Immune Therapy for Liver Cancers


Abstract
1. Introduction
2. Hepatocellular Carcinoma
2.1. Biological Rationale for Immune Therapies
2.1.1. Liver Is an Immunological Organ
2.1.2. Rationale for Using ICI in HCC
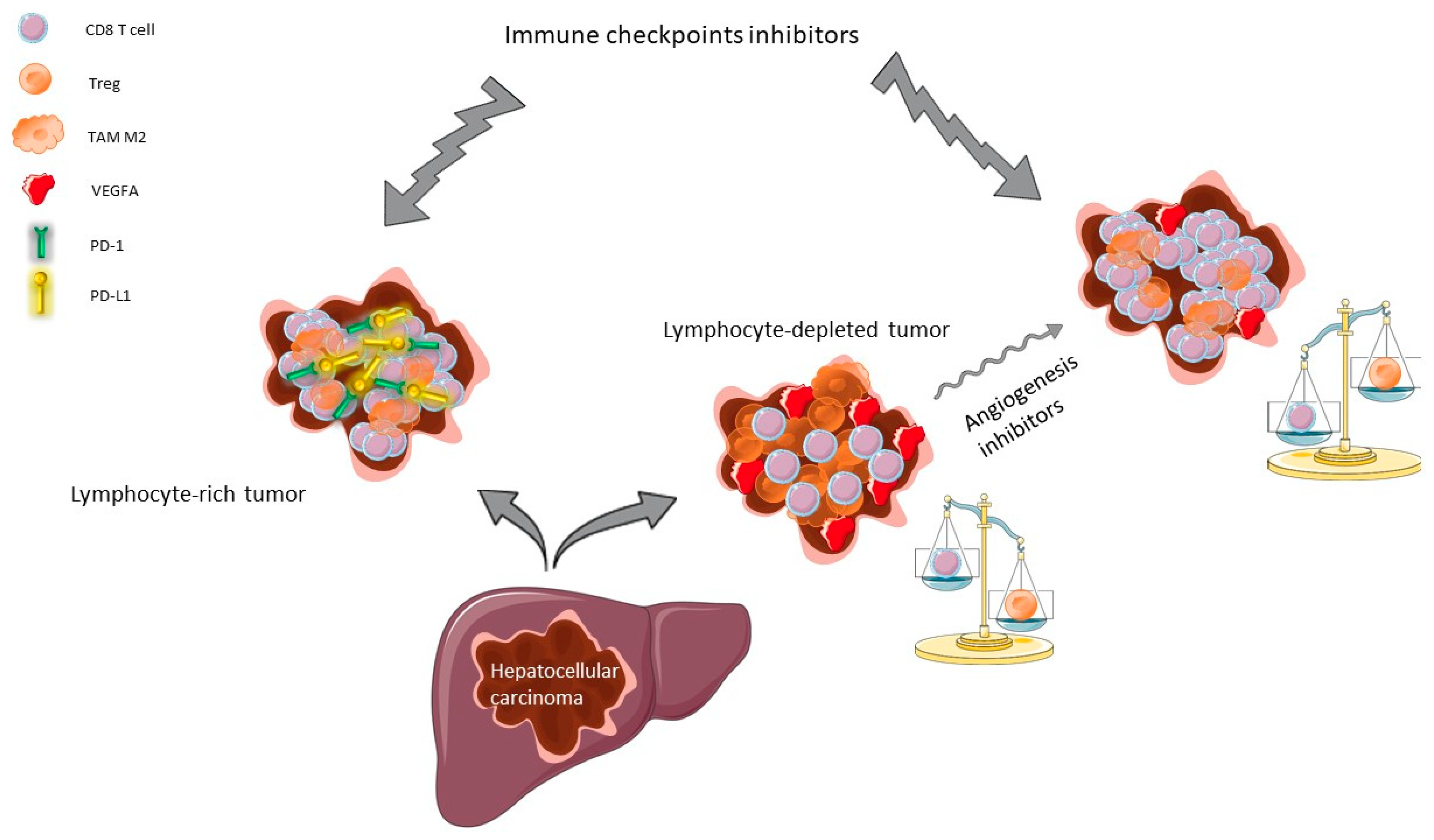
2.1.3. Rationale for Combining Angiogenesis Inhibitors and ICI in HCC
2.2. Current State of Immune Therapies Clinical Development
2.2.1. ICI Monotherapy
2.2.2. Combinations of ICIs and Other Immunotherapies
2.2.3. Combinations of Angiogenesis Inhibitors and ICI
3. Biliary Tract Cancers
3.1. Biological Rationale for Targeted Therapies and Immune Therapies
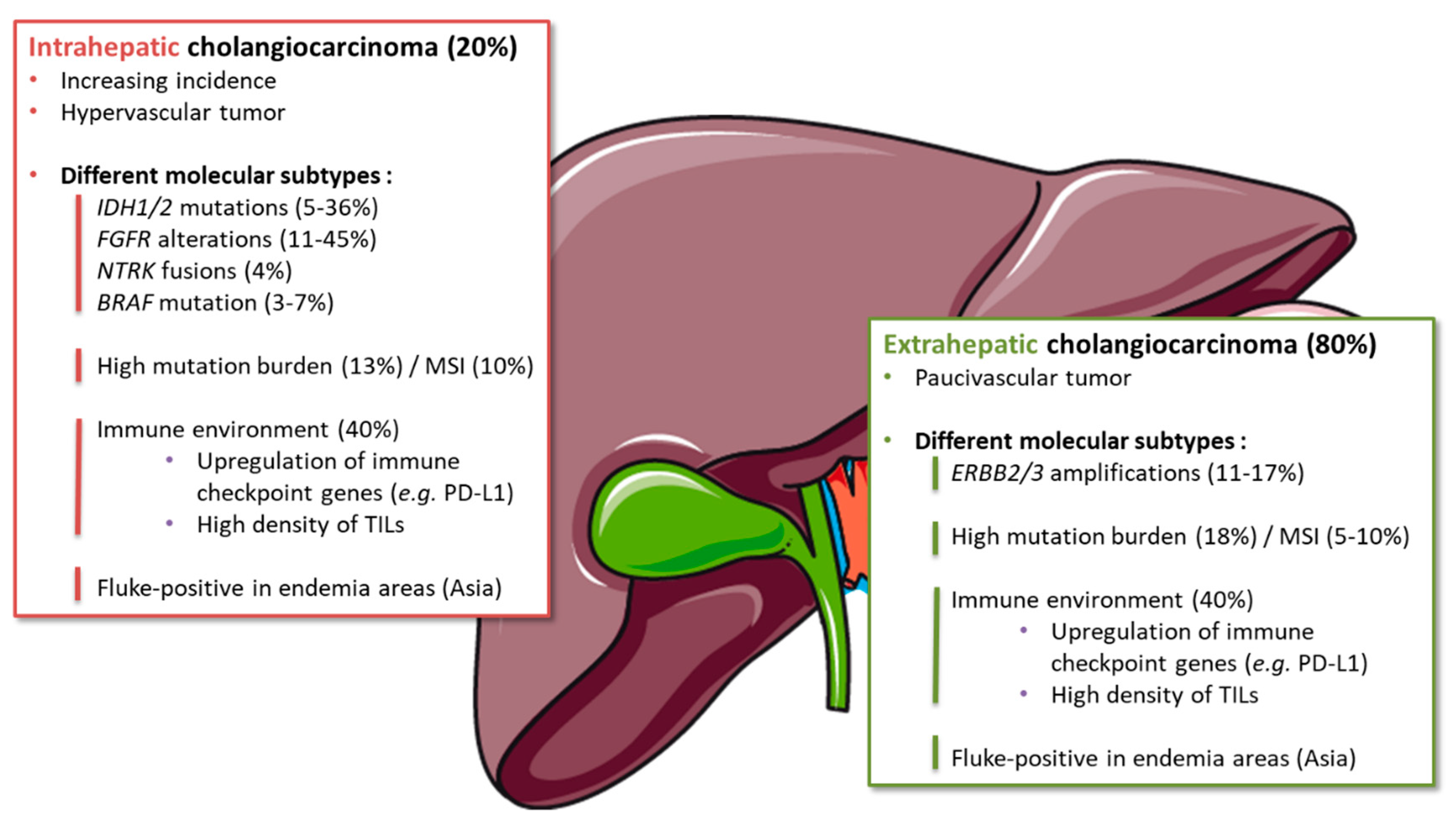
3.1.1. Molecular Alterations
3.1.2. Angiogenesis
3.1.3. Immune Microenvironment
3.1.4. Subgroups Sensitive to Immune Therapies
3.2. Current State of Immune Therapies Clinical Development
3.2.1. ICI Monotherapy
3.2.2. ICI in Combination and Other Immunotherapies
3.3. Predictive Value of Immune Signatures
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004, 12, 96–102. [Google Scholar] [CrossRef]
- Trepo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127, S87–S96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Vanni, E.; Marchesini, G. NASH and the risk of cirrhosis and hepatocellular carcinoma in type 2 diabetes. Curr. Diabetes Rep. 2007, 7, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.; Capocaccia, R.; Hackl, M.; Lemmens, V.; Molina, E.; Pierannunzio, D.; Sant, M.; Trama, A.; Faivre, J. Survival in patients with primary liver cancer, gallbladder and extrahepatic biliary tract cancer and pancreatic cancer in Europe 1999–2007: Results of EUROCARE-5. Eur. J. Cancer 2015, 51, 2169–2178. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D. Biliary cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 21, v59–v64. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Fontugne, J.; Augustin, J.; Pujals, A.; Compagnon, P.; Rousseau, B.; Luciani, A.; Tournigand, C.; Cherqui, D.; Azoulay, D.; Pawlotsky, J.M.; et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 2017, 8, 24644–24651. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Kumar, A.; Le, D.T. Hepatocellular carcinoma regression after cessation of immunosuppressive therapy. J. Clin. Oncol. 2016, 34, 90–92. [Google Scholar] [CrossRef]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, Y.; Okano, S.; Matsumoto, Y.; Nakagawara, H.; Matono, R.; Yoshiya, S.; Yamashita, Y.; Yoshizumi, T.; Ikegami, T.; Soejima, Y.; et al. Prognostic impact of programmed cell death 1 ligand 1 expression in human leukocyte antigen class I-positive hepatocellular carcinoma after curative hepatectomy. J. Gastroenterol. 2015, 50, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Venkatesh, S.K.; Torbenson, M.S.; Roberts, L.R. Clinical implications of basic research in hepatocellular carcinoma. J. Hepatol. 2016, 64, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, e16018. [Google Scholar] [CrossRef]
