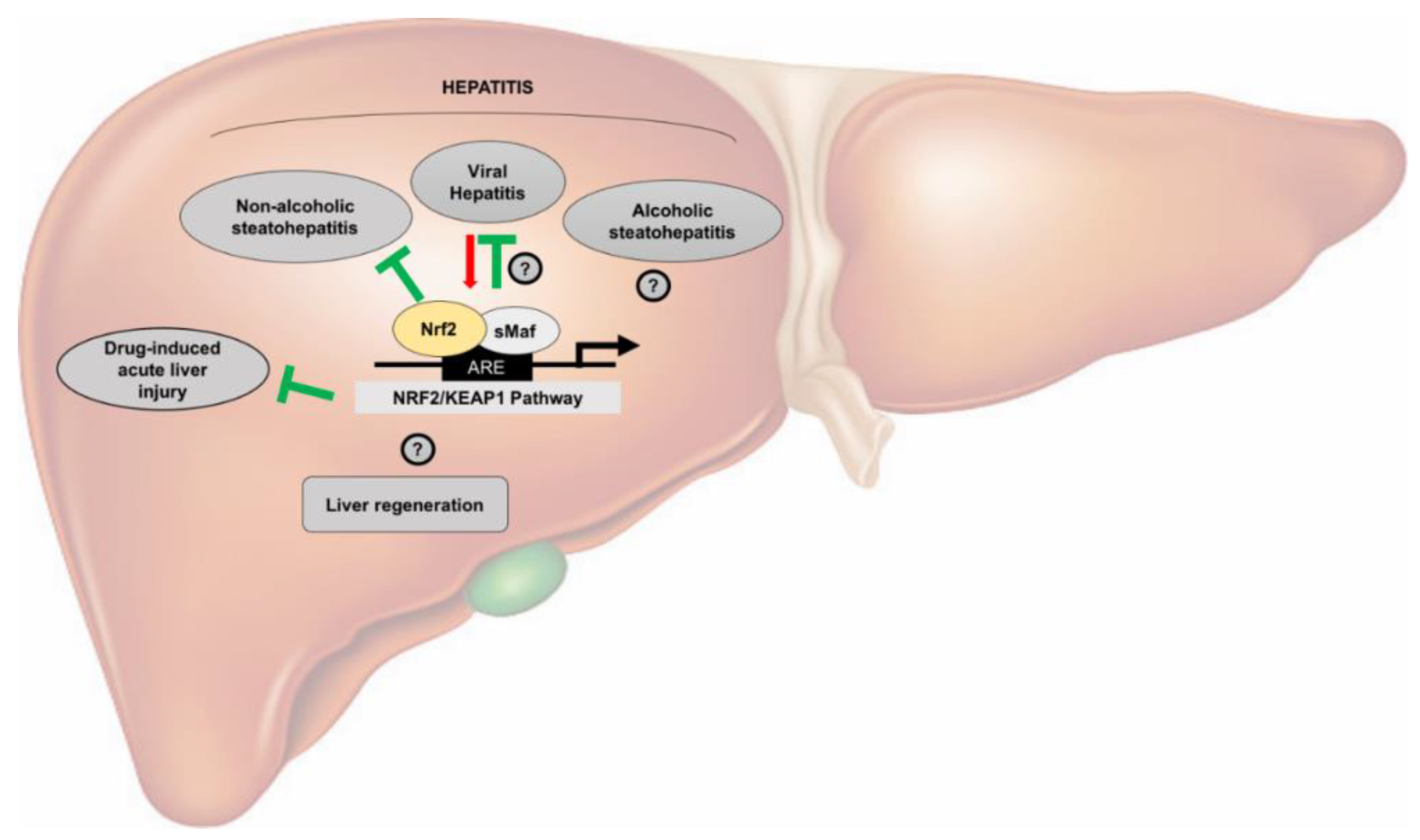
2. Nrf2 in Hepatic Regeneration
3. Nrf2 in Acute Hepatotoxicity
4. Nrf2 in Chronic Liver Injury
4.1. Nrf2 in Viral Hepatitis
4.2. Nrf2 in Alcoholic Fatty Liver Disease (AFLD)
4.3. Nrf2 in Non-Alcoholic Fatty Liver Disease (NAFLD)
5. Nrf2 and Hepatocellular Carcinoma (HCC)
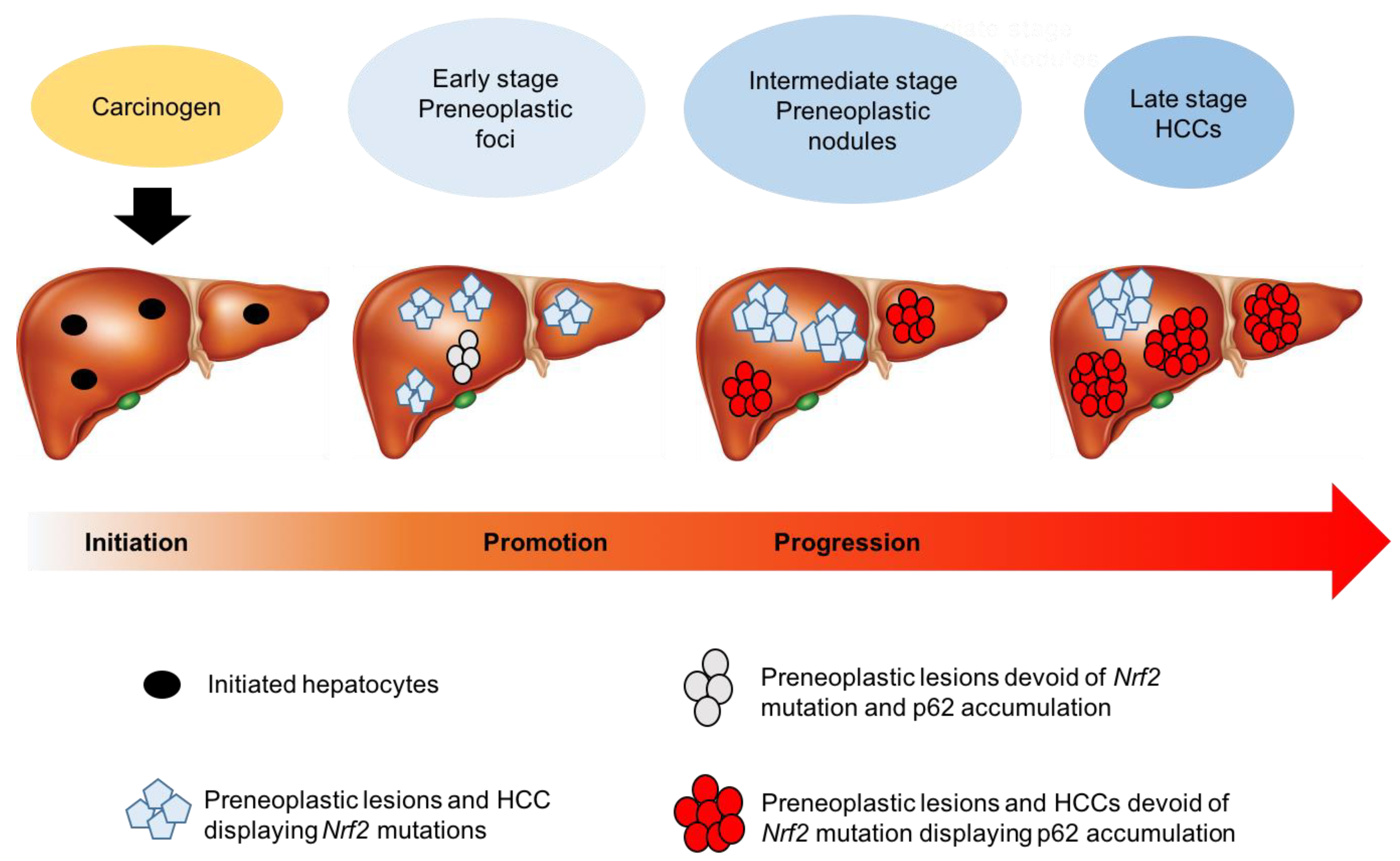
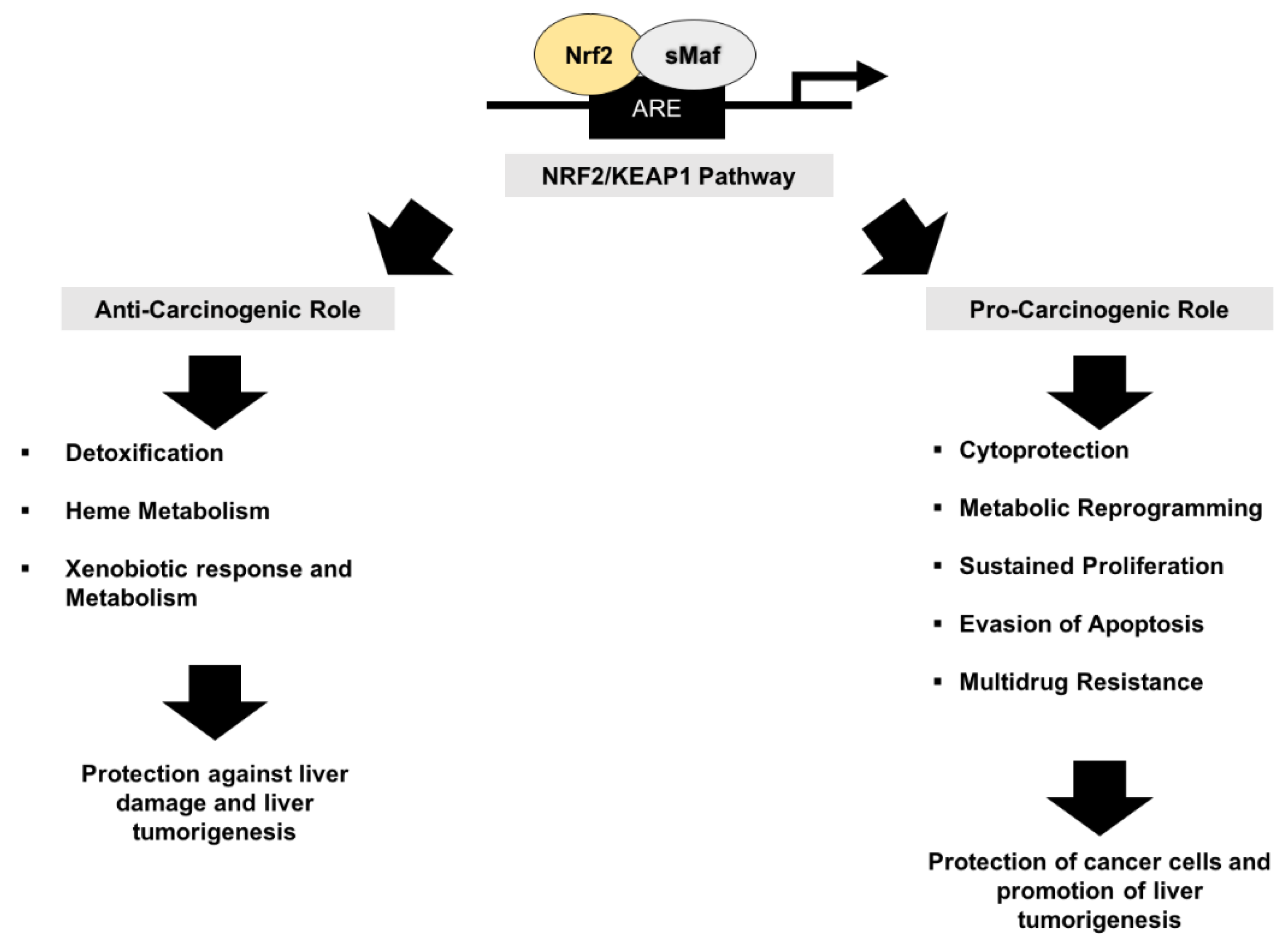
5.1. Nrf2 and Cancer Prevention
5.2. The Other Face of Nrf2
6. Mechanisms of Nrf2 Activation in HCC
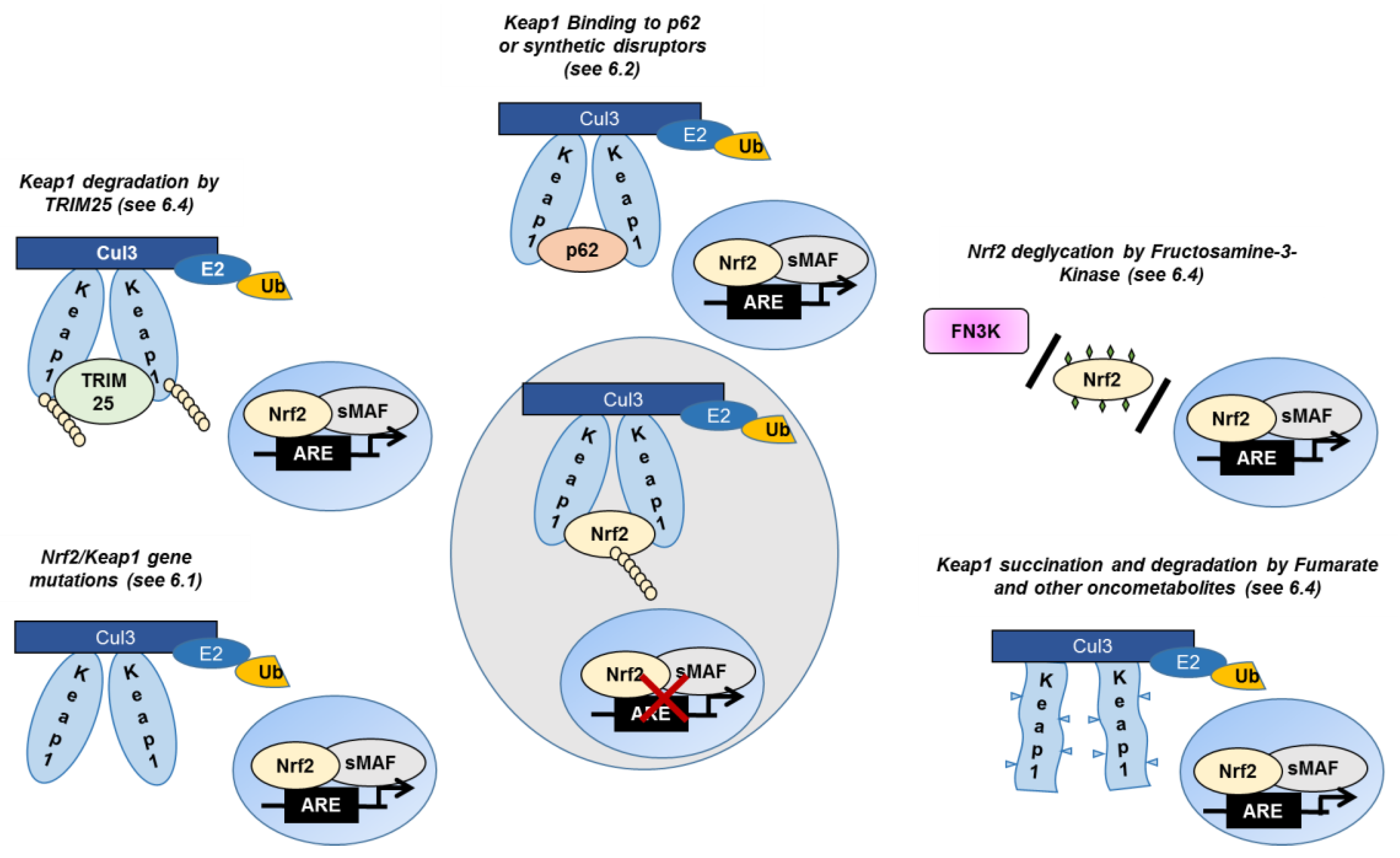
6.1. Somatic Mutations in Keap1 and Nrf2 Genes
6.2. The Crosstalk between Nrf2 and p62
6.3. Nrf2 Regulation by MicroRNAs
6.4. Posttranslational Keap1/Nrf2 Modulation
7. Nrf2 as a Mediator of Resistance
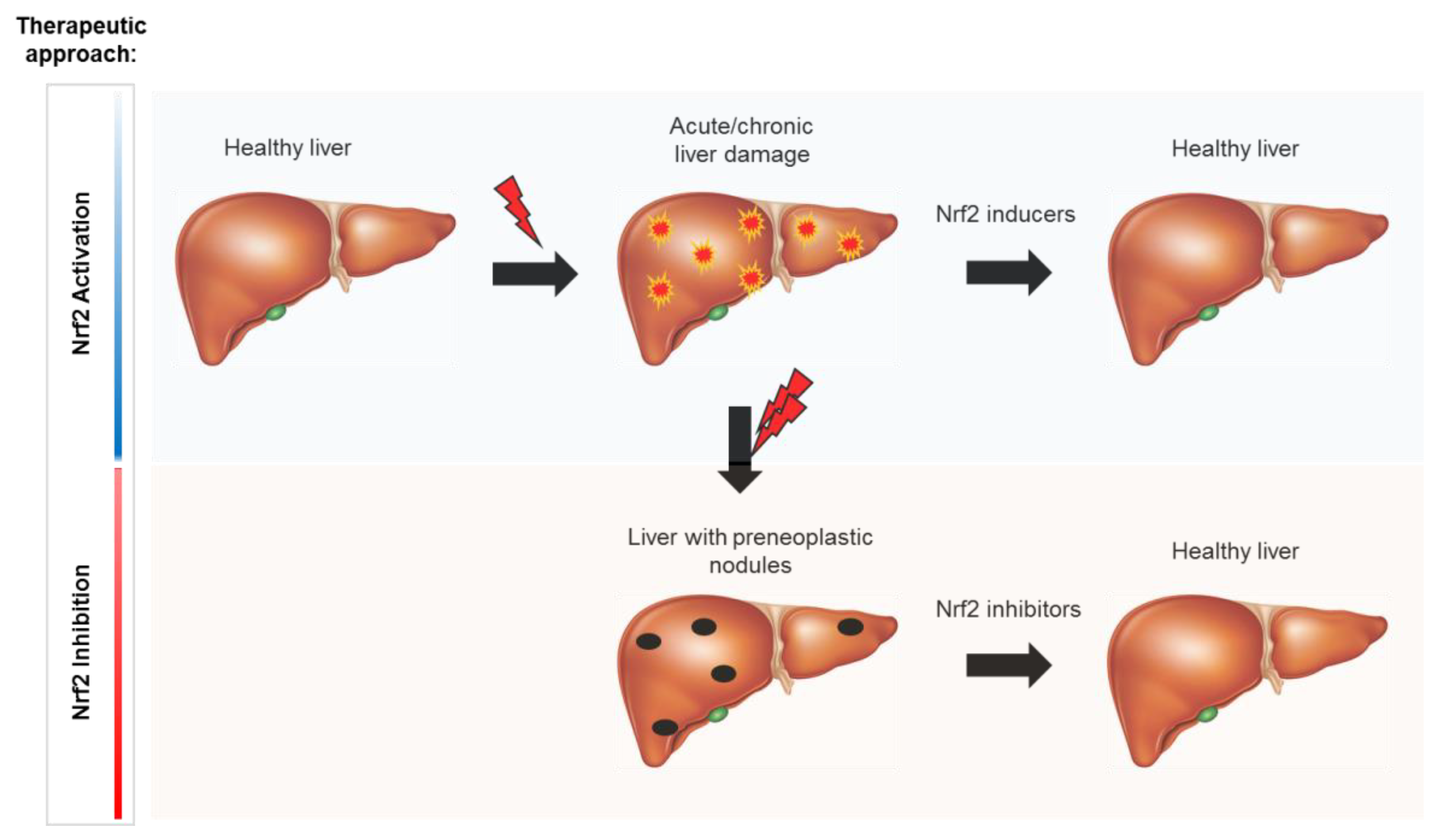
8. Nrf2 as a Molecular Target
8.1. Nrf2 Activators
8.2. Nrf2 Inhibitors
8.3. Additional Strategies to Inhibit the Keap1/Nrf2 Pathway
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Kensler, T.W. Nrf2 in liver toxicology. Arch. Pharm. Res. 2020, 43, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Clarner, T.; Fragoulis, A.; Kan, Y.W.; Pufe, T.; Streetz, K.; Wruck, C.J. Nrf2 in health and disease: Current and future clinical implications. Clin. Sci. 2015, 129, 989–999. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Karp, S.J. Clinical implications of advances in the basic science of liver repair and regeneration. Am. J. Transplant. 2009, 9, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Bucher, N.L.R.; Swaffield, M.N.; DiTroia, J.F. The influence of age upon the incorporation of thymidine-1-C14 into the DNA of regenerating rat liver. Cancer Res. 1964, 24, 509–512. [Google Scholar]
- Wang, X.; Quail, E.; Hung, N.J.; Tan, Y.; Ye, H.; Costa, R.H. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevent age-related proliferation defects in regenerating liver. Proc. Natl. Acad. Sci. USA 2001, 98, 11468–11473. [Google Scholar] [CrossRef]
- Higgins, G.M.A. Experimental pathology of the liver. Restoration of the liver of the white rat following partial surgical removal. Arch. Pathol. 1931, 12, 186–202. [Google Scholar]
- Michalopoulos, G.K. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am. J. Pathol. 2010, 176, 2–13. [Google Scholar] [CrossRef]
- Beyer, T.A.; Xu, W.; Teupser, D.; Auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef]
- Kohler, U.A.; Kurinna, S.; Schwitter, D.; Marti, A.; Schafer, M.; Hellerbrand, C.; Speicher, T.; Werner, S. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology 2014, 60, 670–678. [Google Scholar] [CrossRef]
- Hu, M.; Zou, Y.; Nambiar, S.M.; Lee, J.; Yang, Y.; Dai, G. Keap1 modulates the redox cycle and hepatocyte cell cycle in regenerating liver. Cell Cycle 2014, 13, 2349–2358. [Google Scholar] [CrossRef]
- Zou, Y.; Hu, M.; Lee, J.; Nambiar, S.M.; Garcia, V.; Bao, Q.; Chan, J.Y.; Dai, G. Nrf2 is essential for timely M phase entry of replicating hepatocytes during liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G262–G268. [Google Scholar] [CrossRef]
- Skoko, J.J.; Wakabayashi, N.; Noda, K.; Kimura, S.; Tobita, K.; Shigemura, N.; Tsujita, T.; Yamamoto, M.; Kensler, T.W. Loss of Nrf2 in mice evokes a congenital intrahepatic shunt that alters hepatic oxygen and protein expression gradients and toxicity. Toxicol. Sci. 2014, 141, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Koyama, S.; Tsukada, K.; Hatakeyama, K. Acute portal hypertension reflecting shear stress as a trigger of liver regeneration following partial hepatectomy. Surg. Today 1997, 27, 518–526. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.W.; Columbano, A.; Ledda-Columbano, G.M.; Sarma, D.S. Lead nitrate induces certain biochemical properties characteristic of hepatocyte nodules. Carcinogenesis 1986, 7, 1643–1646. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef] [PubMed]
