Protein Translation Inhibition is Involved in the Activity of the Pan-PIM Kinase Inhibitor PIM447 in Combination with Pomalidomide-Dexamethasone in Multiple Myeloma

 , ,
, ,  , ,
, , 
 and
and 
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Triple Combination of PIM447 + Pomalidomide + Dexamethasone (PIM-Pd) is Synergistic in Vitro and Overcomes the Protective Effect Conferred by BM-MSCs
2.2. The Triple Combination PIM-Pd Delays Tumor Growth and Prolongs Survival in Human Plasmacytoma Murine Models
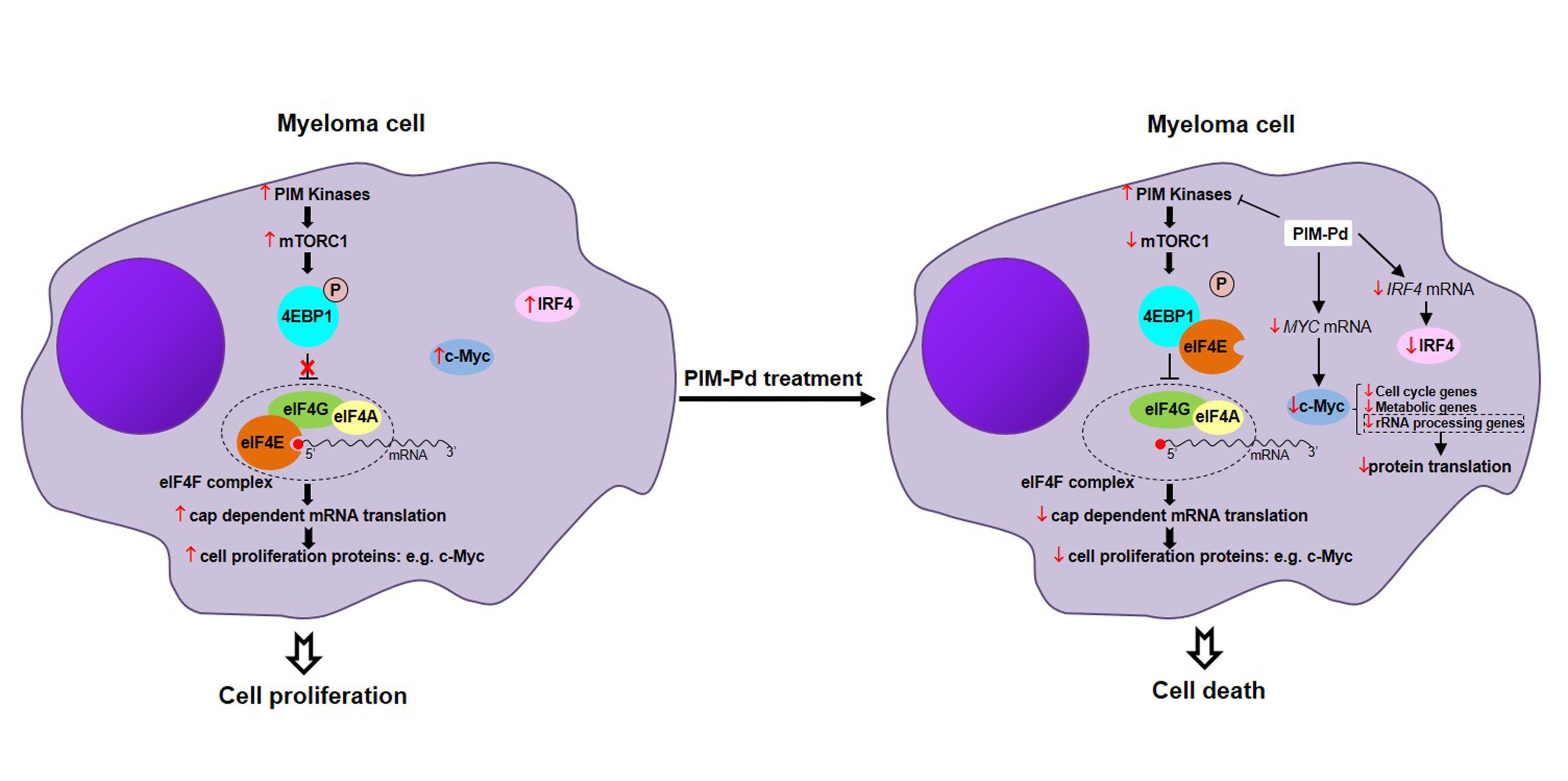
2.3. The PIM-Pd Combination Inhibits Global Protein Synthesis in Myeloma Cells by Targeting the mTORC1 Pathway and Impairing eIF4E Function
2.4. Treatment of Myeloma Cells with PIM-Pd Reduces the Expression of the Survival Factors c-Myc and IRF4
2.5. The PIM-Pd Combination Deregulates Pathways Involved in the Cell Cycle, Gene Expression, Metabolism of RNA and Energy Metabolism
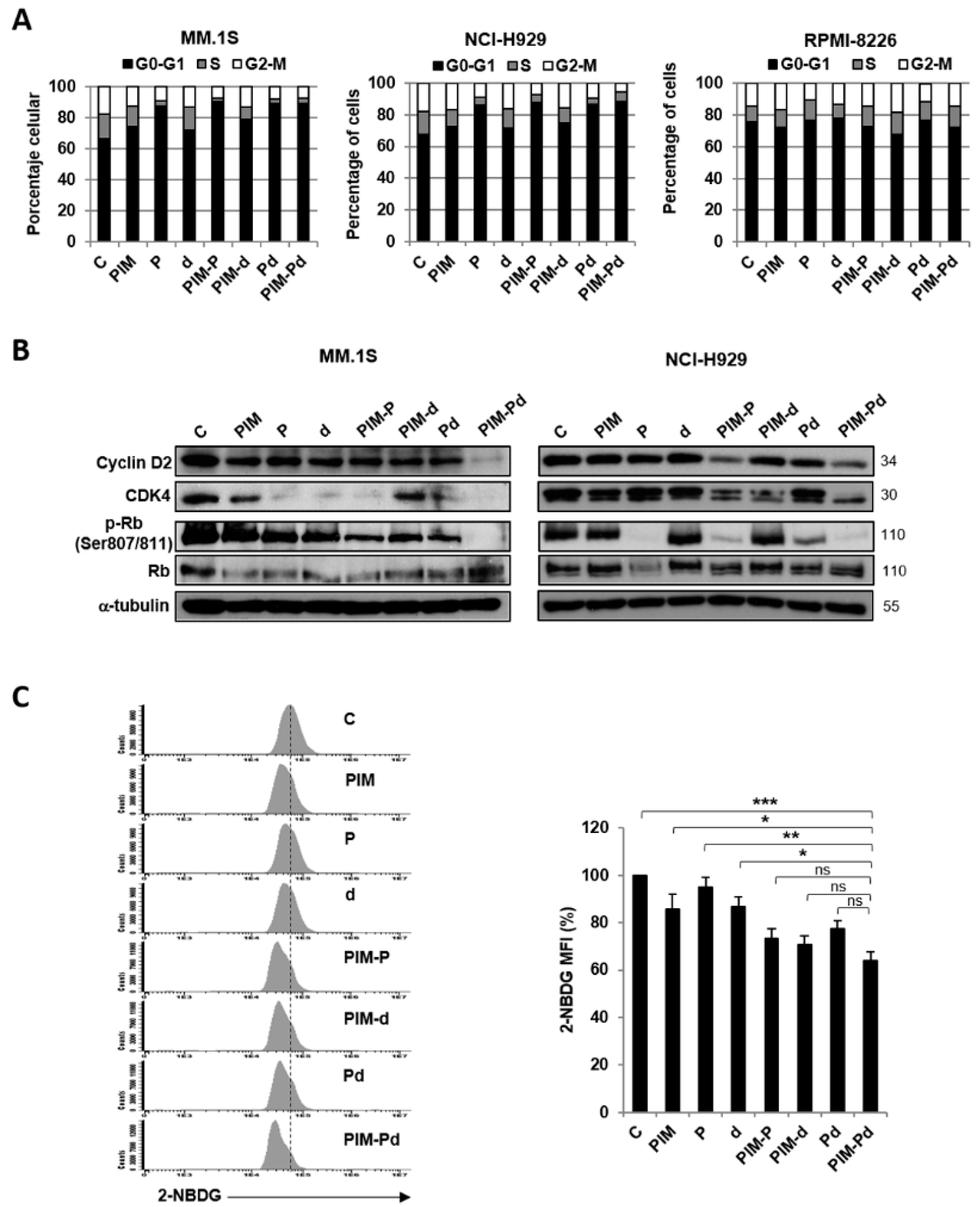
2.6. Treatment of Myeloma Cells with PIM-Pd Induces Cell Cycle Arrest by Modifying the Levels of G0/G1-Transition Regulators and Reduces Glucose Uptake
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines and Cultures
4.3. MTT, Cell Cycle, and Apoptosis Assays
4.4. Western Blot and Immunoprecipitation
4.5. Determination of Protein Biosynthesis Levels
4.6. Transcriptome Profiling Microarrays
4.7. Reverse Transcription Quantitative PCR (RT-qPCR)
4.8. 2-NBDG Assay
4.9. Analysis of Drug Toxicity in Hematopoietic Populations
4.10. Human Plasmacytoma Murine Model
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef]
- Garcia, P.D.; Langowski, J.L.; Wang, Y.; Chen, M.; Castillo, J.; Fanton, C.; Ison, M.; Zavorotinskaya, T.; Dai, Y.; Lu, J.; et al. Pan-PIM Kinase Inhibition Provides a Novel Therapy for Treating Hematologic Cancers. Clin. Cancer Res. 2014, 20, 1834–1845. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Mian, M. Pim kinases in hematological malignancies: Where are we now and where are we going? J. Hematol. Oncol. 2014, 7. [Google Scholar] [CrossRef]
- Asano, J.; Nakano, A.; Oda, A.; Amou, H.; Hiasa, M.; Takeuchi, K.; Miki, H.; Nakamura, S.; Harada, T.; Fujii, S.; et al. The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells. Leukemia 2011, 25, 1182–1188. [Google Scholar] [CrossRef]
- Lu, J.; Zavorotinskaya, T.; Dai, Y.; Niu, X.-H.; Castillo, J.; Sim, J.; Yu, J.; Wang, Y.; Langowski, J.L.; Holash, J.; et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood 2013, 122, 1610–1620. [Google Scholar] [CrossRef]