- Horwitz, E.; Stein, I.; Andreozzi, M.; Nemeth, J.; Shoham, A.; Pappo, O.; Schweitzer, N.; Tornillo, L.; Kanarek, N.; Quagliata, L.; et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 2014, 4, 730–743. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Suzuki, H.; Onishi, H.; Wada, J.; Yamasaki, A.; Tanaka, H.; Nakano, K.; Morisaki, T.; Katano, M. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur. J. Immunol. 2010, 40, 197–203. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- Manzoni, M.; Rovati, B.; Ronzoni, M.; Loupakis, F.; Mariucci, S.; Ricci, V.; Gattoni, E.; Salvatore, L.; Tinelli, C.; Villa, E.; et al. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology 2010, 79, 187–196. [Google Scholar] [CrossRef]
- Martino, E.C.; Misso, G.; Pastina, P.; Costantini, S.; Vanni, F.; Gandolfo, C.; Botta, C.; Capone, F.; Lombardi, A.; Pirtoli, L.; et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov. 2016, 2, e16025. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, e12624. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.-Y.; Breder, V.V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Results of KEYNOTE-240: Phase 3 study of pembrolizumab (Pembro) vs best supportive care (BSC) for second line therapy in advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2019, 37, 4004. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. LBA38_PRCheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Liu, Y.; Xiong, J.; Ren, Z.; Meng, Z.; Gu, S.; Wang, L.; Zou, J. A phase II study of anti-PD-1 antibody camrelizumab plus FOLFOX4 or GEMOX systemic chemotherapy as first-line therapy for advanced hepatocellular carcinoma or biliary tract cancer. J. Clin. Oncol. 2019, 37, 4074. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Formenti, S.; Al-Rajabi, R.; Papadopoulos, K.P.; Stankevich, E.; Feng, M.; Li, J.; Kroog, G.; Lowy, I.; Mathias, M.; et al. 1151PCemiplimab, a human monoclonal anti-PD-1, in patients (pts) with advanced or metastatic hepatocellular carcinoma (HCC): Data from an expansion cohort in a phase I study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Segal, N.H.; Jaeger, D.; Lee, K.-H.; Marshall, J.; Antonia, S.J.; Butler, M.; Sanborn, R.E.; Nemunaitis, J.J.; Carlson, C.A.; et al. Safety and clinical activity of durvalumab monotherapy in patients with hepatocellular carcinoma (HCC). J. Clin. Oncol. 2017, 35, 4071. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.; Ducreux, M.; Zhu, A.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; et al. LBA3IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Llovet, J.; Shepard, K.V.; Finn, R.S.; Ikeda, M.; Sung, M.