- Orrù, C.; Szydlowska, M.; Taguchi, K.; Zavattari, P.; Perra, A.; Yamamoto, M.; Columbano, A. Genetic inactivation of Nrf2 prevents clonal expansion of initiated cells in a nutritional model of rat hepatocarcinogenesis. J. Hepatol. 2018, 69, 635–643. [Google Scholar] [CrossRef]
- Enomoto, A.; Itoh, K.; Nagayoshi, E.; Haruta, J.; Kimura, T.; O’Connor, T.; Harada, T.; Yamamoto, M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 2001, 59, 169–177. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, Y.; Su, Z.; Kou, R.; Xie, K.; Song, F. Activation of p62-keap1-Nrf2 antioxidant pathway in the early stage of acetaminophen-induced acute liver injury in mice. Chem. Biol. Interact. 2018, 282, 22–28. [Google Scholar] [CrossRef]
- Noh, J.R.; Kim, Y.H.; Hwang, J.H.; Choi, D.H.; Kim, K.S.; Oh, W.K.; Lee, C.H. Sulforaphane protects against acetaminophen-induced hepatotoxicity. Food Chem. Toxicol. 2015, 80, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Wang, H.; Li, L.; Zhu, J.; Chen, F.; Chen, Y.; Liu, C.; Fu, J.; Yang, B.; Zhang, Q.; et al. Hepatocyte-specific deficiency of Nrf2 exacerbates carbon tetrachloride-induced liver fibrosis via aggravated hepatocyte injury and subsequent inflammatory and fibrogenic responses. Free Radic. Biol. Med. 2020, 150, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Liu, J.J.; Klaassen, C.D. Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol. Appl. Pharmacol. 2012, 263, 14–20. [Google Scholar] [CrossRef]
- Qu, Q.; Liu, J.; Zhou, H.H.; Klaassen, C.D. Nrf2 protects against furosemide-induced hepatotoxicity. Toxicology 2014, 324, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.F.; Liu, J.; Wu, K.C.; Qu, Q.; Fan, F.; Klaassen, C.D. Overexpression of Nrf2 protects against microcystin-induced hepatotoxicity in mice. PLoS ONE 2014, 9, e93013. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Li, S.; Bai, J.; Zhang, Q.; Han, Z. alpha-Lipoic acid protects against microcystin-LR induced hepatotoxicity through regeneration of glutathione via activation of Nrf2. Environ. Toxicol. 2020, 35, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Takaku, M.; Egner, P.A.; Morita, M.; Kaneko, T.; Mashimo, T.; Kensler, T.W.; Yamamoto, M. Generation of a New Model Rat: Nrf2 Knockout Rats Are Sensitive to Aflatoxin B1 Toxicity. Toxicol. Sci. 2016, 152, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Kwak, M.K.; Egner, P.A.; Groopman, J.D.; Bodreddigari, S.; Sutter, T.R.; Baumgartner, K.J.; Roebuck, B.D.; Liby, K.T.; Yore, M.M.; et al. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006, 66, 2488–2494. [Google Scholar] [CrossRef]
- Yang, D.; Tan, X.; Lv, Z.; Liu, B.; Baiyun, R.; Lu, J.; Zhang, Z. Regulation of Sirt1/Nrf2/TNF-alpha signaling pathway by luteolin is critical to attenuate acute mercuric chloride exposure induced hepatotoxicity. Sci. Rep. 2016, 6, 37157. [Google Scholar] [CrossRef]
- Muhammad, I.; Wang, X.; Li, S.; Li, R.; Zhang, X. Curcumin confers hepatoprotection against AFB1-induced toxicity via activating autophagy and ameliorating inflammation involving Nrf2/HO-1 signaling pathway. Mol. Biol. Rep. 2018, 45, 1775–1785. [Google Scholar] [CrossRef]
- Fan, X.; Lv, H.; Wang, L.; Deng, X.; Ci, X. Isoorientin Ameliorates APAP-Induced Hepatotoxicity via Activation Nrf2 Antioxidative Pathway: The Involvement of AMPK/Akt/GSK3beta. Front. Pharmacol. 2018, 9, 1334. [Google Scholar] [CrossRef]
