Molecular Genetics of Renal Cell Tumors: A Practical Diagnostic Approach



Abstract
1. Introduction
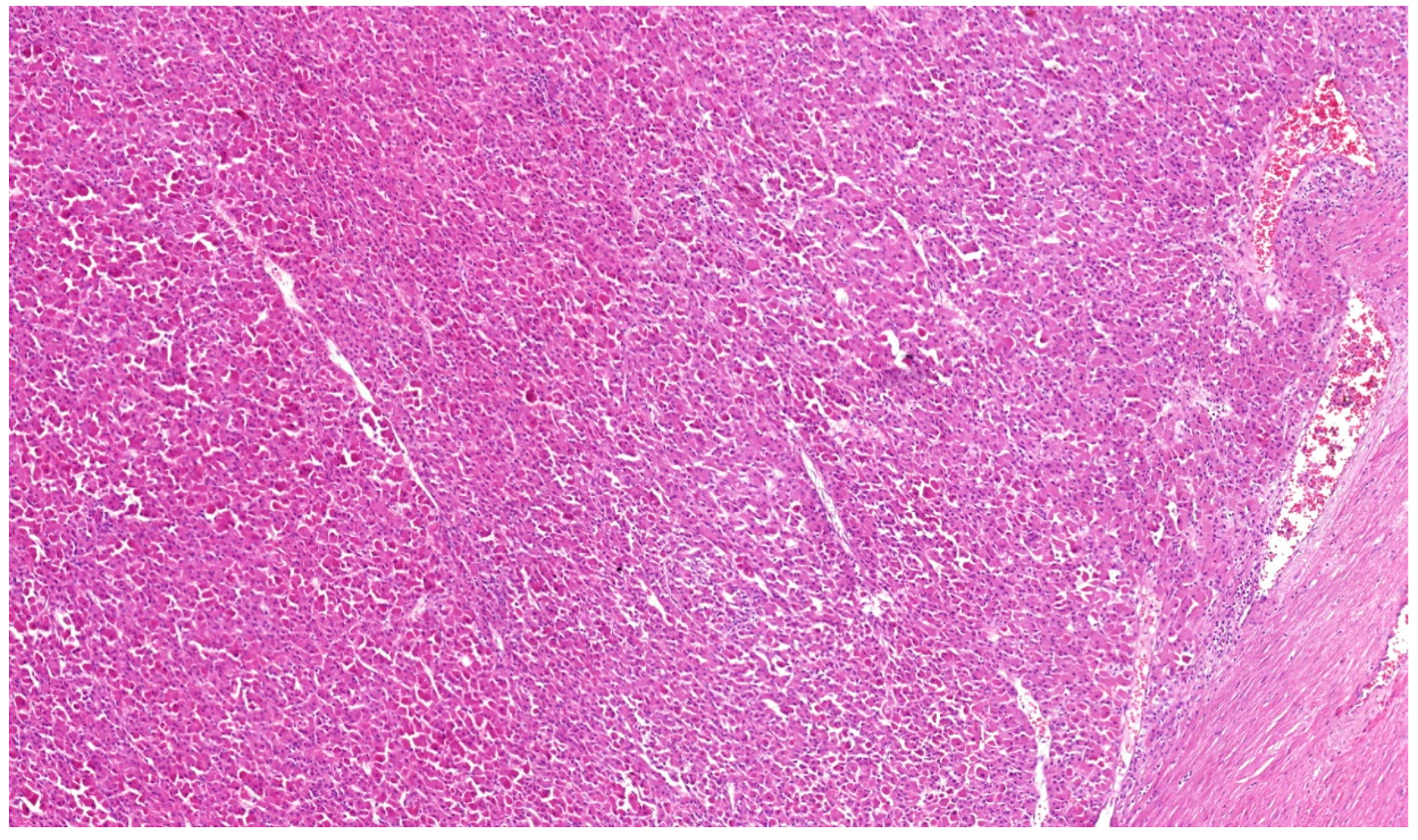
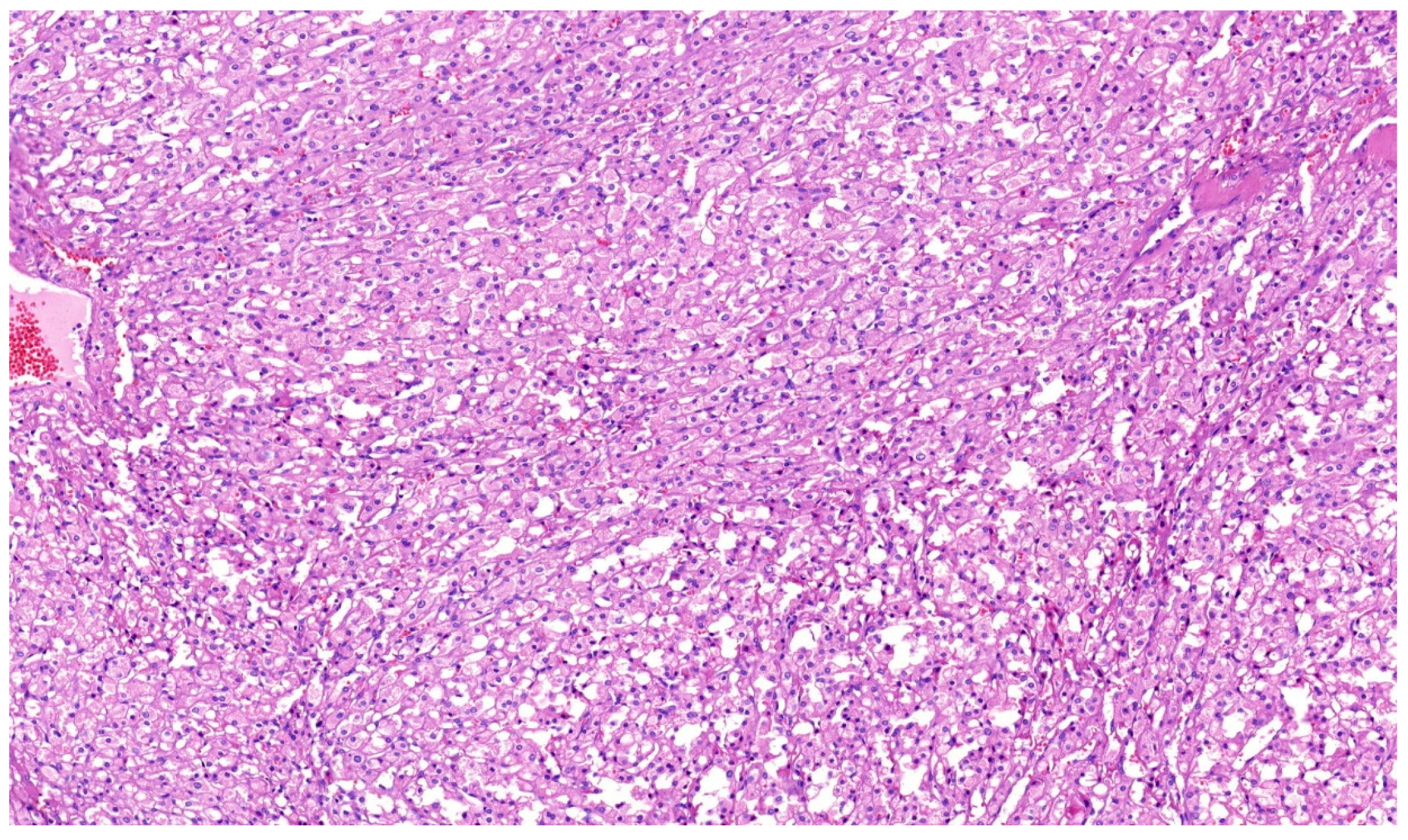
2. Clear Cell Renal Cell Carcinoma (CCRCC)
3. Multilocular Cystic Renal Cell Neoplasm of Low Malignant Potential (MCRCNLMP)
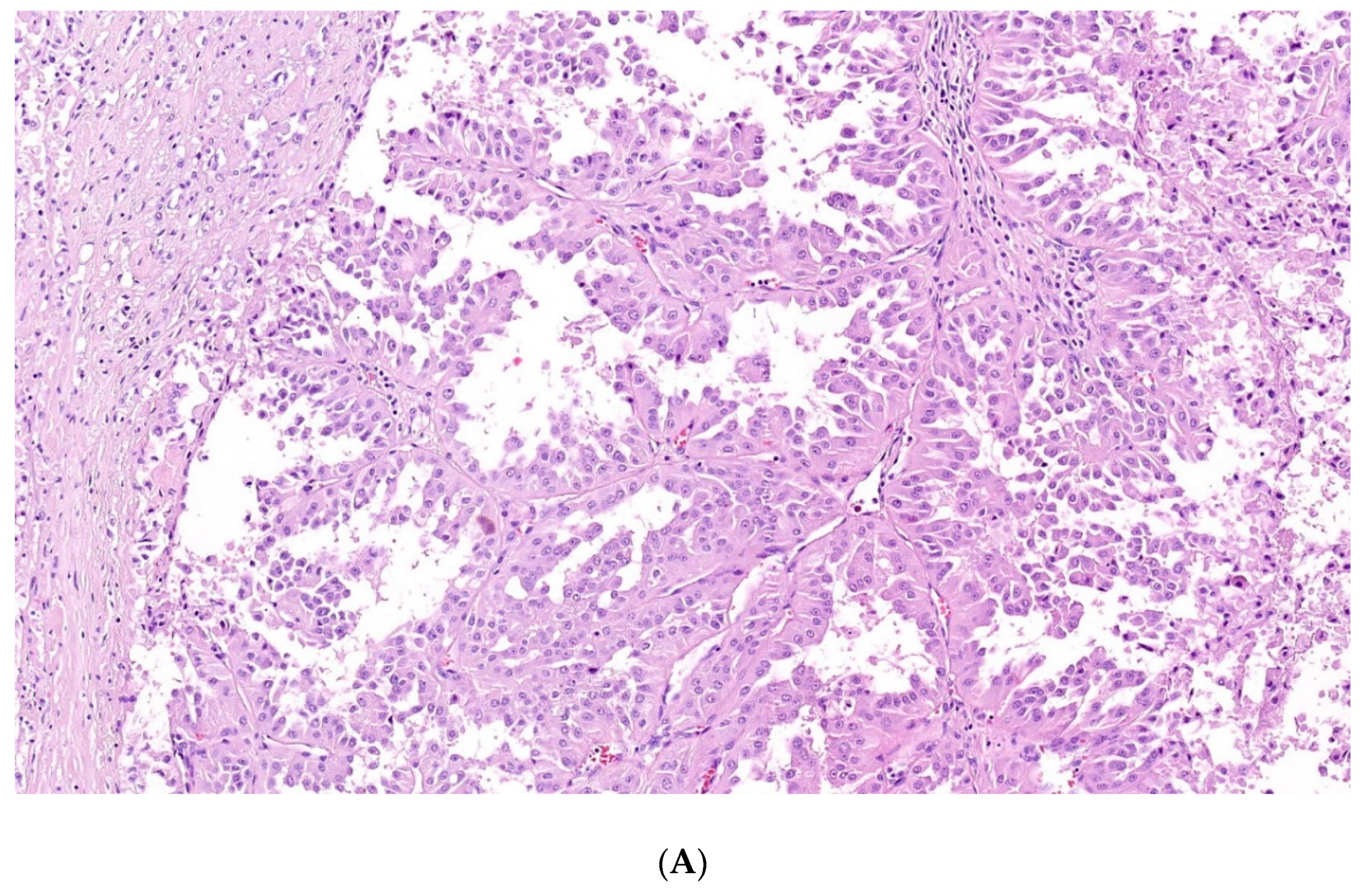
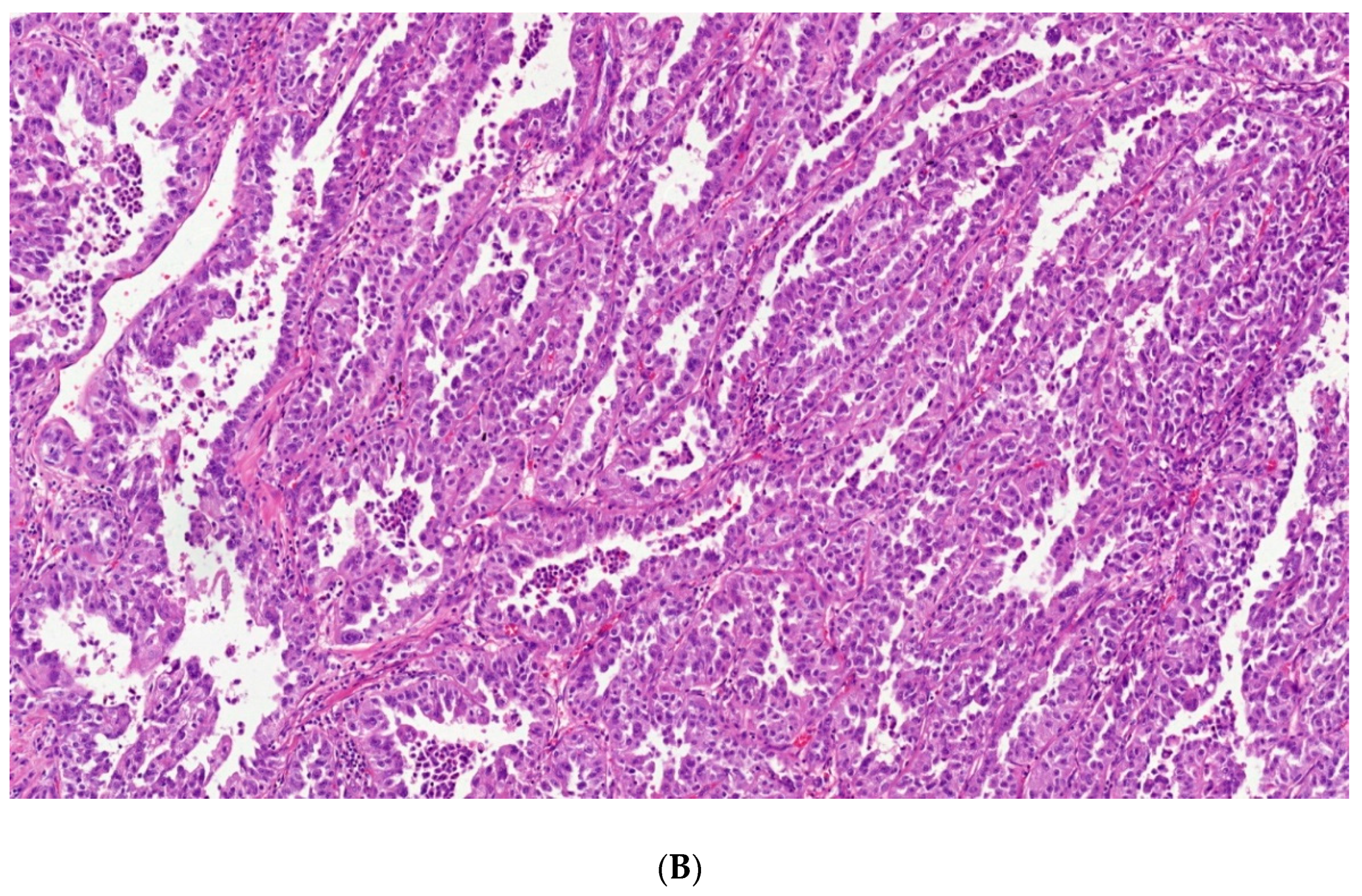
4. Papillary Renal Cell Carcinoma (PRCC)
4.1. Type 1 Papillary RCC
4.2. Type 2 Papillary RCC
4.3. Oncocytic Papillary RCC/Papillary Renal Cell Neoplasm with Reverse Polarity
4.4. Papillary RCC NOS: Other Variants
5. Chromophobe Renal Cell Carcinoma (ChRCC)
6. Oncocytoma
7. Clear Cell Papillary Renal Cell Carcinoma (CCPRCC)
8. MiT Family Translocation-Associated Renal Cell Carcinoma
9. Mucinous Tubular and Spindle Cell Carcinoma (MTSCC)
10. Tubulocystic Renal Cell Carcinoma (TC-RCC)
11. Acquired Cystic Kidney Disease (ACD)-Associated Renal Cell Carcinoma
12. Renal Medullary Carcinoma
13. Collecting Duct Carcinoma (CDC)
14. Succinate Dehydrogenase (SDH)-Deficient Renal Cell Carcinoma
15. Fumarate Hydratase (FH)-Deficient RCC and HLRCC (Hereditary Leiomyomatosis and Renal Cell Carcinoma)
16. New but Perspective Renal Tumors
16.1. Eosinophilic Solid and Cystic (ESC) RCC
16.2. RCC with TSC/MTOR Gene Mutations
16.3. TCEB1-Mutated RCC
16.4. RCC with TFEB/6p21/VEGFA Amplification
16.5. ALK-Rearranged RCC
17. Discussion
18. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kovacs, G.; Akhtar, M.; Beckwith, B.J.; Bugert, P.; Cooper, C.S.; Delahunt, B.; Eble, J.N.; Fleming, S.; Ljungberg, B.; Medeiros, L.J.; et al. The Heidelberg classification of renal cell tumours. J Pathol 1997, 183, 131–133. [Google Scholar] [CrossRef]
- Storkel, S.; Eble, J.N.; Adlakha, K.; Amin, M.; Blute, M.L.; Bostwick, D.G.; Darson, M.; Delahunt, B.; Iczkowski, K. Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 1997, 80, 987–989. [Google Scholar] [CrossRef]
- Eble, J.N.; Sauter, G.; Epstein, J.; Sesterhenn, I. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs, WHO Classification of Tumours, 3rd ed.; WHO: Geneva, Switzerland; IARC Press: Lyon, Switzerland, 2004; Volume 7. [Google Scholar]
- Delahunt, B.; Srigley, J.R.; Montironi, R.; Egevad, L. Advances in renal neoplasia: Recommendations from the 2012 International Society of Urological Pathology Consensus Conference. Urology 2014, 83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Humphrey, P.A.; Ulbright, T.M.; Reuter, V.E. WHO Classification of Tumours of the Urinary System and Male Genital Organs—WHO Classification of Tumours, 4th ed.; WHO: Geneva, Switzerland; IARC Press: Lyon, Switzerland, 2016; Volume 8. [Google Scholar]
- Petersson, F.; Grossmann, P.; Hora, M.; Sperga, M.; Montiel, D.P.; Martinek, P.; Gutierrez, M.E.; Bulimbasic, S.; Michal, M.; Branzovsky, J.; et al. Renal cell carcinoma with areas mimicking renal angiomyoadenomatous tumor/clear cell papillary renal cell carcinoma. Hum. Pathol. 2013, 44, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Somoracz, A.; Kuthi, L.; Micsik, T.; Jenei, A.; Hajdu, A.; Vrabely, B.; Raso, E.; Sapi, Z.; Bajory, Z.; Kulka, J.; et al. Renal Cell Carcinoma with Clear Cell Papillary Features: Perspectives of a Differential Diagnosis. Pathol. Oncol. Res. 2019. [Google Scholar] [CrossRef]
- Carroll, P.R.; Murty, V.V.; Reuter, V.; Jhanwar, S.; Fair, W.R.; Whitmore, W.F.; Chaganti, R.S. Abnormalities at chromosome region 3p12-14 characterize clear cell renal carcinoma. Cancer Genet. Cytogenet. 1987, 26, 253–259. [Google Scholar] [CrossRef]
- Smits, K.M.; Schouten, L.J.; van Dijk, B.A.; Hulsbergen-van de Kaa, C.A.; Wouters, K.A.; Oosterwijk, E.; van Engeland, M.; van den Brandt, P.A. Genetic and epigenetic alterations in the von hippel-lindau gene: The influence on renal cancer prognosis. Clin. Cancer Res. 2008, 14, 782–787. [Google Scholar] [CrossRef]
- Banks, R.E.; Tirukonda, P.; Taylor, C.; Hornigold, N.; Astuti, D.; Cohen, D.; Maher, E.R.; Stanley, A.J.; Harnden, P.; Joyce, A.; et al. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006, 66, 2000–2011. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef]
- Guo, G.; Gui, Y.; Gao, S.; Tang, A.; Hu, X.; Huang, Y.; Jia, W.; Li, Z.; He, M.; Sun, L.; et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat. Genet. 2011, 44, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Suzigan, S.; Lopez-Beltran, A.; Montironi, R.; Drut, R.; Romero, A.; Hayashi, T.; Gentili, A.L.; Fonseca, P.S.; deTorres, I.; Billis, A.; et al. Multilocular cystic renal cell carcinoma: A report of 45 cases of a kidney tumor of low malignant potential. Am. J. Clin. Pathol. 2006, 125, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Halat, S.; Eble, J.N.; Grignon, D.J.; Lopez-Beltran, A.; Montironi, R.; Tan, P.H.; Wang, M.; Zhang, S.; Maclennan, G.T.; et al. Multilocular cystic renal cell carcinoma: Similarities and differences in immunoprofile compared with clear cell renal cell carcinoma. Am. J. Surg. Pathol. 2012, 36, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Halat, S.; Eble, J.N.; Grignon, D.J.; Lopez-Beltran, A.; Montironi, R.; Tan, P.H.; Wang, M.; Zhang, S.; MacLennan, G.T.; Cheng, L. Multilocular cystic renal cell carcinoma is a subtype of clear cell renal cell carcinoma. Mod. Pathol. 2010, 23, 931–936. [Google Scholar] [CrossRef] [PubMed]
- von Teichman, A.; Comperat, E.; Behnke, S.; Storz, M.; Moch, H.; Schraml, P. VHL mutations and dysregulation of pVHL- and PTEN-controlled pathways in multilocular cystic renal cell carcinoma. Mod. Pathol. 2011, 24, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Castiglione, F.; Martignoni, G.; Cheng, L.; Montironi, R.; Lopez-Beltran, A. Unlike in clear cell renal cell carcinoma, KRAS is not mutated in multilocular cystic clear cell renal cell neoplasm of low potential. Virchows Arch. 2015, 467, 687–693. [Google Scholar] [CrossRef]
- Raspollini, M.R.; Castiglione, F.; Cheng, L.; Montironi, R.; Lopez-Beltran, A. Synchronous clear cell renal cell carcinoma and multilocular cystic renal cell neoplasia of low malignant potential: A clinico-pathologic and molecular study. Pathol. Res. Pr. 2016, 212, 471–474. [Google Scholar] [CrossRef]
- Szymanska, K.; Moore, L.E.; Rothman, N.; Chow, W.H.; Waldman, F.; Jaeger, E.; Waterboer, T.; Foretova, L.; Navratilova, M.; Janout, V.; et al. TP53, EGFR, and KRAS mutations in relation to VHL inactivation and lifestyle risk factors in renal-cell carcinoma from central and eastern Europe. Cancer Lett. 2010, 293, 92–98. [Google Scholar] [CrossRef]