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; et al. 747PA phase Ib trial of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC): Updated results. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric, or esophagogastric junction cancer: An open-label, dose escalation and expansion study. Clin. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef]
- Kudo, M.; Motomura, K.; Wada, Y.; Inaba, Y.; Sakamoto, Y.; Kurosaki, M.; Umeyama, Y.; Kamei, Y.; Yoshimitsu, J.; Fujii, Y.; et al. First-line avelumab + axitinib in patients with advanced hepatocellular carcinoma: Results from a phase 1b trial (VEGF Liver 100). J. Clin. Oncol. 2019, 37, 4072. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Mauriello, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Potentiating cancer vaccine efficacy in liver cancer. Oncoimmunology 2018, 7, e1488564. [Google Scholar] [CrossRef]
- Chen, C.; Ma, Y.H.; Zhang, Y.T.; Zhang, F.; Zhou, N.; Wang, X.; Liu, T.; Li, Y.M. Effect of dendritic cell-based immunotherapy on hepatocellular carcinoma: A systematic review and meta-analysis. Cytotherapy 2018, 20, 975–989. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Badrinath, N.; Woo, H.Y.; Heo, J. Oncolytic virus-based immunotherapies for hepatocellular carcinoma. Mediat. Inflamm. 2017, 2017, e5198798. [Google Scholar] [CrossRef]
- Ali, N. Chimeric antigen T cell receptor treatment in hematological malignancies. Blood Res. 2019, 54, 81–83. [Google Scholar] [CrossRef]
- Hendrickson, P.G.; Olson, M.; Luetkens, T.; Weston, S.; Han, T.; Atanackovic, D.; Fine, G.C. The promise of adoptive cellular immunotherapies in hepatocellular carcinoma. Oncoimmunology 2020, 9, e1673129. [Google Scholar] [CrossRef]
- Zhai, B.; Shi, D.; Gao, H.; Qi, X.; Jiang, H.; Zhang, Y.; Chi, J.; Ruan, H.; Wang, H.; Ru, Q.C.; et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). J. Clin. Oncol. 2017, 35, 3049. [Google Scholar] [CrossRef]
- Cheng, H.; Sun, G.; Chen, H.; Li, Y.; Han, Z.; Li, Y.; Zhang, P.; Yang, L.; Li, Y. Trends in the treatment of advanced hepatocellular carcinoma: Immune checkpoint blockade immunotherapy and related combination therapies. Am. J. Cancer Res. 2019, 9, 1536–1545. [Google Scholar] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef]
- Llovet, J.M.; Fuster, J.; Bruix, J. The Barcelona approach: Diagnosis, staging, and treatment of hepatocellular carcinoma. Liver Transplant. 2004, 10, 115–120. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kudo, M.; Cheng, A.-L.; Finn, R.S.; Galle, P.R.; Kaneko, S.; Meyer, T.; Qin, S.; Dutcus, C.E.; Chen, E.; et al. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): Phase 3 LEAP-002 study. J. Clin. Oncol. 