- Aladaileh, S.H.; Abukhalil, M.H.; Saghir, S.A.M.; Hanieh, H.; Alfwuaires, M.A.; Almaiman, A.A.; Bin-Jumah, M.; Mahmoud, A.M. Galangin Activates Nrf2 Signaling and Attenuates Oxidative Damage, Inflammation, and Apoptosis in a Rat Model of Cyclophosphamide-Induced Hepatotoxicity. Biomolecules 2019, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Nyamagoud, S.B.; SreeHarsha, N.; Mishra, A.; Gubbiyappa, S.K.; Singh, Y. Sitagliptin protects liver against aflatoxin B1-induced hepatotoxicity through upregulating Nrf2/ARE/HO-1 pathway. Biofactors 2020, 46, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lu, Z.; Guo, W.; Tumen, B.; He, Y.; Chen, C.; Hu, S.; Xu, K.; Wang, Y.; et al. Cadmium Induces Acute Liver Injury by Inhibiting Nrf2 and the Role of NF-kappaB, NLRP3, and MAPKs Signaling Pathway. Int. J. Environ. Res. Public Health 2019, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Takai, A.; Inuzuka, T.; Marusawa, H. Genetic basis of hepatitis virus-associated hepatocellular carcinoma: Linkage between infection, inflammation, and tumorigenesis. J. Gastroenterol. 2017, 52, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Eng, F.J.; Branch, A.D. HCV and HCC: Clinical update and a review of HCC-associated viral mutations in the core gene. Semin. Liver Dis. 2011, 31, 347–355. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Sugiyama, K.; Ebinuma, H.; Nakamoto, N.; Sakasegawa, N.; Murakami, Y.; Chu, P.S.; Usui, S.; Ishibashi, Y.; Wakayama, Y.; Taniki, N.; et al. Prominent steatosis with hypermetabolism of the cell line permissive for years of infection with hepatitis C virus. PLoS ONE 2014, 9, e94460. [Google Scholar] [CrossRef]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Wang, S.Y.; Chiu, C.C.; Tseng, C.K.; Lin, C.K.; Wang, H.C.; Lee, J.C. Lucidone suppresses hepatitis C virus replication by Nrf2-mediated heme oxygenase-1 induction. Antimicrob. Agents Chemother. 2013, 57, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.K.; Hsu, S.P.; Lin, C.K.; Wu, Y.H.; Lee, J.C.; Young, K.C. Celastrol inhibits hepatitis C virus replication by upregulating heme oxygenase-1 via the JNK MAPK/Nrf2 pathway in human hepatoma cells. Antiviral. Res. 2017, 146, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Chen, W.C.; Tseng, C.K.; Lin, C.K.; Hsu, Y.C.; Chen, Y.H.; Lee, J.C. Sulforaphane Suppresses Hepatitis C Virus Replication by Up-Regulating Heme Oxygenase-1 Expression through PI3K/Nrf2 Pathway. PLoS ONE 2016, 11, e0152236. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.; Hildt, E. Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. Int. J. Mol. Sci. 2019, 20, 4659. [Google Scholar] [CrossRef] [PubMed]
- Livero, F.A.; Acco, A. Molecular basis of alcoholic fatty liver disease: From incidence to treatment. Hepatol. Res. 2016, 46, 111–123. [Google Scholar] [CrossRef]
- Sun, J.; Fu, J.; Li, L.; Chen, C.; Wang, H.; Hou, Y.; Xu, Y.; Pi, J. Nrf2 in alcoholic liver disease. Toxicol. Appl. Pharmacol. 2018, 357, 62–69. [Google Scholar] [CrossRef]
- Lamlé, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.J.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar] [CrossRef]
- Sun, J.; Fu, J.; Zhong, Y.; Li, L.; Chen, C.; Wang, X.; Wang, L.; Hou, Y.; Wang, H.; Zhao, R.; et al. NRF2 mitigates acute alcohol-induced hepatic and pancreatic injury in mice. Food Chem. Toxicol. 2018, 121, 495–503. [Google Scholar] [CrossRef]
- Liu, J.; Wu, K.C.; Lu, Y.F.; Ekuase, E.; Klaassen, C.D. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid. Med. Cell. Longev. 2013, 2013, 305861. [Google Scholar] [CrossRef]
- Dong, J.; Sulik, K.K.; Chen, S.Y. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxid. Redox Signal. 2008, 10, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Lin, J.; Wu, D. Sulforaphane induces Nrf2 and protects against CYP2E1-dependent binge alcohol-induced liver steatosis. Biochim. Biophys. Acta 2014, 1840, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dou, X.; Li, S.; Zhang, X.; Sun, X.; Zhou, Z.; Song, Z. Nuclear factor (erythroid-derived 2)-like 2 activation-induced hepatic very-low-density lipoprotein receptor overexpression in response to oxidative stress contributes to alcoholic liver disease in mice. Hepatology 2014, 59, 1381–1392. [Google Scholar] [CrossRef]