- Gattenlohner, S.; Etschmann, B.; Riedmiller, H.; Muller-Hermelink, H.K. Lack of KRAS and BRAF mutation in renal cell carcinoma. Eur. Urol. 2009, 55, 1490–1491. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, O.; Sen, H.; Bulut, E.; Cengiz, B.; Karakok, M.; Erturhan, S.; Seckiner, I. Evaluation of EGFR, KRAS and BRAF gene mutations in renal cell carcinoma. J. Kidney Cancer VHL 2014, 1, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Delahunt, B.; Algaba, F.; Eble, J.; Cheville, J.; Amin, M.B.; Argani, P.; Martignoni, G.; Moch, H.; Srigley, J.R.; Tan, P.H.; et al. Papillary renal cell carcinoma. In WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; Moch, H., Humphrey, P.A., Ulbright, T.M., Reuter, V.E., Eds.; International Agency for Research on Cancer: Lyon, France, 2016; Volume 8, pp. 23–25. [Google Scholar]
- Cancer Genome Atlas Research, N.; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Pitra, T.; Pivovarcikova, K.; Alaghehbandan, R.; Hes, O. Chromosomal numerical aberration pattern in papillary renal cell carcinoma: Review article. Ann. Diagn. Pathol. 2019, 40, 189–199. [Google Scholar] [CrossRef]
- Zbar, B.; Tory, K.; Merino, M.; Schmidt, L.; Glenn, G.; Choyke, P.; Walther, M.M.; Lerman, M.; Linehan, W.M. Hereditary papillary renal cell carcinoma. J. Urol. 1994, 151, 561–566. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Dharmawardana, P.G.; Giubellino, A.; Bottaro, D.P. Hereditary papillary renal carcinoma type I. Curr. Mol. Med. 2004, 4, 855–868. [Google Scholar] [CrossRef]
- Lefevre, M.; Couturier, J.; Sibony, M.; Bazille, C.; Boyer, K.; Callard, P.; Vieillefond, A.; Allory, Y. Adult papillary renal tumor with oncocytic cells: Clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am. J. Surg. Pathol. 2005, 29, 1576–1581. [Google Scholar] [CrossRef]
- Han, G.; Yu, W.; Chu, J.; Liu, Y.; Jiang, Y.; Li, Y.; Zhang, W. Oncocytic papillary renal cell carcinoma: A clinicopathological and genetic analysis and indolent clinical course in 14 cases. Pathol. Res. Pr. 2017, 213, 1–6. [Google Scholar] [CrossRef]
- Kunju, L.P.; Wojno, K.; Wolf, J.S., Jr.; Cheng, L.; Shah, R.B. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: Expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum. Pathol. 2008, 39, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Hes, O.; Brunelli, M.; Michal, M.; Cossu Rocca, P.; Hora, M.; Chilosi, M.; Mina, M.; Boudova, L.; Menestrina, F.; Martignoni, G. Oncocytic papillary renal cell carcinoma: A clinicopathologic, immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann. Diagn. Pathol. 2006, 10, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Srigley, J.R.; Delahunt, B.; Eble, J.N.; Egevad, L.; Epstein, J.I.; Grignon, D.; Hes, O.; Moch, H.; Montironi, R.; Tickoo, S.K.; et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am. J. Surg. Pathol. 2013, 37, 1469–1489. [Google Scholar] [CrossRef] [PubMed]
- Michalova, K.; Steiner, P.; Alaghehbandan, R.; Trpkov, K.; Martinek, P.; Grossmann, P.; Montiel, D.P.; Sperga, M.; Straka, L.; Prochazkova, K.; et al. Papillary renal cell carcinoma with cytologic and molecular genetic features overlapping with renal oncocytoma: Analysis of 10 cases. Ann. Diagn. Pathol. 2018, 35, 1–6. [Google Scholar] [CrossRef]
- Saleeb, R.M.; Brimo, F.; Farag, M.; Rompre-Brodeur, A.; Rotondo, F.; Beharry, V.; Wala, S.; Plant, P.; Downes, M.R.; Pace, K.; et al. Toward Biological Subtyping of Papillary Renal Cell Carcinoma With Clinical Implications Through Histologic, Immunohistochemical, and Molecular Analysis. Am. J. Surg. Pathol. 2017, 41, 1618–1629. [Google Scholar] [CrossRef]
- Al-Obaidy, K.I.; Eble, J.N.; Cheng, L.; Williamson, S.R.; Sakr, W.A.; Gupta, N.; Idrees, M.T.; Grignon, D.J. Papillary Renal Neoplasm with Reverse Polarity: A Morphologic, Immunohistochemical, and Molecular Study. Am. J. Surg. Pathol. 2019, 43, 1099–1111. [Google Scholar] [CrossRef]
- Al-Obaidy, K.I.; Eble, J.N.; Nassiri, M.; Cheng, L.; Eldomery, M.K.; Williamson, S.R.; Sakr, W.A.; Gupta, N.; Hassan, O.; Idrees, M.T.; et al. Recurrent KRAS mutations in papillary renal neoplasm with reverse polarity. Mod. Pathol. 2019. [Google Scholar] [CrossRef]
- Marsaud, A.; Dadone, B.; Ambrosetti, D.; Baudoin, C.; Chamorey, E.; Rouleau, E.; Lefol, C.; Roussel, J.F.; Fabas, T.; Cristofari, G.; et al. Dismantling papillary renal cell carcinoma classification: The heterogeneity of genetic profiles suggests several independent diseases. Genes Chromosomes Cancer 2015, 54, 369–382. [Google Scholar] [CrossRef]
- Pivovarcikova, K.; Peckova, K.; Martinek, P.; Montiel, D.P.; Kalusova, K.; Pitra, T.; Hora, M.; Skenderi, F.; Ulamec, M.; Daum, O.; et al. “Mucin”-secreting papillary renal cell carcinoma: Clinicopathological, immunohistochemical, and molecular genetic analysis of seven cases. Virchows Arch. 2016, 469, 71–80. [Google Scholar] [CrossRef]
- Peckova, K.; Martinek, P.; Pivovarcikova, K.; Vanecek, T.; Alaghehbandan, R.; Prochazkova, K.; Montiel, D.P.; Hora, M.; Skenderi, F.; Ulamec, M.; et al. Cystic and necrotic papillary renal cell carcinoma: Prognosis, morphology, immunohistochemical, and molecular-genetic profile of 10 cases. Ann. Diagn Pathol. 2017, 26, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ulamec, M.; Skenderi, F.; Trpkov, K.; Kruslin, B.; Vranic, S.; Bulimbasic, S.; Trivunic, S.; Montiel, D.P.; Peckova, K.; Pivovarcikova, K.; et al. Solid papillary renal cell carcinoma: Clinicopathologic, morphologic, and immunohistochemical analysis of 10 cases and review of the literature. Ann. Diagn. Pathol. 2016, 23, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Hes, O.; Condom Mundo, E.; Peckova, K.; Lopez, J.I.; Martinek, P.; Vanecek, T.; Falconieri, G.; Agaimy, A.; Davidson, W.; Petersson, F.; et al. Biphasic Squamoid Alveolar Renal Cell Carcinoma: A Distinctive Subtype of Papillary Renal Cell Carcinoma? Am. J. Surg. Pathol. 2016, 40, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Trpkov, K.; Athanazio, D.; Magi-Galluzzi, C.; Yilmaz, H.; Clouston, D.; Agaimy, A.; Williamson, S.R.; Brimo, F.; Lopez, J.I.; Ulamec, M.; et al. Biphasic papillary renal cell carcinoma is a rare morphological variant with frequent multifocality: A study of 28 cases. Histopathology 2018, 72, 777–785. [Google Scholar] [CrossRef]