2019, 37, TPS4152. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.; Lee, D.; Ma, Y.; et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and function of the PD-L1 checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune exclusion-wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin. Cancer Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Ross, P.; Wasan, H.S.; Hubner, R.A.; McNamara, M.G.; Lopes, A.; Manoharan, P.; Palmer, D.; Bridgewater, J.; Valle, J.W. Advanced intrahepatic cholangiocarcinoma: Post-hoc analysis of the ABC-01, -02 and -03 clinical trials. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, L.; Vivaldi, C.; Cereda, S.; Leone, F.; Aprile, G.; Lonardi, S.; Silvestris, N.; Santini, D.; Milella, M.; Caparello, C.; et al. Second-line chemotherapy in advanced biliary cancer progressed to first-line platinum-gemcitabine combination: A multicenter survey and pooled analysis with published data. J. Exp. Clin. Cancer Res. 2015, 34, e156. [Google Scholar] [CrossRef]
- Goldstein, D.; Lemech, C.; Valle, J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017, 2, e000152. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Neuzillet, C.; Rousseau, B.; Kocher, H.; Bourget, P.; Tournigand, C. Unravelling the pharmacologic opportunities and future directions for targeted therapies in gastro-intestinal cancers Part 1: GI carcinomas. Pharmacol. Ther. 2017, 174, 145–172. [Google Scholar] [CrossRef]
- Mertens, J.C.; Rizvi, S.; Gores, G.J. Targeting cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1454–1460. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New horizons for precision medicine in biliary tract cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla Mercade, T.; Javle, M.; Kelley, R.K.; Lubner, S.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.A.; et al. LBA10_PRClarIDHy: A global, phase III, randomized, double-blind study of ivosidenib (IVO) vs placebo in patients with advanced cholangiocarcinoma (CC) with an isocitrate dehydrogenase 1 (IDH1) mutation. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Vogel, A.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. LBA40FIGHT-202: A phase II study of pemigatinib in patients (pts) with previously treated locally advanced or metastatic cholangiocarcinoma (CCA). Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Guedj, N.; Zhan, Q.; Perigny, M.; Rautou, P.E.; Degos, F.; Belghiti, J.; Farges, O.; Bedossa, P.; Paradis, V. Comparative protein expression profiles of hilar and peripheral hepatic cholangiocarcinomas. J. Hepatol. 2009, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, J.K.; Ruys, A.T.; Groot Koerkamp, B.; Beuers, U.; ten Kate, F.J.; van Gulik, T.M. Differences in immunohistochemical biomarkers between intra- and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2014, 29, 1582–1594. [Google Scholar] [CrossRef]