- Shen, F.; Wang, Z.; Liu, W.; Liang, Y. Ethyl pyruvate can alleviate alcoholic liver disease through inhibiting Nrf2 signaling pathway. Exp. Ther. Med. 2018, 15, 4223–4228. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.H.; Huo, L.J.; Li, T.T. Antioxidant axis Nrf2-keap1-ARE in inhibition of alcoholic liver fibrosis by IL-22. World J. Gastroenterol. 2017, 23, 2002–2011. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Chambel, S.S.; Santos-Goncalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. BioMed Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Zhang, D.D. NRF2 Induction for NASH Treatment: A New Hope Rises. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 422–423. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cui, Y.; Li, C.; Zhang, Y.; Xu, S.; Li, X.; Li, H.; Zhang, X. Nrf2 deletion causes “benign” simple steatosis to develop into nonalcoholic steatohepatitis in mice fed a high-fat diet. Lipids Health Dis. 2013, 12, 165. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Lombardi, B.; Shinozuka, H. Progression of carcinogen-induced foci of gamma-glutamyltranspeptidase-positive hepatocytes to hepatomas in rats fed a choline-deficient diet. Int. J. Cancer 1982, 29, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Choline deficiency. J. Nutr. Biochem. 1990, 1, 332–349. [Google Scholar] [CrossRef]
- Sugimoto, H.; Okada, K.; Shoda, J.; Warabi, E.; Ishige, K.; Ueda, T.; Taguchi, K.; Yanagawa, T.; Nakahara, A.; Hyodo, I.; et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G283–G294. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Ramadori, P.; Drescher, H.; Erschfeld, S.; Fragoulis, A.; Kensler, T.W.; Wruck, C.J.; Cubero, F.J.; Trautwein, C.; Streetz, K.L.; Kroy, D.C. Genetic Nrf2 Overactivation Inhibits the Deleterious Effects Induced by Hepatocyte-Specific c-met Deletion during the Progression of NASH. Oxid. Med. Cell. Longev. 2017, 2017, 3420286. [Google Scholar] [CrossRef] [PubMed]
- Kroy, D.C.; Schumacher, F.; Ramadori, P.; Hatting, M.; Bergheim, I.; Gassler, N.; Boekschoten, M.V.; Muller, M.; Streetz, K.L.; Trautwein, C. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J. Hepatol. 2014, 61, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Gan, L.T.; Yeh, M.M.; Haigh, W.G.; Larter, C.Z.; Ioannou, G.; Teoh, N.C.; Farrell, G.C. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J. Hepatol. 2013, 59, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Han, D.H.; Nam, K.T.; Park, J.S.; Kim, S.H.; Lee, M.; Kim, G.; Min, B.S.; Cha, B.S.; Lee, Y.S.; et al. Ezetimibe, an NPC1L1 inhibitor, is a potent Nrf2 activator that protects mice from diet-induced nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2016, 99, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef]
- Haque, E.; Karim, M.R.; Salam Teeli, A.; Smiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular Mechanisms Underlying Hepatocellular Carcinoma Induction by Aberrant NRF2 Activation-Mediated Transcription Networks: Interaction of NRF2-KEAP1 Controls the Fate of Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef]
- Long, D.J., 2nd; Waikel, R.L.; Wang, X.J.; Perlaky, L.; Roop, D.R.; Jaiswal, A.K. NAD(P)H:quinone oxidoreductase 1 deficiency increases susceptibility to benzo(a)pyrene-induced mouse skin carcinogenesis. Cancer Res. 2000, 60, 5913–5915. [Google Scholar]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Ansher, S.S.; Dolan, P.; Bueding, E. Biochemical effects of dithiolthiones. Food Chem. Toxicol. 1986, 24, 405–415. [Google Scholar] [CrossRef]
- Yates, M.S.; Kensler, T.W. Keap1 eye on the target: Chemoprevention of liver cancer. Acta Pharmacol. Sin. 2007, 28, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef]