- Skenderi, F.; Ulamec, M.; Vanecek, T.; Martinek, P.; Alaghehbandan, R.; Foix, M.P.; Babankova, I.; Montiel, D.P.; Alvarado-Cabrero, I.; Svajdler, M.; et al. Warthin-like papillary renal cell carcinoma: Clinicopathologic, morphologic, immunohistochemical and molecular genetic analysis of 11 cases. Ann. Diagn. Pathol. 2017, 27, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Hes, O.; Vanecek, T.; Perez-Montiel, D.M.; Alvarado Cabrero, I.; Hora, M.; Suster, S.; Lamovec, J.; Curik, R.; Mandys, V.; Michal, M. Chromophobe renal cell carcinoma with microcystic and adenomatous arrangement and pigmentation--a diagnostic pitfall. Morphological, immunohistochemical, ultrastructural and molecular genetic report of 20 cases. Virchows Arch. 2005, 446, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Michal, M.; Hes, O.; Svec, A.; Ludvikova, M. Pigmented microcystic chromophobe cell carcinoma: A unique variant of renal cell carcinoma. Ann. Diagn. Pathol. 1998, 2, 149–153. [Google Scholar] [CrossRef]
- Dundr, P.; Pesl, M.; Povysil, C.; Tvrdik, D.; Pavlik, I.; Soukup, V.; Dvoracek, J. Pigmented microcystic chromophobe renal cell carcinoma. Pathol. Res. Pr. 2007, 203, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Foix, M.P.; Dunatov, A.; Martinek, P.; Mundo, E.C.; Suster, S.; Sperga, M.; Lopez, J.I.; Ulamec, M.; Bulimbasic, S.; Montiel, D.P.; et al. Morphological, immunohistochemical, and chromosomal analysis of multicystic chromophobe renal cell carcinoma, an architecturally unusual challenging variant. Virchows Arch. 2016, 469, 669–678. [Google Scholar] [CrossRef]
- Peckova, K.; Martinek, P.; Ohe, C.; Kuroda, N.; Bulimbasic, S.; Condom Mundo, E.; Perez Montiel, D.; Lopez, J.I.; Daum, O.; Rotterova, P.; et al. Chromophobe renal cell carcinoma with neuroendocrine and neuroendocrine-like features. Morphologic, immunohistochemical, ultrastructural, and array comparative genomic hybridization analysis of 18 cases and review of the literature. Ann. Diagn. Pathol. 2015, 19, 261–268. [Google Scholar] [CrossRef]
- Parada, D.D.; Pena, K.B. Chromophobe renal cell carcinoma with neuroendocrine differentiation. APMIS 2008, 116, 859–865. [Google Scholar] [CrossRef]
- Kuroda, N.; Tamura, M.; Hes, O.; Michal, M.; Gatalica, Z. Chromophobe renal cell carcinoma with neuroendocrine differentiation and sarcomatoid change. Pathol. Int. 2011, 61, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, G.A.; Al-Zahrani, R. Chromophobe renal cell carcinoma of the kidney with neuroendocrine differentiation: A case report with review of literature. Urol. Ann. 2015, 7, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Ohe, C.; Kuroda, N.; Matsuura, K.; Kai, T.; Moriyama, M.; Sugiguchi, S.; Terahata, S.; Hosaka, N.; Hes, O.; Michal, M.; et al. Chromophobe renal cell carcinoma with neuroendocrine differentiation/morphology: A clinicopathological and genetic study of three cases. Hum. Pathol. Case Rep. 2014, 1, 31–39. [Google Scholar] [CrossRef]
- Kuroda, N.; Tanaka, A.; Yamaguchi, T.; Kasahara, K.; Naruse, K.; Yamada, Y.; Hatanaka, K.; Shinohara, N.; Nagashima, Y.; Mikami, S.; et al. Chromophobe renal cell carcinoma, oncocytic variant: A proposal of a new variant giving a critical diagnostic pitfall in diagnosing renal oncocytic tumors. Med. Mol. Morphol. 2013, 46, 49–55. [Google Scholar] [CrossRef]
- Speicher, M.R.; Schoell, B.; du Manoir, S.; Schrock, E.; Ried, T.; Cremer, T.; Storkel, S.; Kovacs, A.; Kovacs, G. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am. J. Pathol. 1994, 145, 356–364. [Google Scholar]
- Paner, G.; Amin, M.B.; Moch, H.; Störkel, S. Chromophobe renal cell carcinoma. In WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; Moch, H., Humphrey, P.A., Ulbright, T.M., Reuter, V.E., Eds.; International Agency for Research on Cancer: Lyon, France, 2016; Volume 8, pp. 27–28. [Google Scholar]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef]
- Vieira, J.; Henrique, R.; Ribeiro, F.R.; Barros-Silva, J.D.; Peixoto, A.; Santos, C.; Pinheiro, M.; Costa, V.L.; Soares, M.J.; Oliveira, J.; et al. Feasibility of differential diagnosis of kidney tumors by comparative genomic hybridization of fine needle aspiration biopsies. Genes Chromosomes Cancer 2010, 49, 935–947. [Google Scholar] [CrossRef]
- Sperga, M.; Martinek, P.; Vanecek, T.; Grossmann, P.; Bauleth, K.; Perez-Montiel, D.; Alvarado-Cabrero, I.; Nevidovska, K.; Lietuvietis, V.; Hora, M.; et al. Chromophobe renal cell carcinoma--chromosomal aberration variability and its relation to Paner grading system: An array CGH and FISH analysis of 37 cases. Virchows Arch. 2013, 463, 563–573. [Google Scholar] [CrossRef]
- Tan, M.H.; Wong, C.F.; Tan, H.L.; Yang, X.J.; Ditlev, J.; Matsuda, D.; Khoo, S.K.; Sugimura, J.; Fujioka, T.; Furge, K.A.; et al. Genomic expression and single-nucleotide polymorphism profiling discriminates chromophobe renal cell carcinoma and oncocytoma. BMC Cancer 2010, 10, 196. [Google Scholar] [CrossRef]
- Crotty, T.B.; Lawrence, K.M.; Moertel, C.A.; Bartelt, D.H., Jr.; Batts, K.P.; Dewald, G.W.; Farrow, G.M.; Jenkins, R.B. Cytogenetic analysis of six renal oncocytomas and a chromophobe cell renal carcinoma. Evidence that -Y, -1 may be a characteristic anomaly in renal oncocytomas. Cancer Genet Cytogenet. 1992, 61, 61–66. [Google Scholar] [CrossRef]
- Fuzesi, L.; Gunawan, B.; Braun, S.; Boeckmann, W. Renal oncocytoma with a translocation t(9;11)(p23;q13). J. Urol. 1994, 152, 471–472. [Google Scholar] [CrossRef]
- Paner, G.P.; Lindgren, V.; Jacobson, K.; Harrison, K.; Cao, Y.; Campbell, S.C.; Flanigan, R.C.; Picken, M.M. High incidence of chromosome 1 abnormalities in a series of 27 renal oncocytomas: Cytogenetic and fluorescence in situ hybridization studies. Arch. Pathol. Lab. Med. 2007, 131, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, V.; Paner, G.P.; Omeroglu, A.; Campbell, S.C.; Waters, W.B.; Flanigan, R.C.; Picken, M.M. Cytogenetic analysis of a series of 13 renal oncocytomas. J. Urol. 2004, 171, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M.; Chyna, B.; Flanigan, R.C.; Lee, J.M. Analysis of chromosome 1p abnormalities in renal oncocytomas by loss of heterozygosity studies: Correlation with conventional cytogenetics and fluorescence in situ hybridization. Am. J. Clin. Pathol. 2008, 129, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.B.; Lipsky, M.; Nandula, S.V.; Freeman, C.E.; Matthews, T.; Walsh, C.E.; Li, G.; Szabolcs, M.; Mansukhani, M.M.; McKiernan, J.M.; et al. Cytogenetic analysis of 130 renal oncocytomas identify three distinct and mutually exclusive diagnostic classes of chromosome aberrations. Genes Chromosomes Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Tolkunov, D.; Aviv, H.; Hakimi, A.A.; Yao, M.; Hsieh, J.J.; Ganesan, S.; Chan, C.S.; White, E. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep. 2015, 13, 1895–1908. [Google Scholar] [CrossRef]
