RNA Sequencing of Collecting Duct Renal Cell Carcinoma Suggests an Interaction between miRNA and Target Genes and a Predominance of Deregulated Solute Carrier Genes


 ,
,  , , ,
, , ,  ,
, 

Abstract
1. Introduction
2. Results
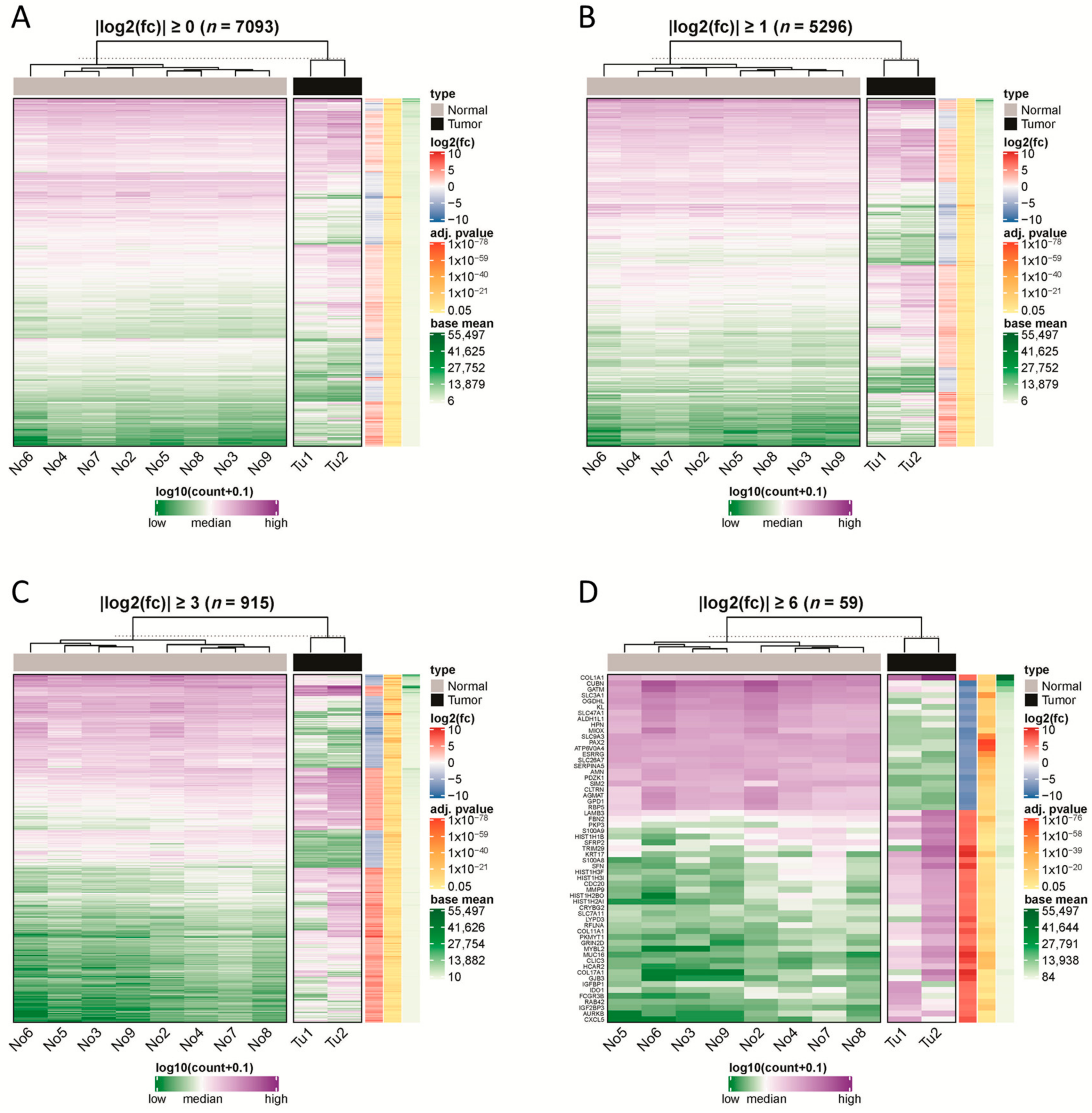
2.1. RNA Sequencing Revealed Up- and Downregulated Genes
2.2. Pathway Analyses
2.3. Investigation of SNPs and Mutations
2.4. Correlations of miRNAs and Target mRNA Expression
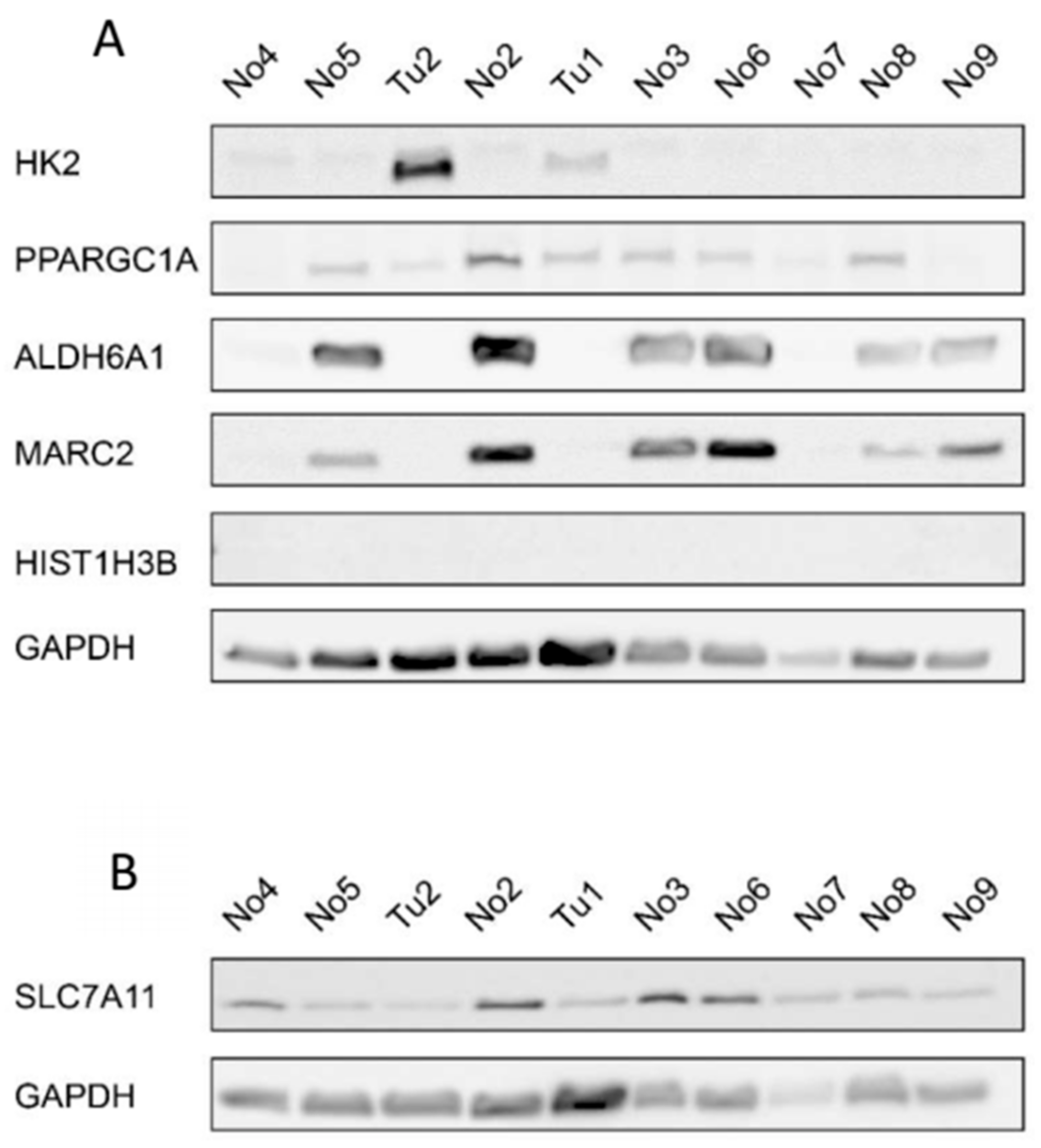
2.5. Protein Expression of miRNA Target Genes
2.6. Solute Carrier Genes
2.7. Survival Analysis of Deregulated Genes
3. Discussion
4. Material and Methods
4.1. Patients and Tumor Material
4.2. RNA and Protein Isolation
4.3. Quantitative Real-Time PCR
4.4. Western Blotting
4.5. RNA Sequencing Data Processing
4.6. Differential Gene Expression Analysis
4.7. Gene Enrichment Analyses
4.8. Survival Analysis
4.9. miRNA Target Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDC | collecting duct renal cell carcinoma |
| SLC | solute carrier |
| SLC7A11 | solute carrier family 7, member 11 |
| SLC47A1 | solute carrier family 47, member 1 |
References
- Ciszewski, S.; Jakimow, A.; Smolska-Ciszewska, B. Collecting (Bellini) duct carcinoma: A clinical study of a rare tumour and review of the literature. Can. Urol. Assoc. J. 2015, 9, E589–E593. [Google Scholar] [CrossRef]
- Ito, K. Recent advances in the systemic treatment of metastatic non-clear cell renal cell carcinomas. Int. J. Urol. 2019, 26, 868–877. [Google Scholar] [CrossRef]
- Pagani, F.; Colecchia, M.; Sepe, P.; Apollonio, G.; Claps, M.; Verzoni, E.; de Braud, F.; Procopio, G. Collecting ducts carcinoma: An orphan disease. Literature overview and future perspectives. Cancer Treat. Rev. 2019, 79, e101891. [Google Scholar] [CrossRef]
- Mancilla-Jimenez, R.; Stanley, R.J.; Blath, R.A. Papillary renal cell carcinoma: A clinical, radiologic, and pathologic study of 34 cases. Cancer 1976, 38, 2469–2480. [Google Scholar] [CrossRef]
- Cromie, W.J.; Davis, C.J.; DeTure, F.A. Atypical carcinoma of kidney: Possibly originating from collecting duct epithelium. Urology 1979, 1, 315–317. [Google Scholar] [CrossRef]