- Kitamura, Y.; Umemura, T.; Kanki, K.; Kodama, Y.; Kitamoto, S.; Saito, K.; Itoh, K.; Yamamoto, M.; Masegi, T.; Nishikawa, A.; et al. Increased susceptibility to hepatocarcinogenicity of Nrf2-deficient mice exposed to 2-amino-3-methylimidazo[4,5-f]quinoline. Cancer Sci. 2007, 98, 19–24. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef]
- Kensler, T.W.; Egner, P.A.; Agyeman, A.S.; Visvanathan, K.; Groopman, J.D.; Chen, J.G.; Chen, T.Y.; Fahey, J.W.; Talalay, P. Keap1-nrf2 signaling: A target for cancer prevention by sulforaphane. Top Curr. Chem. 2013, 329, 163–177. [Google Scholar] [CrossRef]
- Liu, P.; Wang, W.; Zhou, Z.; Smith, A.J.O.; Bowater, R.P.; Wormstone, I.M.; Chen, Y.; Bao, Y. Chemopreventive Activities of Sulforaphane and Its Metabolites in Human Hepatoma HepG2 Cells. Nutrients 2018, 10, 585. [Google Scholar] [CrossRef]
- Kensler, T.W.; Egner, P.A.; Dolan, P.M.; Groopman, J.D.; Roebuck, B.D. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res. 1987, 47, 4271–4277. [Google Scholar]
- Roebuck, B.D.; Liu, Y.L.; Rogers, A.E.; Groopman, J.D.; Kensler, T.W. Protection against aflatoxin B1-induced hepatocarcinogenesis in F344 rats by 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione (oltipraz): Predictive role for short-term molecular dosimetry. Cancer Res. 1991, 51, 5501–5506. [Google Scholar] [PubMed]
- Wang, J.S.; Shen, X.; He, X.; Zhu, Y.R.; Zhang, B.C.; Wang, J.B.; Qian, G.S.; Kuang, S.Y.; Zarba, A.; Egner, P.A.; et al. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl. Cancer Inst. 1999, 91, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Schor, N.A. The use of the D-T diaphorase for the detection of foci of early neoplastic transformation in rat liver. Cancer Lett. 1978, 5, 167–171. [Google Scholar] [CrossRef]
- Bannasch, P.; Mayer, D.; Hacker, H.J. Hepatocellular glycogenosis and hepatocarcinogenesis. Biochim. Biophys. Acta 1980, 605, 217–245. [Google Scholar] [CrossRef]
- Pickett, C.B.; Williams, J.B.; Lu, A.Y.; Cameron, R.G. Regulation of glutathione transferase and DT-diaphorase mRNAs in persistent hepatocyte nodules during chemical hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1984, 81, 5091–5095. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Nishi, S.; Sakai, M. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem. J. 2004, 380, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, A.; Satoh, K.; Nishimura, K.; Ishikawa, T.; Ruike, K.; Sato, K.; Tsuda, H.; Ito, N. Changes in molecular forms of rat hepatic glutathione S-transferase during chemical hepatocarcinogenesis. Cancer Res. 1984, 44, 2698–2703. [Google Scholar]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Lee, D.; Xu, I.M.; Chiu, D.K.; Lai, R.K.; Tse, A.P.; Lan Li, L.; Law, C.T.; Tsang, F.H.; Wei, L.L.; Chan, C.Y.; et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J. Clin. Investig. 2017, 127, 1856–1872. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Sugimoto, M.; Arias, E.; Kasashima, H.; Cordes, T.; Linares, J.F.; Duran, A.; Nakanishi, Y.; Nakanishi, N.; L’Hermitte, A.; et al. PKClambda/iota Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 2020, 38, 247–262. e11. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 1977, 88, 595–618. [Google Scholar] [PubMed]
- Andersen, J.B.; Loi, R.; Perra, A.; Factor, V.M.; Ledda-Columbano, G.M.; Columbano, A.; Thorgeirsson, S.S. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology 2010, 51, 1401–1409. [Google Scholar] [CrossRef]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not beta-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef]
- Orrù, C.; Perra, A.; Kowalik, M.A.; Rizzolio, S.; Puliga, E.; Cabras, L.; Giordano, S.; Columbano, A. Distinct Mechanisms Are Responsible for Nrf2-Keap1 Pathway Activation at Different Stages of Rat Hepatocarcinogenesis. Cancers 2020, 12, 2305. [Google Scholar] [CrossRef]