- Sukov, W.R.; Ketterling, R.P.; Lager, D.J.; Carlson, A.W.; Sinnwell, J.P.; Chow, G.K.; Jenkins, R.B.; Cheville, J.C. CCND1 rearrangements and cyclin D1 overexpression in renal oncocytomas: Frequency, clinicopathologic features, and utility in differentiation from chromophobe renal cell carcinoma. Hum. Pathol. 2009, 40, 1296–1303. [Google Scholar] [CrossRef]
- Williamson, S.R.; Eble, J.N.; Cheng, L.; Grignon, D.J. Clear cell papillary renal cell carcinoma: Differential diagnosis and extended immunohistochemical profile. Mod. Pathol. 2013, 26, 697–708. [Google Scholar] [CrossRef]
- Mantilla, J.G.; Antic, T.; Tretiakova, M.S. GATA-3 Is a Specific Marker for Clear Cell Papillary Renal Cell Carcinoma. Mod. Pathol. 2017, 30, 241A. [Google Scholar]
- Martignoni, G.; Brunelli, M.; Segala, D.; Munari, E.; Gobbo, S.; Cima, L.; Borze, I.; Wirtanen, T.; Sarhadi, V.K.; Atanesyan, L.; et al. Validation of 34betaE12 immunoexpression in clear cell papillary renal cell carcinoma as a sensitive biomarker. Pathology 2017, 49, 10–18. [Google Scholar] [CrossRef][Green Version]
- Hes, O.; Comperat, E.M.; Rioux-Leclercq, N. Clear cell papillary renal cell carcinoma, renal angiomyoadenomatous tumor, and renal cell carcinoma with leiomyomatous stroma relationship of 3 types of renal tumors: A review. Ann. Diagn. Pathol. 2016, 21, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.S.; Malik, F.; Amin, M.B.; Argani, P.; Bahrami, A. MiT family translocation renal cell carcinomas: A 15th anniversary update. Histol. Histopathol. 2019, 18159. [Google Scholar] [CrossRef]
- Argani, P.; Antonescu, C.R.; Illei, P.B.; Lui, M.Y.; Timmons, C.F.; Newbury, R.; Reuter, V.E.; Garvin, A.J.; Perez-Atayde, A.R.; Fletcher, J.A.; et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: A distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am. J. Pathol. 2001, 159, 179–192. [Google Scholar] [CrossRef]
- Argani, P.; Lui, M.Y.; Couturier, J.; Bouvier, R.; Fournet, J.C.; Ladanyi, M. A novel CLTC-TFE3 gene fusion in pediatric renal adenocarcinoma with t(X;17)(p11.2;q23). Oncogene 2003, 22, 5374–5378. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Ladanyi, M. Renal carcinomas associated with Xp11.2 translocations / TFE3 gene fusions. In Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs, 1st ed.; Eble, J.N., Sauter, G., Epstein, J.I., Sesterhenn, I.A., Eds.; IARC Press: Lyon, France, 2004; pp. 37–38. [Google Scholar]
- Argani, P.; Olgac, S.; Tickoo, S.K.; Goldfischer, M.; Moch, H.; Chan, D.Y.; Eble, J.N.; Bonsib, S.M.; Jimeno, M.; Lloreta, J.; et al. Xp11 translocation renal cell carcinoma in adults: Expanded clinical, pathologic, and genetic spectrum. Am. J. Surg. Pathol. 2007, 31, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Hicks, J.; De Marzo, A.M.; Albadine, R.; Illei, P.B.; Ladanyi, M.; Reuter, V.E.; Netto, G.J. Xp11 translocation renal cell carcinoma (RCC): Extended immunohistochemical profile emphasizing novel RCC markers. Am. J. Surg. Pathol. 2010, 34, 1295–1303. [Google Scholar] [CrossRef]
- Ellis, C.L.; Eble, J.N.; Subhawong, A.P.; Martignoni, G.; Zhong, M.; Ladanyi, M.; Epstein, J.I.; Netto, G.J.; Argani, P. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: Impact of fusion subtype, age, and stage. Mod. Pathol. 2014, 27, 875–886. [Google Scholar] [CrossRef]
- Argani, P. MiT family translocation renal cell carcinoma. Semin. Diagn. Pathol. 2015, 32, 103–113. [Google Scholar] [CrossRef]
- Argani, P.; Zhong, M.; Reuter, V.E.; Fallon, J.T.; Epstein, J.I.; Netto, G.J.; Antonescu, C.R. TFE3-Fusion Variant Analysis Defines Specific Clinicopathologic Associations Among Xp11 Translocation Cancers. Am. J. Surg. Pathol. 2016, 40, 723–737. [Google Scholar] [CrossRef]
- Hayes, M.; Peckova, K.; Martinek, P.; Hora, M.; Kalusova, K.; Straka, L.; Daum, O.; Kokoskova, B.; Rotterova, P.; Pivovarcikova, K.; et al. Molecular-genetic analysis is essential for accurate classification of renal carcinoma resembling Xp11.2 translocation carcinoma. Virchows Arch. 2015, 466, 313–322. [Google Scholar] [CrossRef]
- Kato, I.; Furuya, M.; Baba, M.; Kameda, Y.; Yasuda, M.; Nishimoto, K.; Oyama, M.; Yamasaki, T.; Ogawa, O.; Niino, H.; et al. RBM10-TFE3 renal cell carcinoma characterised by paracentric inversion with consistent closely split signals in break-apart fluorescence in-situ hybridisation: Study of 10 cases and a literature review. Histopathology 2019, 75, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Malouf, G.G.; Su, X.; Yao, H.; Gao, J.; Xiong, L.; He, Q.; Comperat, E.; Couturier, J.; Molinie, V.; Escudier, B.; et al. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clin. Cancer Res. 2014, 20, 4129–4140. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; du Parcq, J.; Larkin, J.M.; Fisher, C.; Livni, N. Metastatic renal mucinous tubular and spindle cell carcinoma. Atypical behavior of a rare, morphologically bland tumor. Ann. Diagn. Pathol. 2012, 16, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, J.; Amin, M.B.; Selbs, E.; Turi, G.K.; Paner, G.P.; Reuter, V.E. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid change. Am. J. Surg. Pathol. 2009, 33, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Bulimbasic, S.; Ljubanovic, D.; Sima, R.; Michal, M.; Hes, O.; Kuroda, N.; Persec, Z. Aggressive high-grade mucinous tubular and spindle cell carcinoma. Hum. Pathol. 2009, 40, 906–907. [Google Scholar] [CrossRef] [PubMed]
- Paner, G.P.; Srigley, J.R.; Radhakrishnan, A.; Cohen, C.; Skinnider, B.F.; Tickoo, S.K.; Young, A.N.; Amin, M.B. Immunohistochemical analysis of mucinous tubular and spindle cell carcinoma and papillary renal cell carcinoma of the kidney: Significant immunophenotypic overlap warrants diagnostic caution. Am J Surg. Pathol. 2006, 30, 13–19. [Google Scholar] [CrossRef]
- Ren, Q.; Wang, L.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Sirintrapun, S.J.; Tickoo, S.K.; Reuter, V.E.; Chen, Y.B. Distinct Genomic Copy Number Alterations Distinguish Mucinous Tubular and Spindle Cell Carcinoma of the Kidney From Papillary Renal Cell Carcinoma With Overlapping Histologic Features. Am. J. Surg. Pathol. 2018, 42, 767–777. [Google Scholar] [CrossRef]