- Paíno, T.; Garcia-Gomez, A.; González-Méndez, L.; San-Segundo, L.; Hernández-García, S.; López-Iglesias, A.-A.; Algarín, E.M.; Martín-Sánchez, M.; Corbacho, D.; Ortiz-de-Solorzano, C.; et al. The Novel Pan-PIM Kinase Inhibitor, PIM447, Displays Dual Antimyeloma and Bone-Protective Effects, and Potently Synergizes with Current Standards of Care. Clin. Cancer Res. 2017, 23, 225–238. [Google Scholar] [CrossRef]
- Nair, J.R.; Caserta, J.; Belko, K.; Howell, T.; Fetterly, G.; Baldino, C.; Lee, K.P. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia 2017, 31, 1715–1726. [Google Scholar] [CrossRef]
- Koblish, H.; Li, Y.; Shin, N.; Hall, L.; Wang, Q.; Wang, K.; Covington, M.; Marando, C.; Bowman, K.; Boer, J.; et al. Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS ONE 2018, 13, e0199108. [Google Scholar] [CrossRef]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.-T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef]
- Huang, S.-M.A.; Wang, A.; Greco, R.; Li, Z.; Barberis, C.; Tabart, M.; Patel, V.; Schio, L.; Hurley, R.; Cheng, H.; et al. Combination of PIM and JAK2 inhibitors synergistically suppresses cell proliferation and overcomes drug resistance of myeloproliferative neoplasms. Oncotarget 2014, 5, 3362–3374. [Google Scholar] [CrossRef]
- Harada, M.; Benito, J.; Yamamoto, S.; Kaur, S.; Arslan, D.; Ramirez, S.; Jacamo, R.; Platanias, L.; Matsushita, H.; Fujimura, T.; et al. The novel combination of dual mTOR inhibitor AZD2014 and pan-PIM inhibitor AZD1208 inhibits growth in acute myeloid leukemia via HSF pathway suppression. Oncotarget 2015, 6, 37930–37947. [Google Scholar] [CrossRef]
- Kapoor, S.; Natarajan, K.; Baldwin, P.R.; Doshi, K.A.; Lapidus, R.G.; Mathias, T.J.; Scarpa, M.; Trotta, R.; Davila, E.; Kraus, M.; et al. Concurrent Inhibition of Pim and FLT3 Kinases Enhances Apoptosis of FLT3-ITD Acute Myeloid Leukemia Cells through Increased Mcl-1 Proteasomal Degradation. Clin. Cancer Res. 2018, 24, 234–247. [Google Scholar] [CrossRef]
- Zheng, J.; Sha, Y.; Roof, L.; Foreman, O.; Lazarchick, J.; Venkta, J.K.; Kozlowski, C.; Gasparetto, C.; Chao, N.; Ebens, A.; et al. Pan-PIM kinase inhibitors enhance Lenalidomide’s anti-myeloma activity via cereblon-IKZF1/3 cascade. Cancer Lett. 2019, 440–441, 1–10. [Google Scholar] [CrossRef]
- Thoreen, C.C. The molecular basis of mTORC1-regulated translation. Biochem. Soc. Trans. 2017, 45, 213–221. [Google Scholar] [CrossRef]