- Neuzillet, C.; Seitz, J.-F.; Fartoux, L.; Malka, D.; Lledo, G.; Tijeras-Raballand, A.; Gramont, A.D.; Ronot, M.; Bouattour, M.; Dreyer, C.; et al. Sunitinib as second-line treatment in patients with advanced intrahepatic cholangiocarcinoma (SUN-CK phase II trial): Safety, efficacy, and updated translational results. J. Clin. Oncol. 2015, 33, 343. [Google Scholar] [CrossRef]
- Guion-Dusserre, J.F.; Lorgis, V.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 2096–2101. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Patel, A.; Normolle, D.; Patel, K.; Ohr, J.; Lee, J.J.; Bahary, N.; Chu, E.; Streeter, N.; Drummond, S. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer 2019, 125, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Demols, A.; Borbath, I.; den Eynde, M.V.; Houbiers, G.; Peeters, M.; Marechal, R.; Delaunoit, T.; Goeminne, J.; Laurent, S.; Holbrechts, S.; et al. O-003Exploratory analysis based on tumor location of REACHIN, a randomized, double-blinded, placebo-controlled phase 2 trial of regorafenib after failure of gemcitabine and platinum-based chemotherapy for advanced/metastatic biliary tract tumors. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, M.; Zhang, Q.; Wang, J.; Yang, X.; Zhou, F.; Li, L.; Yuan, Z.; Jin, H.; Qian, Q. Activation or suppression of the immune response mediators in biliary tract cancer (BTC) patients: A systematic review and meta-analysis. J. Cancer 2017, 8, 74–84. [Google Scholar] [CrossRef][Green Version]
- Kitano, Y.; Okabe, H.; Yamashita, Y.I.; Nakagawa, S.; Saito, Y.; Umezaki, N.; Tsukamoto, M.; Yamao, T.; Yamamura, K.; Arima, K.; et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br. J. Cancer 2018, 118, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.M.; Gao, Q.; Shi, G.M.; Zhang, X.; Wang, J.; Jiang, J.H.; Wang, X.Y.; Shi, Y.H.; Ding, Z.B.; Fan, J.; et al. Intratumoral IL-17(+) cells and neutrophils show strong prognostic significance in intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 2012, 19, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Stenzinger, A.; Andrulis, M.; Klauschen, F.; Joehrens, K.; Warth, A.; Renner, M.; Mehrabi, A.; et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br. J. Cancer 2013, 109, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Tsukagoshi, M.; Wada, S.; Yokobori, T.; Altan, B.; Ishii, N.; Watanabe, A.; Kubo, N.; Saito, F.; Araki, K.; Suzuki, H.; et al. Overexpression of natural killer group 2 member D ligands predicts favorable prognosis in cholangiocarcinoma. Cancer Sci. 2016, 107, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Oshikiri, T.; Miyamoto, M.; Shichinohe, T.; Suzuoki, M.; Hiraoka, K.; Nakakubo, Y.; Shinohara, T.; Itoh, T.; Kondo, S.; Katoh, H. Prognostic value of intratumoral CD8+ T lymphocyte in extrahepatic bile duct carcinoma as essential immune response. J. Surg. Oncol. 