- Malouf, G.G.; Compérat, E.; Yao, H.; Mouawad, R.; Lindner, V.; Rioux-Leclercq, N.; Verkarre, V.; Leroy, X.; Dainese, L.; Classe, M.; et al. Unique transcriptomic profile of collecting duct carcinomas relative to upper tract urothelial carcinomas and other kidney carcinomas. Sci. Rep. 2016, 6, e30988. [Google Scholar] [CrossRef]
- Pal, S.K.; Choueiri, T.K.; Wang, K.; Khaira, D.; Karam, J.A.; Van Allen, E.; Palma, N.A.; Stein, M.N.; Johnson, A.; Squillace, R.; et al. Characterization of clinical cases of collecting duct carcinoma of the kidney assessed by comprehensive genomic profiling. Eur. Urol. 2015. [Google Scholar] [CrossRef]
- Wang, J.; Papanicolau-Sengos, A.; Chintala, S.; Wei, L.; Liu, B.; Hu, Q.; Miles, K.M.; Conroy, J.M.; Glenn, S.T.; Costantini, M.; et al. Collecting duct carcinoma of the kidney is associated with CDKN2A deletion and SLC family gene up-regulation. Oncotarget 2016, 7, 29901–29915. [Google Scholar] [CrossRef]
- Kröger, N.; Merseburger, A.S.; Bedke, J. Current recommendations for the systemic treatment of metastatic renal cell carcinoma. Aktuelle Urol. 2019. [Google Scholar] [CrossRef]
- Stukalin, I.; Wells, J.C.; Graham, J.; Yuasa, T.; Beuselinck, B.; Kollmansberger, C.; Ernst, D.S.; Agarwal, N.; Le, T.; Donskov, F.; et al. Real-world outcomes of nivolumab and cabozantinib in metastatic renal cell carcinoma: Results from the international metastatic renal cell carcinoma database consortium. Curr. Oncol. 2019, 26, e175–e179. [Google Scholar] [CrossRef]
- Chintala, S.; Pili, R. Genomic profiling of collecting duct renal carcinoma. Aging 2016, 8, 2260–2261. [Google Scholar] [CrossRef][Green Version]
- Nykjaer, A.; Fyfe, J.C.; Kozyraki, R.; Leheste, J.-R.; Jacobsen, C.; Nielsen, M.S.; Verroust, P.J.; Aminoff, M.; de la Chapelle, A.; Moestrup, S.K.; et al. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D3. Proc. Natl. Acad. Sci. USA 2001, 98, 13895–13900. [Google Scholar] [CrossRef]
- Kim, S.; Wong, P.; Coulombe, P.A. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006, 441, 362–365. [Google Scholar] [CrossRef]
- Lalli, A.; Tilakaratne, W.M.; Ariyawardana, A.; Fitchett, C.; Leigh, I.M.; Hagi-Pavli, E.; Cruchley, A.T.; Parkinson, E.K.; Teh, M.T.; Fortune, F.; et al. An altered keratinocyte phenotype in oral submucous fibrosis: Correlation of keratin K17 expression with disease severity. J. Oral. Pathol. Med. 2008, 37, 211–220. [Google Scholar] [CrossRef]