- Flynn, A.; Hogarty, M. Myc, Oncogenic Protein Translation, and the Role of Polyamines. Med. Sci. 2018, 6, 41. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Rychak, E.; Mendy, D.; Shi, T.; Ning, Y.; Leisten, J.; Lu, L.; Miller, K.; Narla, R.K.; Orlowski, R.Z.; Raymon, H.K.; et al. Pomalidomide in combination with dexamethasone results in synergistic anti-tumour responses in pre-clinical models of lenalidomide-resistant multiple myeloma. Br. J. Haematol. 2016, 172, 889–901. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef]
- Huang, X.; Di Liberto, M.; Jayabalan, D.; Liang, J.; Ely, S.; Bretz, J.; Shaffer, A.L.; Louie, T.; Chen, I.; Randolph, S.; et al. Prolonged early G1 arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood 2012, 120, 1095–1106. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef]
- Claudio, J.O.; Masih-Khan, E.; Tang, H.; Gonçalves, J.; Voralia, M.; Li, Z.H.; Nadeem, V.; Cukerman, E.; Francisco-Pabalan, O.; Liew, C.C.; et al. A molecular compendium of genes expressed in multiple myeloma. Blood 2002, 100, 2175–2186. [Google Scholar] [CrossRef]
- Stumpf, C.R.; Ruggero, D. The cancerous translation apparatus. Curr. Opin. Genet. Dev. 2011, 21, 474–483. [Google Scholar] [CrossRef]
- Peters, T.L.; Li, L.; Tula-Sanchez, A.A.; Pongtornpipat, P.; Schatz, J.H. Control of translational activation by PIM kinase in activated B-cell diffuse large B-cell lymphoma confers sensitivity to inhibition by PIM447. Oncotarget 2016, 7, 63362–63373. [Google Scholar] [CrossRef]
- Merrick, W.C. Cap-dependent and cap-independent translation in eukaryotic systems. Gene 2004, 332, 1–11. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; LeBacquer, O.; Sonenberg, N. mTOR, translation initiation and cancer. Oncogene 2006, 25, 6416–6422. [Google Scholar] [CrossRef]
- Jia, Y.; Polunovsky, V.; Bitterman, P.B.; Wagner, C.R. Cap-Dependent Translation Initiation Factor eIF4E: An Emerging Anticancer Drug Target. Med. Res. Rev. 2012, 32, 786–814. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013, 23, 345–356. [Google Scholar] [CrossRef]
- Descamps, G.; Gomez-Bougie, P.; Tamburini, J.; Green, A.; Bouscary, D.; Maïga, S.; Moreau, P.; Le Gouill, S.; Pellat-Deceunynck, C.; Amiot, M. The cap-translation inhibitor 4EGI-1 induces apoptosis in multiple myeloma through Noxa induction. Br. J. Cancer 2012, 106, 1660–1667. [Google Scholar] [CrossRef]