- Peckova, K.; Martinek, P.; Sperga, M.; Montiel, D.P.; Daum, O.; Rotterova, P.; Kalusova, K.; Hora, M.; Pivovarcikova, K.; Rychly, B.; et al. Mucinous spindle and tubular renal cell carcinoma: Analysis of chromosomal aberration pattern of low-grade, high-grade, and overlapping morphologic variant with papillary renal cell carcinoma. Ann. Diagn. Pathol. 2015, 19, 226–231. [Google Scholar] [CrossRef]
- Sadimin, E.T.; Chen, Y.B.; Wang, L.; Argani, P.; Epstein, J.I. Chromosomal abnormalities of high-grade mucinous tubular and spindle cell carcinoma of the kidney. Histopathology 2017, 71, 719–724. [Google Scholar] [CrossRef]
- Cossu-Rocca, P.; Eble, J.N.; Delahunt, B.; Zhang, S.; Martignoni, G.; Brunelli, M.; Cheng, L. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod. Pathol. 2006, 19, 488–493. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, X.J.; Lopez, J.I.; Shah, R.B.; Hes, O.; Shen, S.S.; Li, R.; Yang, Y.; Lin, F.; Elson, P.; et al. Renal tubulocystic carcinoma is closely related to papillary renal cell carcinoma: Implications for pathologic classification. Am. J. Surg. Pathol. 2009, 33, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Jones, C.L.; Williamson, S.R.; Eble, J.N.; Grignon, D.J.; Zhang, S.; Wang, M.; Baldridge, L.A.; Wang, L.; Montironi, R.; et al. Tubulocystic renal cell carcinoma is an entity that is immunohistochemically and genetically distinct from papillary renal cell carcinoma. Histopathology 2016, 68, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Zhou, M.; Hes, O.; Shen, S.; Li, R.; Lopez, J.; Shah, R.B.; Yang, Y.; Chuang, S.T.; Lin, F.; et al. Tubulocystic carcinoma of the kidney: Clinicopathologic and molecular characterization. Am. J. Surg. Pathol. 2008, 32, 177–187. [Google Scholar] [CrossRef]
- Sarungbam, J.; Mehra, R.; Tomlins, S.A.; Smith, S.C.; Jayakumaran, G.; Al-Ahmadie, H.; Gopalan, A.; Sirintrapun, S.J.; Fine, S.W.; Zhang, Y.; et al. Tubulocystic renal cell carcinoma: A distinct clinicopathologic entity with a characteristic genomic profile. Mod. Pathol. 2019, 32, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussain, T.O.; Cheng, L.; Zhang, S.; Epstein, J.I. Tubulocystic carcinoma of the kidney with poorly differentiated foci: A series of 3 cases with fluorescence in situ hybridization analysis. Hum. Pathol. 2013, 44, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Trpkov, K.; Chen, Y.B.; Mehra, R.; Sirohi, D.; Ohe, C.; Cani, A.K.; Hovelson, D.H.; Omata, K.; McHugh, J.B.; et al. Tubulocystic Carcinoma of the Kidney With Poorly Differentiated Foci: A Frequent Morphologic Pattern of Fumarate Hydratase-deficient Renal Cell Carcinoma. Am. J. Surg. Pathol. 2016, 40, 1457–1472. [Google Scholar] [CrossRef]
- Kuroda, N.; Ohe, C.; Mikami, S.; Hes, O.; Michal, M.; Brunelli, M.; Martignoni, G.; Sato, Y.; Yoshino, T.; Kakehi, Y.; et al. Review of acquired cystic disease-associated renal cell carcinoma with focus on pathobiological aspects. Histol. Histopathol. 2011, 26, 1215–1218. [Google Scholar] [CrossRef]
- Kuroda, N.; Shiotsu, T.; Hes, O.; Michal, M.; Shuin, T.; Lee, G.H. Acquired cystic disease-associated renal cell carcinoma with gain of chromosomes 3, 7, and 16, gain of chromosome X, and loss of chromosome Y. Med. Mol. Morphol. 2010, 43, 231–234. [Google Scholar] [CrossRef]
- Kuroda, N.; Tamura, M.; Hamaguchi, N.; Mikami, S.; Pan, C.C.; Brunelli, M.; Martignoni, G.; Hes, O.; Michal, M.; Lee, G.H. Acquired cystic disease-associated renal cell carcinoma with sarcomatoid change and rhabdoid features. Ann. Diagn. Pathol. 2011, 15, 462–466. [Google Scholar] [CrossRef]
- Kuroda, N.; Yamashita, M.; Kakehi, Y.; Hes, O.; Michal, M.; Lee, G.H. Acquired cystic disease-associated renal cell carcinoma: An immunohistochemical and fluorescence in situ hybridization study. Med. Mol. Morphol. 2011, 44, 228–232. [Google Scholar] [CrossRef]
- Cossu-Rocca, P.; Eble, J.N.; Zhang, S.; Martignoni, G.; Brunelli, M.; Cheng, L. Acquired cystic disease-associated renal tumors: An immunohistochemical and fluorescence in situ hybridization study. Mod. Pathol. 2006, 19, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.; Yusenko, M.V.; Nagy, A.; Kovacs, G. Oligoarray comparative genomic hybridization of renal cell tumors that developed in patients with acquired cystic renal disease. Hum. Pathol. 2010, 41, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Ohe, C.; Smith, S.C.; Sirohi, D.; Divatia, M.; de Peralta-Venturina, M.; Paner, G.P.; Agaimy, A.; Amin, M.B.; Argani, P.; Chen, Y.B.; et al. Reappraisal of Morphologic Differences Between Renal Medullary Carcinoma, Collecting Duct Carcinoma, and Fumarate Hydratase-deficient Renal Cell Carcinoma. Am. J. Surg. Pathol. 2018, 42, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Carlo, M.I.; Khan, H.; Nanjangud, G.J.; Rana, S.; Cimera, R.; Zhang, Y.; Hakimi, A.A.; Verma, A.K.; Al-Ahmadie, H.A.; et al. Distinctive mechanisms underlie the loss of SMARCB1 protein expression in renal medullary carcinoma: Morphologic and molecular analysis of 20 cases. Mod. Pathol. 2019, 32, 1329–1343. [Google Scholar] [CrossRef]
- Calderaro, J.; Masliah-Planchon, J.; Richer, W.; Maillot, L.; Maille, P.; Mansuy, L.; Bastien, C.; de la Taille, A.; Boussion, H.; Charpy, C.; et al. Balanced Translocations Disrupting SMARCB1 Are Hallmark Recurrent Genetic Alterations in Renal Medullary Carcinomas. Eur. Urol. 2016, 69, 1055–1061. [Google Scholar] [CrossRef]
- Carlo, M.I.; Chaim, J.; Patil, S.; Kemel, Y.; Schram, A.M.; Woo, K.; Coskey, D.; Nanjangud, G.J.; Voss, M.H.; Feldman, D.R.; et al. Genomic Characterization of Renal Medullary Carcinoma and Treatment Outcomes. Clin. Genitourin. Cancer 2017, 15, e987–e994. [Google Scholar] [CrossRef]
- Calderaro, J.; Moroch, J.; Pierron, G.; Pedeutour, F.; Grison, C.; Maille, P.; Soyeux, P.; de la Taille, A.; Couturier, J.; Vieillefond, A.; et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology 2012, 61, 428–435. [Google Scholar] [CrossRef]
- Liu, Q.; Galli, S.; Srinivasan, R.; Linehan, W.M.; Tsokos, M.; Merino, M.J. Renal medullary carcinoma: Molecular, immunohistochemistry, and morphologic correlation. Am. J. Surg. Pathol. 2013, 37, 368–374. [Google Scholar] [CrossRef]
- Cheng, J.X.; Tretiakova, M.; Gong, C.; Mandal, S.; Krausz, T.; Taxy, J.B. Renal medullary carcinoma: Rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod. Pathol. 2008, 21, 647–652. [Google Scholar] [CrossRef]