- Khanom, R.; Nguyen, C.T.; Kayamori, K.; Zhao, X.; Morita, K.; Miki, Y.; Katsube, K.; Yamaguchi, A.; Sakamoto, K. Keratin 17 is induced in oral cancer and facilitates tumor growth. PLoS ONE 2016, 11, e0161163. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, e3698. [Google Scholar] [CrossRef]
- Gremel, G.; Djureinovic, D.; Niinivirta, M.; Laird, A.; Ljungqvist, O.; Johannesson, H.; Bergman, J.; Edqvist, P.H.; Navani, S.; Khan, N.; et al. A systematic search strategy identifies cubilin as independent prognostic marker for renal cell carcinoma. BMC Cancer 2017, 17, e9. [Google Scholar] [CrossRef]
- Niinivirta, M.; Enblad, G.; Edqvist, P.H.; Pontén, F.; Dragomir, A.; Ullenhag, G.J. Tumoral cubilin is a predictive marker for treatment of renal cancer patients with sunitinib and sorafenib. J. Cancer Res. Clin. Oncol. 2017, 143, 961–970. [Google Scholar] [CrossRef][Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. New Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Schrödter, S.; Braun, M.; Syring, I.; Klümper, N.; Deng, M.; Schmidt, D.; Perner, S.; Müller, S.C.; Ellinger, J. Identification of the dopamine transporter SLC6A3 as a biomarker for patients with renal cell carcinoma. Mol. Cancer 2016, 15, e10. [Google Scholar] [CrossRef]
- Hansson, J.; Lindgren, D.; Nilsson, H.; Johansson, E.; Johansson, M.; Gustavsson, L.; Axelson, H. Overexpression of functional SLC6A3 in clear cell renal cell carcinoma. Clin. Cancer Res. 2017, 23, 2105–2115. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P.; Methner, A. Regulation of xCT expression and system x (c) (-) function in neuronal cells. Amino Acids 2012, 42, 171–179. [Google Scholar] [CrossRef]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef]
- Staud, F.; Cerveny, L.; Ahmadimoghaddam, D.; Ceckova, M. Multidrug and toxin extrusion proteins (MATE/SLC47); role in pharmacokinetics. Int. J. Biochem. Cell Biol. 2013, 45, 2007–2011. [Google Scholar] [CrossRef]
- Shuch, B.; Linehan, W.M.; Srinivasan, R. Aerobic glycolysis: A novel target in kidney cancer. Expert Rev. Anticancer Ther. 2013, 13, 711–719. [Google Scholar] [CrossRef]