- Angioni, M.M.; Bellofatto, K.; Merlin, S.; Menegon, S.; Perra, A.; Petrelli, A.; Sulas, P.; Giordano, S.; Columbano, A.; Follenzi, A. A long term, non-tumorigenic rat hepatocyte cell line and its malignant counterpart, as tools to study hepatocarcinogenesis. Oncotarget 2017, 8, 15716–15731. [Google Scholar] [CrossRef]
- Taguchi, K.; Maher, J.M.; Suzuki, T.; Kawatani, Y.; Motohashi, H.; Yamamoto, M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 2010, 30, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef]
- Shimokawa, M.; Yoshizumi, T.; Itoh, S.; Iseda, N.; Sakata, K.; Yugawa, K.; Toshima, T.; Harada, N.; Ikegami, T.; Mori, M. Modulation of Nqo1 activity intercepts anoikis resistance and reduces metastatic potential of hepatocellular carcinoma. Cancer Sci. 2020, 111, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, Y.; Ji, T.; Ma, R.; Chen, M.; Li, G.; Li, F.; Ding, Q.; Kang, Q.; Huang, D.; et al. Clinical implication of Keap1 and phosphorylated Nrf2 expression in hepatocellular carcinoma. Cancer Med. 2016, 5, 2678–2687. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef]
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; de Stanchina, E.; et al. The Oncogenic Action of NRF2 Depends on De-glycation by Fructosamine-3-Kinase. Cell 2019, 178, 807–819. e21. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Perra, A.; Ledda-Columbano, G.M.; Ippolito, G.; Piacentini, M.; Columbano, A.; Falasca, L. Induction of autophagy promotes the growth of early preneoplastic rat liver nodules. Oncotarget 2016, 7, 5788–5799. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Shimizu, T.; Inoue, K.; Hachiya, H.; Shibuya, N.; Aoki, T.; Kubota, K. Accumulation of phosphorylated p62 is associated with NF-E2-related factor 2 activation in hepatocellular carcinoma. J. Hepato Biliary Pancreat. Sci. 2016, 23, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Ohe, T.; Takahashi, K.; Imamura, R.; Kojima, H.; Okabe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Nagano, T.; et al. Inhibitors of the protein-protein interaction between phosphorylated p62 and Keap1 attenuate chemoresistance in a human hepatocellular carcinoma cell line. Free Radic. Res. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-Mediated Autophagy in the Liver: Good or Bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. BioMed Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Aydin, Y.; Stephens, C.M.; Chava, S.; Heidari, Z.; Panigrahi, R.; Williams, D.D.; Wiltz, K.; Bell, A.; Wilson, W.; Reiss, K.; et al. Chaperone-Mediated Autophagy Promotes Beclin1 Degradation in Persistently Infected Hepatitis C Virus Cell Culture. Am. J. Pathol. 2018, 188, 2339–2355. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Chen, X.; Yu, F.; Zheng, F.; Lin, X. MiR-141 Activates Nrf2-Dependent Antioxidant Pathway via Down-Regulating the Expression of Keap1 Conferring the Resistance of Hepatocellular Carcinoma Cells to 5-Fluorouracil. Cell. Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef]
- Zhou, S.; Ye, W.; Zhang, Y.; Yu, D.; Shao, Q.; Liang, J.; Zhang, M. miR-144 reverses chemoresistance of hepatocellular carcinoma cell lines by targeting Nrf2-dependent antioxidant pathway. Am. J. Transl. Res. 2016, 8, 2992–3002. [Google Scholar] [PubMed]