- Rao, P.; Tannir, N.M.; Tamboli, P. Expression of OCT3/4 in renal medullary carcinoma represents a potential diagnostic pitfall. Am. J. Surg. Pathol. 2012, 36, 583–588. [Google Scholar] [CrossRef]
- Sirohi, D.; Smith, S.C.; Ohe, C.; Colombo, P.; Divatia, M.; Dragoescu, E.; Rao, P.; Hirsch, M.S.; Chen, Y.B.; Mehra, R.; et al. Renal cell carcinoma, unclassified with medullary phenotype: Poorly differentiated adenocarcinomas overlapping with renal medullary carcinoma. Hum. Pathol. 2017, 67, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Smith, S.C.; Agaimy, A.; Argani, P.; Comperat, E.M.; Delahunt, B.; Epstein, J.I.; Eble, J.N.; Grignon, D.J.; Hartmann, A.; et al. Collecting duct carcinoma versus renal medullary carcinoma: An appeal for nosologic and biological clarity. Am. J. Surg. Pathol. 2014, 38, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 2012, 44, 285–292. [Google Scholar] [CrossRef]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2010, 41, 805–814. [Google Scholar] [CrossRef] [PubMed]
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.; Sleddens, H.F.; Derkx, P.; Riviere, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef]
- Merino, M.J.; Linehan, W.M. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC)-associated renal cell carcinoma. In WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; Moch, H., Humphrey, P.A., Ulbright, T.M., Reuter, V.E., Eds.; International Agency for Research on Cancer: Lyon, France, 2016; Volume 8, pp. 25–26. [Google Scholar]
- Trpkov, K.; Hes, O.; Agaimy, A.; Bonert, M.; Martinek, P.; Magi-Galluzzi, C.; Kristiansen, G.; Luders, C.; Nesi, G.; Comperat, E.; et al. Fumarate Hydratase-deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am. J. Surg. Pathol. 2016, 40, 865–875. [Google Scholar] [CrossRef]
- Pivovarcikova, K.; Martinek, P.; Grossmann, P.; Trpkov, K.; Alaghehbandan, R.; Magi-Galluzzi, C.; Pane Foix, M.; Condom Mundo, E.; Berney, D.; Gill, A.; et al. Fumarate hydratase deficient renal cell carcinoma: Chromosomal numerical aberration analysis of 12 cases. Ann. Diagn. Pathol. 2019, 39, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.D.; Chan, E.; Fan, A.C.; Kunder, C.A.; Williamson, S.R.; Zhou, M.; Idrees, M.T.; Maclean, F.M.; Gill, A.J.; Kao, C.S. A Clinicopathologic and Molecular Analysis of Fumarate Hydratase-Deficient Renal Cell Carcinoma in 32 Patients. Am. J. Surg. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shyu, I.; Mirsadraei, L.; Wang, X.; Robila, V.; Mehra, R.; McHugh, J.B.; Chen, Y.B.; Udager, A.M.; Gill, A.J.; Cheng, L.; et al. Clues to recognition of fumarate hydratase-deficient renal cell carcinoma: Findings from cytologic and limited biopsy samples. Cancer Cytopathol. 2018. [Google Scholar] [CrossRef]
- Chen, Y.B.; Brannon, A.R.; Toubaji, A.; Dudas, M.E.; Won, H.H.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Frizzell, N.; Voss, M.H.; et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: Recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am. J. Surg. Pathol. 2014, 38, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Guillaud-Bataille, M.; Salleron, J.; Genestie, C.; Deveaux, S.; Slama, A.; de Paillerets, B.B.; Richard, S.; Benusiglio, P.R.; Ferlicot, S. Pattern multiplicity and fumarate hydratase (FH)/S-(2-succino)-cysteine (2SC) staining but not eosinophilic nucleoli with perinucleolar halos differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas from kidney tumors without FH gene alteration. Mod. Pathol. 2018, 31, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Harrison, W.J.; Andrici, J.; Maclean, F.; Madadi-Ghahan, R.; Farzin, M.; Sioson, L.; Toon, C.W.; Clarkson, A.; Watson, N.; Pickett, J.; et al. Fumarate Hydratase-deficient Uterine Leiomyomas Occur in Both the Syndromic and Sporadic Settings. Am. J. Surg. Pathol. 2016, 40, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tretiakova, M.S.; Troxell, M.L.; Osunkoya, A.O.; Fadare, O.; Sangoi, A.R.; Shen, S.S.; Lopez-Beltran, A.; Mehra, R.; Heider, A.; et al. Tuberous sclerosis-associated renal cell carcinoma: A clinicopathologic study of 57 separate carcinomas in 18 patients. Am. J. Surg. Pathol. 2014, 38, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; Jiang, Z.; Oliva, E.; Jozwiak, S.; Nussbaum, R.L.; et al. Renal cell carcinoma in tuberous sclerosis complex. Am. J. Surg. Pathol. 2014, 38, 895–909. [Google Scholar] [CrossRef] [PubMed]
- Trpkov, K.; Hes, O.; Bonert, M.; Lopez, J.I.; Bonsib, S.M.; Nesi, G.; Comperat, E.; Sibony, M.; Berney, D.M.; Martinek, P.; et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am. J. Surg. Pathol. 2016, 40, 60–71. [Google Scholar] [CrossRef]
- Mehra, R.; Vats, P.; Cao, X.; Su, F.; Lee, N.D.; Lonigro, R.; Premkumar, K.; Trpkov, K.; McKenney, J.K.; Dhanasekaran, S.M.; et al. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur. Urol. 2018, 74, 483–486. [Google Scholar] [CrossRef]
- Palsgrove, D.N.; Li, Y.; Pratilas, C.A.; Lin, M.T.; Pallavajjalla, A.; Gocke, C.; De Marzo, A.M.; Matoso, A.; Netto, G.J.; Epstein, J.I.; et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas Harbor TSC Mutations: Molecular Analysis Supports an Expanding Clinicopathologic Spectrum. Am. J. Surg. Pathol. 2018, 42, 1166–1181. [Google Scholar] [CrossRef]
- Parilla, M.; Kadri, S.; Patil, S.A.; Ritterhouse, L.; Segal, J.; Henriksen, K.J.; Antic, T. Are Sporadic Eosinophilic Solid and Cystic Renal Cell Carcinomas Characterized by Somatic Tuberous Sclerosis Gene Mutations? Am. J. Surg. Pathol. 2018, 42, 911–917. [Google Scholar] [CrossRef]
- Tretiakova, M.S. Eosinophilic solid and cystic renal cell carcinoma mimicking epithelioid angiomyolipoma: Series of 4 primary tumors and 2 metastases. Hum. Pathol. 2018, 80, 65–75. [Google Scholar] [CrossRef]
- Li, Y.; Reuter, V.E.; Matoso, A.; Netto, G.J.; Epstein, J.I.; Argani, P. Re-evaluation of 33 ‘unclassified’ eosinophilic renal cell carcinomas in young patients. Histopathology 2018, 72, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R. Renal cell carcinomas with a mesenchymal stromal component: What do we know so far? Pathology 2019, 51, 453–462. [Google Scholar] [CrossRef]