2003, 84, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Chan-On, W.; Nairismagi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Silva, V.W.; Askan, G.; Daniel, T.D.; Lowery, M.; Klimstra, D.S.; Abou-Alfa, G.K.; Shia, J. Biliary carcinomas: Pathology and the role of DNA mismatch repair deficiency. Chin. Clin. Oncol. 2016, 5, e62. [Google Scholar] [CrossRef]
- Diaz, L.A.; Le, D.; Maio, M.; Ascierto, P.A.; Geva, R.; Motola-Kuba, D.; André, T.; Van Cutsem, E.; Gottfried, M.; Elez, E.; et al. 1174OPembrolizumab in microsatellite instability high cancers: Updated analysis of the phase II KEYNOTE-164 and KEYNOTE-158 studies. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Shah, U.A.; Nandikolla, A.G.; Rajdev, L. Immunotherapeutic approaches to biliary cancer. Curr. Treat. Options Oncol. 2017, 18, e44. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Piha-Paul, S.A.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Pembrolizumab (pembro) for advanced biliary adenocarcinoma: Results from the KEYNOTE-028 (KN028) and KEYNOTE-158 (KN158) basket studies. J. Clin. Oncol. 2019, 37, 4079. [Google Scholar] [CrossRef]
- Ueno, M.; Chung, H.C.; Nagrial, A.; Marabelle, A.; Kelley, R.K.; Xu, L.; Mahoney, J.; Pruitt, S.K.; Oh, D.-Y. 625PDPembrolizumab for advanced biliary adenocarcinoma: Results from the multicohort, phase II KEYNOTE-158 study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Alshari, O.M.; Dawaymeh, T.A.; Tashtush, N.A.; Aleshawi, A.J.; Al Manasra, A.R.A.; Obeidat, K.A. Completely resolved advanced biliary tract cancer after treatment by pembrolizumab: A report of two cases. Onco Targets Ther. 2019, 12, 5293–5298. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.D.; Kim, D.W.; Alese, O.B.; Li, D.; Shah, N.; Schell, M.J.; Zhou, J.M.; Chung, V. A phase II study of nivolumab in patients with advanced refractory biliary tract cancers (BTC). J. Clin. Oncol. 2019, 37, 4097. [Google Scholar] [CrossRef]
- Ioka, T.; Ueno, M.; Oh, D.-Y.; Fujiwara, Y.; Chen, J.-S.; Doki, Y.; Mizuno, N.; Park, K.; Asagi, A.; Hayama, M.; et al. Evaluation of safety and tolerability of durvalumab (D) with or without tremelimumab (T) in patients (pts) with biliary tract cancer (BTC). J. Clin. Oncol. 2019, 37, 387. [Google Scholar] [CrossRef]
- Ueno, M.; Ikeda, M.; Morizane, C.; Kobayashi, S.; Ohno, I.; Kondo, S.; Okano, N.; Kimura, K.; Asada, S.; Namba, Y.; et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: A non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 611–621. [Google Scholar] [CrossRef]