- Shi, L.; Chen, Z.G.; Wu, L.L.; Zheng, J.J.; Yang, J.R.; Chen, X.F.; Chen, Z.Q.; Liu, C.L.; Chi, S.Y.; Zheng, J.Y.; et al. miR-340 reverses cisplatin resistance of hepatocellular carcinoma cell lines by targeting Nrf2-dependent antioxidant pathway. Asian Pac. J. Cancer Prev. 2014, 15, 10439–10444. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.; Grishin, N.V.; Brugarolas, J. Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell 2011, 20, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, J.; Deng, H.; Wang, Y.; Lei, C.; Wang, Q.; Xiang, J.; Liang, L.; Xia, J.; Pan, X.; et al. GSTZ1-1 Deficiency Activates NRF2/IGF1R Axis in HCC via Accumulation of Oncometabolite Succinylacetone. EMBO J. 2019, 38, e101964. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xu, J.; Zheng, Q.; He, J.; Zhou, W.; Wang, K.; Huang, X.; Fan, Q.; Ma, J.; Cheng, J.; et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett. 2019, 466, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Lu, Z.N.; Guo, X.L. Regulation and role of nuclear factor-E2-related factor 2 (Nrf2) in multidrug resistance of hepatocellular carcinoma. Chem. Biol. Interact. 2018, 280, 70–76. [Google Scholar] [CrossRef]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef]
- Zhou, S.; Ye, W.; Duan, X.; Zhang, M.; Wang, J. The noncytotoxic dose of sorafenib sensitizes Bel-7402/5-FU cells to 5-FU by down-regulating 5-FU-induced Nrf2 expression. Dig. Dis. Sci. 2013, 58, 1615–1626. [Google Scholar] [CrossRef]
- Chen, F.; Wang, H.; Zhu, J.; Zhao, R.; Xue, P.; Zhang, Q.; Bud Nelson, M.; Qu, W.; Feng, B.; Pi, J. Camptothecin suppresses NRF2-ARE activity and sensitises hepatocellular carcinoma cells to anticancer drugs. Br. J. Cancer 2017, 117, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.W.; Lau, E.Y.T.; Leung, C.O.N.; Lei, M.M.L.; Mok, E.H.K.; Ma, V.W.S.; Cho, W.C.S.; Ng, I.O.L.; Yun, J.P.; Cai, S.H.; et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepatocellular carcinoma. Cancer Lett. 2020, 476, 48–56. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Maher, J.M.; Cheng, X.; Slitt, A.L.; Dieter, M.Z.; Klaassen, C.D. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab. Dispos. 2005, 33, 956–962. [Google Scholar] [CrossRef]
- Maher, J.M.; Dieter, M.Z.; Aleksunes, L.M.; Slitt, A.L.; Guo, G.; Tanaka, Y.; Scheffer, G.L.; Chan, J.Y.; Manautou, J.E.; Chen, Y.; et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology 2007, 46, 1597–1610. [Google Scholar] [CrossRef]
- Kwak, M.K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef]
- Hoxtermann, S.; Nuchel, C.; Altmeyer, P. Fumaric acid esters suppress peripheral CD4- and CD8-positive lymphocytes in psoriasis. Dermatology 1998, 196, 223–230. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rezk, A.; Ghadiri, M.; Luessi, F.; Zipp, F.; Li, H.; Giacomini, P.S.; Antel, J.; Bar-Or, A. Dimethyl Fumarate Treatment Mediates an Anti-Inflammatory Shift in B Cell Subsets of Patients with Multiple Sclerosis. J. Immunol. 2017, 198, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Kobayashi, Y.; Souda, K.; Kawamura, K.; Sumiyoshi, S.; Takahashi, Y.; Noritake, H.; Watanabe, S.; Suehiro, T.; Nakamura, H. Enhanced hepatic Nrf2 activation after ursodeoxycholic acid treatment in patients with primary biliary cirrhosis. Antioxid. Redox Signal. 2010, 13, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345. e22. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lu, M.C.; You, Q.D. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Inhibition: An Emerging Strategy in Cancer Therapy. J. Med. Chem. 2019, 62, 3840–3856. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, S.; Ma, T.P.; Lee, J.; Haverty, P.M.; Kirkpatrick, D.S.; Yu, K.; Stokoe, D. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Mol. Cell. Proteomics 2016, 15, 1220–1231. [Google Scholar] [CrossRef]
- Harder, B.; Tian, W.; La Clair, J.J.; Tan, A.C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Chian, S.; Thapa, R.; Chi, Z.; Wang, X.J.; Tang, X. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochem. Biophys. Res. Commun. 2014, 447, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Wang, Y.; Zhong, Y.; Tang, J.; Zhang, J.; Li, Z.; Wang, Q.; Hu, R. Wogonin-enhanced reactive oxygen species-induced apoptosis and potentiated cytotoxic effects of chemotherapeutic agents by suppression Nrf2-mediated signaling in HepG2 cells. Free Radic. Res. 2014, 48, 607–621. [Google Scholar] [CrossRef]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef]