- Williamson, S.R.; Hornick, J.L.; Eble, J.N.; Gupta, N.S.; Rogers, C.G.; True, L.; Grignon, D.J.; Cheng, L. Renal cell carcinoma with angioleiomyoma-like stroma and clear cell papillary renal cell carcinoma: Exploring SDHB protein immunohistochemistry and the relationship to tuberous sclerosis complex. Hum. Pathol. 2018, 75, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Parilla, M.; Alikhan, M.; Al-Kawaaz, M.; Patil, S.; Kadri, S.; Ritterhouse, L.L.; Segal, J.; Fitzpatrick, C.; Antic, T. Genetic Underpinnings of Renal Cell Carcinoma With Leiomyomatous Stroma. Am. J. Surg. Pathol. 2019, 43, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Verkarre, V.; Mensah, A.; Leroy, X.; Sibony, M.; Vasiliu, V.; Comperat, E.; Richard, S.; Mejean, A. A Clinico-Pathologic Study of 17 Patients with Renal Cell Carcinoma Associated With Leiomyomatous Stroma Identifies a Strong Association With Tuberous Sclerosis. Lab. Invest 2015, 95, 266A. [Google Scholar]
- Jia, L.; Jayakumaran, G.; Al-Ahmadie, H.; Fine, S.W.; Gopalan, A.; Sirintrapun, S.J.; Tickoo, S.; Reuter, V.; Cheng, Y.B. Expanding the Morphologic Spectrum of Sporadic Renal Cell Carcinoma (RCC) Harboring Somatic TSC or MTOR Alterations: Analysis of 8 Cases with Clear Cytoplasm and Leiomyomatous Stroma. Mod. Pathol. 2019, 32, 78–79. [Google Scholar]
- Chen, Y.B.; Mirsadraei, L.; Jayakumaran, G.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Sirintrapun, S.J.; Tickoo, S.K.; Reuter, V.E. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma with Eosinophilic and Vacuolated Cytoplasm. Am. J. Surg. Pathol. 2019, 43, 121–131. [Google Scholar] [CrossRef]
- He, H.; Trpkov, K.; Martinek, P.; Isikci, O.T.; Maggi-Galuzzi, C.; Alaghehbandan, R.; Gill, A.J.; Tretiakova, M.; Lopez, J.I.; Williamson, S.R.; et al. “High-grade oncocytic renal tumor”: Morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018, 473, 725–738. [Google Scholar] [CrossRef]
- Trpkov, K.; Bonert, M.; Gao, Y.; Kapoor, A.; He, H.; Yilmaz, A.; Gill, A.J.; Williamson, S.R.; Comperat, E.; Tretiakova, M.; et al. High-grade oncocytic tumour (HOT) of kidney in a patient with tuberous sclerosis complex. Histopathology 2019, 75, 440–442. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Tickoo, S.K.; Jacobsen, A.; Sarungbam, J.; Sfakianos, J.P.; Sato, Y.; Morikawa, T.; Kume, H.; Fukayama, M.; Homma, Y.; et al. TCEB1-mutated renal cell carcinoma: A distinct genomic and morphological subtype. Mod. Pathol. 2015, 28, 845–853. [Google Scholar] [CrossRef]
- Favazza, L.; Chitale, D.A.; Barod, R.; Rogers, C.G.; Kalyana-Sundaram, S.; Palanisamy, N.; Gupta, N.S.; Williamson, S.R. Renal cell tumors with clear cell histology and intact VHL and chromosome 3p: A histological review of tumors from the Cancer Genome Atlas database. Mod. Pathol. 2017, 30, 1603–1612. [Google Scholar] [CrossRef]
- Hirsch, M.S.; Barletta, J.A.; Gorman, M.; Dal Cin, P. Renal Cell Carcinoma With Monosomy 8 and CAIX Expression: A Distinct Entity or Another Member or the Clear Cell Tubulopapillary RCC/RAT Family? Mod. Pathol. 2015, 28, 229A. [Google Scholar]
- Williamson, S.R.; Grignon, D.J.; Cheng, L.; Favazza, L.; Gondim, D.D.; Carskadon, S.; Gupta, N.S.; Chitale, D.A.; Kalyana-Sundaram, S.; Palanisamy, N. Renal Cell Carcinoma With Chromosome 6p Amplification Including the TFEB Gene: A Novel Mechanism of Tumor Pathogenesis? Am. J. Surg. Pathol. 2017, 41, 287–298. [Google Scholar] [CrossRef]
- Argani, P.; Reuter, V.E.; Zhang, L.; Sung, Y.S.; Ning, Y.; Epstein, J.I.; Netto, G.J.; Antonescu, C.R. TFEB-amplified Renal Cell Carcinomas: An Aggressive Molecular Subset Demonstrating Variable Melanocytic Marker Expression and Morphologic Heterogeneity. Am. J. Surg. Pathol. 2016, 40, 1484–1495. [Google Scholar] [CrossRef]
- Gupta, S.; Johnson, S.H.; Vasmatzis, G.; Porath, B.; Rustin, J.G.; Rao, P.; Costello, B.A.; Leibovich, B.C.; Thompson, R.H.; Cheville, J.C.; et al. TFEB-VEGFA (6p21.1) co-amplified renal cell carcinoma: A distinct entity with potential implications for clinical management. Mod. Pathol. 2017, 30, 998–1012. [Google Scholar] [CrossRef]
- Peckova, K.; Vanecek, T.; Martinek, P.; Spagnolo, D.; Kuroda, N.; Brunelli, M.; Vranic, S.; Djuricic, S.; Rotterova, P.; Daum, O.; et al. Aggressive and nonaggressive translocation t(6;11) renal cell carcinoma: Comparative study of 6 cases and review of the literature. Ann. Diagn. Pathol. 2014, 18, 351–357. [Google Scholar] [CrossRef]
- Skala, S.L.; Xiao, H.; Udager, A.M.; Dhanasekaran, S.M.; Shukla, S.; Zhang, Y.; Landau, C.; Shao, L.; Roulston, D.; Wang, L.; et al. Detection of 6 TFEB-amplified renal cell carcinomas and 25 renal cell carcinomas with MITF translocations: Systematic morphologic analysis of 85 cases evaluated by clinical TFE3 and TFEB FISH assays. Mod. Pathol. 2018, 31, 179–197. [Google Scholar] [CrossRef]
- Durinck, S.; Stawiski, E.W.; Pavia-Jimenez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; Zhang, N.; Toffessi-Tcheuyap, V.; Nguyen, T.T.; Pahuja, K.B.; et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef]
- Gupta, S.; Argani, P.; Jungbluth, A.A.; Chen, Y.B.; Tickoo, S.K.; Fine, S.W.; Gopalan, A.; Al-Ahmadie, H.A.; Sirintrapun, S.J.; Sanchez, A.; et al. TFEB Expression Profiling in Renal Cell Carcinomas: Clinicopathologic Correlations. Am. J. Surg. Pathol. 2019, 43, 1445–1461. [Google Scholar] [CrossRef]
- Andeen, N.K.; Qu, X.; Antic, T.; Tykodi, S.S.; Fang, M.; Tretiakova, M.S. Clinical Utility of Chromosome Genomic Array Testing for Unclassified and Advanced-Stage Renal Cell Carcinomas. Arch. Pathol. Lab. Med. 2019, 143, 494–504. [Google Scholar] [CrossRef]
- Moch, H.; Amin, M.; Argani, P.; Cheville, J.; Delahunt, B.; Martignoni, G.; Srigley, J.; Tan, P.; Tickoo, S. Renal cell tumors. In WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; Moch, H., Humphrey, P.A., Ulbright, T.M., Reuter, V.E., Eds.; International Agency for Research on Cancer: Lyon, France, 2016; pp. 14–17. [Google Scholar]
- Debelenko, L.V.; Raimondi, S.C.; Daw, N.; Shivakumar, B.R.; Huang, D.; Nelson, M.; Bridge, J.A. Renal cell carcinoma with novel VCL-ALK fusion: New representative of ALK-associated tumor spectrum. Mod. Pathol. 2011, 24, 430–442. [Google Scholar] [CrossRef]
- Sugawara, E.; Togashi, Y.; Kuroda, N.; Sakata, S.; Hatano, S.; Asaka, R.; Yuasa, T.; Yonese, J.; Kitagawa, M.; Mano, H.; et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: Large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer 2012, 118, 4427–4436. [Google Scholar] [CrossRef]