- Smith, T.A. Mammalian hexokinases and their abnormal expression in cancer. Br. J. Biomed. Sci. 2000, 57, 170–178. [Google Scholar]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Nam, H.Y.; Chandrashekar, D.S.; Kundu, A.; Shelar, S.; Kho, E.Y.; Sonpavde, G.; Naik, G.; Ghatalia, P.; Livi, C.B.; Varambally, S.; et al. Integrative epigenetic and gene expression analysis of renal tumor progression to metastasis. Mol. Cancer Res. 2019, 17, 84–96. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1-alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1α is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef]
- Kedishvili, N.Y.; Popov, K.M.; Rougraff, P.M.; Zhao, Y.; Crabb, D.W.; Harris, R.A. CoA-dependent methylmalonate-semialdehyde dehydrogenase, a unique member of the aldehyde dehydrogenase superfamily: CDNA cloning, evolutionary relationships, and tissue distribution. J Biol. Chem. 1992, 267, 19724–19729. [Google Scholar]
- Zhang, B.; Wu, Q.; Wang, Z.; Xu, R.; Hu, X.; Sun, Y.; Wang, Q.; Ju, F.; Ren, S.; Zhang, C.; et al. The promising novel biomarkers and candidate small molecule drugs in kidney renal clear cell carcinoma: Evidence from bioinformatics analysis of high-throughput data. Mol. Genet. Genom. Med. 2019, 7, e607. [Google Scholar] [CrossRef]
- Kotthaus, J.; Wahl, B.; Havemeyer, A.; Kotthaus, J.; Schade, D.; Garbe-Schonberg, D.; Mendel, R.; Bittner, F.; Clement, B. Reduction of N(omega)-hydroxy-L-arginine by the mitochondrial amidoxime reducing component (mARC). Biochem. J. 2011, 433, 383–391. [Google Scholar] [CrossRef]
- Rixen, S.; Havemeyer, A.; Tyl-Bielicka, A.; Pysniak, K.; Gajewska, M.; Kulecka, M.; Ostrowski, J.; Mikula, M.; Clement, B. Mitochondrial amidoxime-reducing component 2 (mARC2) has a significant role in N-reductive activity and energy metabolism. J. Biol. Chem. 2019, 294, 17593–17602. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Zhang, Z.; Zhao, Z.; Lv, Q. A four-gene signature predicts the efficacy of paclitaxel-based neoadjuvant therapy in human epidermal growth factor receptor 2-negative breast cancer. J. Cell. Biochem. 2019, 120, 6046–6056. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 January 2019).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, e550. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, e128. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Mélius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Target Gene | Correlation Coefficient | p-Value | log2fold Change of Target Genes | qRT-PCR of Target Genes |