- Zeller, K.I.; Jegga, A.G.; Aronow, B.J.; Dang, C.V. An integrated database of genes responsive to the Myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003, 4, R69. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef]
- Pelletier, J.; Graff, J.; Ruggero, D.; Sonenberg, N. Targeting the eIF4F Translation Initiation Complex: A Critical Nexus for Cancer Development. Cancer Res. 2015, 75, 250–263. [Google Scholar] [CrossRef]
- Rodriguez-Corona, U.; Sobol, M.; Rodriguez-Zapata, L.C.; Hozak, P.; Castano, E. Fibrillarin from Archaea to human: Review on fibrillarin. Biol. Cell 2015, 107, 159–174. [Google Scholar] [CrossRef]
- Schlosser, I. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003, 31, 6148–6156. [Google Scholar] [CrossRef]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef]
- Manier, S.; Huynh, D.; Shen, Y.J.; Zhou, J.; Yusufzai, T.; Salem, K.Z.; Ebright, R.Y.; Shi, J.; Park, J.; Glavey, S.V.; et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci. Transl. Med. 2017, 9, eaal2668. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Fernando, R.C.; de Carvalho, F.; Mazzotti, D.R.; Evangelista, A.F.; Braga, W.M.T.; de Lourdes Chauffaille, M.; Leme, A.F.P.; Colleoni, G.W.B. Multiple myeloma cell lines and primary tumors proteoma: Protein biosynthesis and Immune system as potential therapeutic targets. Genes Cancer 2015, 6, 462–471. [Google Scholar] [CrossRef]
- Affer, M.; Chesi, M.; Chen, W.D.; Keats, J.J.; Demchenko, Y.N.; Tamizhmani, K.; Garbitt, V.M.; Riggs, D.L.; Brents, L.A.; Roschke, A.V.; et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef]
- Chng, W.-J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef]
- Chiecchio, L.; Dagrada, G.P.; Protheroe, R.K.M.; Stockley, D.M.; Smith, A.G.; Orchard, K.H.; Cross, N.C.P.; Harrison, C.J.; Ross, F.M. Loss of 1p and rearrangement of MYC are associated with progression of smouldering myeloma to myeloma: Sequential analysis of a single case. Haematologica 2009, 94, 1024–1028. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Quwaider, D.; Canavese, M.; Ocio, E.M.; Tian, Z.; Blanco, J.F.; Berger, A.J.; Ortiz-de-Solorzano, C.; Hernandez-Iglesias, T.; Martens, A.C.M.; et al. Preclinical Activity of the Oral Proteasome Inhibitor MLN9708 in Myeloma Bone Disease. Clin. Cancer Res. 2014, 20, 1542–1554. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Ocio, E.M.; Maiso, P.; Chen, X.; Garayoa, M.; Álvarez-Fernández, S.; San-Segundo, L.; Vilanova, D.; López-Corral, L.; Montero, J.C.; Hernández-Iglesias, T.; et al. Zalypsis: A novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood 2009, 113, 3781–3791. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Hernández-García, S.; San-Segundo, L.; González-Méndez, L.; Corchete, L.A.; Misiewicz-Krzeminska, I.; Martín-Sánchez, M.; López-Iglesias, A.-A.; Algarín, E.M.; Mogollón, P.; Díaz-Tejedor, A.; et al. The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma. Haematologica 2017, 102, 2113–2124. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.; Miller, S.; Curwen, J.; Dymond, M. Design, analysis and reporting of tumor models. Lab Anim. 2017, 46, 207–211. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paíno, T.; González-Méndez, L.; San-Segundo, L.; Corchete, L.A.; Hernández-García, S.; Díaz-Tejedor, A.; Algarín, E.M.; Mogollón, P.; Martín-Sánchez, M.; Gutiérrez, N.C.; et al. Protein Translation Inhibition is Involved in the Activity of the Pan-PIM Kinase Inhibitor PIM447 in Combination with Pomalidomide-Dexamethasone in Multiple Myeloma. Cancers 2020, 12, 2743. https://doi.org/10.3390/cancers12102743
Paíno T, González-Méndez L, San-Segundo L, Corchete LA, Hernández-García S, Díaz-Tejedor A, Algarín EM, Mogollón P, Martín-Sánchez M, Gutiérrez NC, et al. Protein Translation Inhibition is Involved in the Activity of the Pan-PIM Kinase Inhibitor PIM447 in Combination with Pomalidomide-Dexamethasone in Multiple Myeloma. Cancers. 2020; 12(10):2743. https://doi.org/10.3390/cancers12102743
Chicago/Turabian StylePaíno, Teresa, Lorena González-Méndez, Laura San-Segundo, Luis A. Corchete, Susana Hernández-García, Andrea Díaz-Tejedor, Esperanza M. Algarín, Pedro Mogollón, Montserrat Martín-Sánchez, Norma C. Gutiérrez, and et al. 2020. "Protein Translation Inhibition is Involved in the Activity of the Pan-PIM Kinase Inhibitor PIM447 in Combination with Pomalidomide-Dexamethasone in Multiple Myeloma" Cancers 12, no. 10: 2743. https://doi.org/10.3390/cancers12102743
APA StylePaíno, T., González-Méndez, L., San-Segundo, L., Corchete, L. A., Hernández-García, S., Díaz-Tejedor, A., Algarín, E. M., Mogollón, P., Martín-Sánchez, M., Gutiérrez, N. C., Mateos, M.-V., Garayoa, M., & Ocio, E. M. (2020). Protein Translation Inhibition is Involved in the Activity of the Pan-PIM Kinase Inhibitor PIM447 in Combination with Pomalidomide-Dexamethasone in Multiple Myeloma. Cancers, 12(10), 2743. https://doi.org/10.3390/cancers12102743