- Sukov, W.R.; Hodge, J.C.; Lohse, C.M.; Akre, M.K.; Leibovich, B.C.; Thompson, R.H.; Cheville, J.C. ALK alterations in adult renal cell carcinoma: Frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod. Pathol. 2012, 25, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Bodokh, Y.; Ambrosetti, D.; Kubiniek, V.; Tibi, B.; Durand, M.; Amiel, J.; Pertuit, M.; Barlier, A.; Pedeutour, F. ALK-TPM3 rearrangement in adult renal cell carcinoma: Report of a new case showing loss of chromosome 3 and literature review. Cancer Genet. 2018, 221, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Cajaiba, M.M.; Jennings, L.J.; George, D.; Perlman, E.J. Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer 2016, 55, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Cajaiba, M.M.; Jennings, L.J.; Rohan, S.M.; Perez-Atayde, A.R.; Marino-Enriquez, A.; Fletcher, J.A.; Geller, J.I.; Leuer, K.M.; Bridge, J.A.; Perlman, E.J. ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer 2016, 55, 442–451. [Google Scholar] [CrossRef]
- Hodge, J.C.; Pearce, K.E.; Sukov, W.R. Distinct ALK-rearranged and VCL-negative papillary renal cell carcinoma variant in two adults without sickle cell trait. Mod. Pathol. 2013, 26, 604–605. [Google Scholar] [CrossRef][Green Version]
- Jeanneau, M.; Gregoire, V.; Desplechain, C.; Escande, F.; Tica, D.P.; Aubert, S.; Leroy, X. ALK rearrangements-associated renal cell carcinoma (RCC) with unique pathological features in an adult. Pathol. Res. Pr. 2016, 212, 1064–1066. [Google Scholar] [CrossRef]
- Kuroda, N.; Liu, Y.; Tretiakova, M.; Ulamec, M.; Takeuchi, K.; Przybycin, C.; Magi-Galluzzi, C.; Agaimy, A.; Yilmaz, A.; Trpkov, K.; et al. Clinicopathological Study of Seven Cases of ALK-positive Renal Tumor Identification of New Fusion Partners including CLIP1 and KIF5B Genes. Mod. Pathol. 2019, 32, 85. [Google Scholar]
- Kuroda, N.; Sugawara, E.; Kusano, H.; Yuba, Y.; Yorita, K.; Takeuchi, K. A review of ALK-rearranged renal cell carcinomas with a focus on clinical and pathobiological aspects. Pol. J. Pathol. 2018, 69, 109–113. [Google Scholar] [CrossRef]
- Kusano, H.; Togashi, Y.; Akiba, J.; Moriya, F.; Baba, K.; Matsuzaki, N.; Yuba, Y.; Shiraishi, Y.; Kanamaru, H.; Kuroda, N.; et al. Two Cases of Renal Cell Carcinoma Harboring a Novel STRN-ALK Fusion Gene. Am. J. Surg. Pathol. 2016, 40, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, J.W.; Suh, J.H.; Nam, K.H.; Moon, K.C. ALK-Positive Renal Cell Carcinoma in a Large Series of Consecutively Resected Korean Renal Cell Carcinoma Patients. Korean J. Pathol. 2013, 47, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Marino-Enriquez, A.; Ou, W.B.; Weldon, C.B.; Fletcher, J.A.; Perez-Atayde, A.R. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer 2011, 50, 146–153. [Google Scholar] [CrossRef]
- Pal, S.K.; Bergerot, P.; Dizman, N.; Bergerot, C.; Adashek, J.; Madison, R.; Chung, J.H.; Ali, S.M.; Jones, J.O.; Salgia, R. Responses to Alectinib in ALK-rearranged Papillary Renal Cell Carcinoma. Eur. Urol. 2018, 74, 124–128. [Google Scholar] [CrossRef]
- Smith, N.E.; Deyrup, A.T.; Marino-Enriquez, A.; Fletcher, J.A.; Bridge, J.A.; Illei, P.B.; Netto, G.J.; Argani, P. VCL-ALK renal cell carcinoma in children with sickle-cell trait: The eighth sickle-cell nephropathy? Am. J. Surg. Pathol. 2014, 38, 858–863. [Google Scholar] [CrossRef]
- Thorner, P.S.; Shago, M.; Marrano, P.; Shaikh, F.; Somers, G.R. TFE3-positive renal cell carcinomas are not always Xp11 translocation carcinomas: Report of a case with a TPM3-ALK translocation. Pathol. Res. Pr. 2016, 212, 937–942. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Y.; Jiang, Y.; Zhang, W.; Li, Y. Genetic analysis and clinicopathological features of ALK-rearranged renal cell carcinoma in a large series of resected Chinese renal cell carcinoma patients and literature review. Histopathology 2017, 71, 53–62. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, J.; Han, Y.; Wang, X.; Ye, X.; Meng, Y.; Parwani, A.; Han, Z.; Feng, Q.; Huang, K. Integrative Analysis of Histopathological Images and Genomic Data Predicts Clear Cell Renal Cell Carcinoma Prognosis. Cancer Res. 2017, 77, e91–e100. [Google Scholar] [CrossRef]
- Shao, W.; Han, Z.; Cheng, J.; Cheng, L.; Wang, T.; Sun, L.; Lu, Z.; Zhang, J.; Zhang, D.; Huang, K. Integrative analysis of pathological images and multi-dimensional genomic data for early-stage cancer prognosis. Ieee Trans. Med. Imaging 2019. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | IHC | Mutation | Method |
|---|---|---|---|
| CCRCC | Carbonic Anhydrase (CA) IX, Vimentin | VHL (von Hippel Lindau) inactivation | Sequencing (NGS/classical)/methylation specific PCR (polymerase chain reaction) |
| MCRCNLMP | NS | VHL | NR |
| PRCC type 1 hereditary syndrome | CK7, AMACR | MET | Sequencing (NGS/classical) |
| PRCC type 1 conventional type | CK7, AMACR | Gain of 7,17 | aCGH/FISH |
| PRCC type 2 | NS | NS | |
| OPRCC | AMACR, Vimentin | KRAS | Sequencing (NGS/classical) |
| FH-deficient RCC | FH, 2SC | FH mutation/LOH analysis | Sequencing (NGS/classical)/fragment analysis |
| ChRCCC | CK7, CD117 | NS | |
| “Hybrid” On/Ch tumors | NS | FLCN ** | Sequencing (NGS/classical) |
| Oncocytoma | CK7, CD117 | NS | |
| Clear cell PRCC | CK7, AMACR | VHL | Sequencing (NGS/classical) |
| MiT RCC | TFE3, Cathepsin K | TFE3, TFEB | FISH/NGS |
| MTSCC | AMACR, EMA | CNV pattern analysis | aCGH |
| TC-RCC | CK7, AMACR | CNV pattern analysis | aCGH |
| ACD-associated RCC | CK7, AMACR | NS | |
| RMC | INI1 | SMARCB1 | Sequencing (NGS/classical) |
| CDC | 34betaE12, Ck7 | NS | |
| SDH-deficient renal cell carcinoma | SDHB | SDHB | Sequencing (NGS, classical)/IHC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaghehbandan, R.; Perez Montiel, D.; Luis, A.S.; Hes, O. Molecular Genetics of Renal Cell Tumors: A Practical Diagnostic Approach. Cancers 2020, 12, 85. https://doi.org/10.3390/cancers12010085
Alaghehbandan R, Perez Montiel D, Luis AS, Hes O. Molecular Genetics of Renal Cell Tumors: A Practical Diagnostic Approach. Cancers. 2020; 12(1):85. https://doi.org/10.3390/cancers12010085
Chicago/Turabian StyleAlaghehbandan, Reza, Delia Perez Montiel, Ana Silvia Luis, and Ondrej Hes. 2020. "Molecular Genetics of Renal Cell Tumors: A Practical Diagnostic Approach" Cancers 12, no. 1: 85. https://doi.org/10.3390/cancers12010085
APA StyleAlaghehbandan, R., Perez Montiel, D., Luis, A. S., & Hes, O. (2020). Molecular Genetics of Renal Cell Tumors: A Practical Diagnostic Approach. Cancers, 12(1), 85. https://doi.org/10.3390/cancers12010085