- Bollong, M.J.; Yun, H.; Sherwood, L.; Woods, A.K.; Lairson, L.L.; Schultz, P.G. A Small Molecule Inhibits Deregulated NRF2 Transcriptional Activity in Cancer. ACS Chem. Biol. 2015, 10, 2193–2198. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Liu, H.Y.; Tuckett, A.Z.; Fennell, M.; Garippa, R.; Zakrzewski, J.L. Repurposing of the CDK inhibitor PHA-767491 as a NRF2 inhibitor drug candidate for cancer therapy via redox modulation. Investig. New Drugs 2018, 36, 590–600. [Google Scholar] [CrossRef]

 stress promoting stimuli.
stress promoting stimuli.
stress promoting stimuli.
stress promoting stimuli.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Target | Mechanism of Action | Effect | Experimental Model | Use | References |
|---|---|---|---|---|---|---|
| Sulforaphane | Keap1 | Interferes with Keap1-Nrf2 binding | Nrf2 activation | in vitro | Liver Cancer | [99] |
| DMF | Keap1 | Interferes with Keap1-Nrf2 binding | Nrf2 activation | in vitro/in vivo | Multiple Sclerosis | [173] |
| Oltipraz | Keap1 | Interferes with Keap1-Nrf2 binding | Nrf2 activation | in vivo | Gastric Cancer | [91] |
| Bladder Cancer | [94] | |||||
| Liver Cancer | [100,101,102] | |||||
| CDDO-Im | Keap1 | Interferes with Keap1-Nrf2 binding | Nrf2 activation | in vivo | Liver Cancer | [37,38] |
| Curcumin | Keap1 p38 | Interferes with Keap1-Nrf2 binding | Nrf2 activation | in vitro | Renal epithelial cells | [175] |
| Ursodiol | Not specified | Not specified | Nrf2 activation | Human biopsies | Primary biliary cirrhosis | [174] |
| Apigenin | PI3K/Akt | Decreases Nrf2 mRNA and protein levels | Nrf2 inhibition | in vitro/in vivo | Liver Cancer | [162] |
| Camptotechin | Nrf2 | Decreases NRF2 mRNA and protein levels | Nrf2 inhibition | in vitro | Liver Cancer | [161] |
| Brusatol | Overall protein translation | Decreases Nrf2 protein levels | Nrf2 inhibition | in vitro/in vivo | Lung Cancer | [180] |
| Halofuginone | Overall protein translation | Decreases Nrf2 protein levels | Nrf2 inhibition | in vitro/in vivo | Lung Cancer Esophageal squamous Carcinoma | [186] |
| Wogonin | Nrf2 | Decreases Nrf2 protein levels Increases Keap1 protein levels | Nrf2 inhibition | in vitro | Liver Cancer | [185] |
| Luteolin | Nrf2 | Decreases Nrf2 transcriptional activity | Nrf2 inhibition | in vivo | Lung Cancer | [184] |
| ATRA | Neh7 domain of Nrf2 | Decreases Nrf2 transcriptional activity | Nrf2 inhibition | in vitro | Breast Cancer | [187] |
| ML385 | Neh1 domain of NRF2 | Decreases Nrf2 transcriptional activity | Nrf2 inhibition | in vitro/in vivo | Lung Cancer | [189] |
| AEM1 | Nrf2 | Decreases Nrf2 transcriptional activity | Nrf2 inhibition | in vitro | Lung Cancer | [188] |
| PHA767491 | Nrf2 | Decreases Nrf2 transcriptional activity | Nrf2 inhibition | in vitro | Liver Cancer | [190] |
| K67 | Keap1 | Interferes with Keap1-P62 binding | Nrf2 inhibition | in vitro | Liver Cancer | [49] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orrù, C.; Giordano, S.; Columbano, A. Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers 2020, 12, 2932. https://doi.org/10.3390/cancers12102932
Orrù C, Giordano S, Columbano A. Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers. 2020; 12(10):2932. https://doi.org/10.3390/cancers12102932
Chicago/Turabian StyleOrrù, Claudia, Silvia Giordano, and Amedeo Columbano. 2020. "Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases" Cancers 12, no. 10: 2932. https://doi.org/10.3390/cancers12102932
APA StyleOrrù, C., Giordano, S., & Columbano, A. (2020). Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers, 12(10), 2932. https://doi.org/10.3390/cancers12102932