|---|---|---|---|---|---|
| miR-374b-5p | SLC7A11 | −0.67 | 0.034 | 6.41 | up |
| miR-374b-5p | HIST1H3B | −0.71 | 0.021 | 5.87 | up |
| miR-374b-5p | HK2 | −0.74 | 0.013 | 5.72 | up |
| miR-26b-5p | PPARGC1A | −0.70 | 0.020 | −4.87 | down |
| miR-26b-5p | ALDH6A1 | −0.66 | 0.039 | −4.71 | down |
| miR-26b-5p | MARC2 | −0.68 | 0.030 | −4.08 | down |
| miR-26b-5p | SLC7A11 | +0.82 | 0.004 | 6.41 | up |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wach, S.; Taubert, H.; Weigelt, K.; Hase, N.; Köhn, M.; Misiak, D.; Hüttelmaier, S.; Stöhr, C.G.; Kahlmeyer, A.; Haller, F.; et al. RNA Sequencing of Collecting Duct Renal Cell Carcinoma Suggests an Interaction between miRNA and Target Genes and a Predominance of Deregulated Solute Carrier Genes. Cancers 2020, 12, 64. https://doi.org/10.3390/cancers12010064
Wach S, Taubert H, Weigelt K, Hase N, Köhn M, Misiak D, Hüttelmaier S, Stöhr CG, Kahlmeyer A, Haller F, et al. RNA Sequencing of Collecting Duct Renal Cell Carcinoma Suggests an Interaction between miRNA and Target Genes and a Predominance of Deregulated Solute Carrier Genes. Cancers. 2020; 12(1):64. https://doi.org/10.3390/cancers12010064
Chicago/Turabian StyleWach, Sven, Helge Taubert, Katrin Weigelt, Nora Hase, Marcel Köhn, Danny Misiak, Stefan Hüttelmaier, Christine G. Stöhr, Andreas Kahlmeyer, Florian Haller, and et al. 2020. "RNA Sequencing of Collecting Duct Renal Cell Carcinoma Suggests an Interaction between miRNA and Target Genes and a Predominance of Deregulated Solute Carrier Genes" Cancers 12, no. 1: 64. https://doi.org/10.3390/cancers12010064
APA StyleWach, S., Taubert, H., Weigelt, K., Hase, N., Köhn, M., Misiak, D., Hüttelmaier, S., Stöhr, C. G., Kahlmeyer, A., Haller, F., Vera, J., Hartmann, A., Wullich, B., & Lai, X. (2020). RNA Sequencing of Collecting Duct Renal Cell Carcinoma Suggests an Interaction between miRNA and Target Genes and a Predominance of Deregulated Solute Carrier Genes. Cancers, 12(1), 64. https://doi.org/10.3390/cancers12010064