- Yoo, C.; Oh, D.-Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.-T.; Osada, M.; Helwig, C.; Dussault, I.; et al. 757PM7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF-β, in Asian patients with pretreated biliary tract cancer: Preliminary results from a phase I trial. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Long-term vaccination with multiple peptides derived from cancer-testis antigens can maintain a specific T-cell response and achieve disease stability in advanced biliary tract cancer. Clin. Cancer Res. 2013, 19, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Phase I clinical trial of multiple-peptide vaccination for patients with advanced biliary tract cancer. J. Transl. Med. 2014, 12, e61. [Google Scholar] [CrossRef]
- Shirahama, T.; Muroya, D.; Matsueda, S.; Yamada, A.; Shichijo, S.; Naito, M.; Yamashita, T.; Sakamoto, S.; Okuda, K.; Itoh, K.; et al. A randomized phase II trial of personalized peptide vaccine with low dose cyclophosphamide in biliary tract cancer. Cancer Sci. 2017, 108, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Sakabe, T.; Abe, H.; Tanii, M.; Takahashi, H.; Chiba, A.; Yanagida, E.; Shibamoto, Y.; Ogasawara, M.; Tsujitani, S.; et al. Dendritic cell-based immunotherapy targeting synthesized peptides for advanced biliary tract cancer. J. Gastrointest. Surg. 2013, 17, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E.; Campbell, D.J.; Dumur, C.I. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2011, 27, 276–284. [Google Scholar] [CrossRef]
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef]
- Abdel-Wahab, R.; Ali, S.M.; Borad, M.J.; Shroff, R.T.; Kwong, L.; Vauthey, J.-N.; Koay, E.J.; Zuo, M.; Rashid, A.; Schrock, A.B.; et al. Variations in DNA repair genomic alterations and tumor mutation burden in biliary tract cancer (BTC) subtypes. J. Clin. Oncol. 2018, 36, 263. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, 542–551. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the Era of checkpoint inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Immunotherapy | Molecules | Trial | Phase | n | Population | mOS | mPFS | ORR | DCR |
|---|---|---|---|---|---|---|---|---|---|
| Anti-CTLA-4 | Tremelimumab | Sangro et al. [34] | II | 20 | Pre-treated | 8.2 mo | 6.5 mo | 17.6% | 76.4% |
| Duffy et al. [33] | II | 32 | Pre-treated | 12.3 mo | 7.4 mo | 26.3% | 63% | ||
| Ant-PD1 | Pembrolizumab | Finn et al. [35] | III | 413 | Pre-treated | 12.9 mo | 4.9 mo | 16.9% | NA |
| Nivolumab | Yau et al. [37] | III | 371 | Naive | 16.4 mo | 3.7 mo | 15%, 4% CR | NA | |
| El-Khoueiry et al. [37] | I/II | 262 | Pre-treated and naive | NR | 4 mo | 20%, 1% CR | 64% | ||
| Cemiplimab | Pishvaian et al. [39] | I | 26 | Pre-treated | NR | 3.7 mo | 19.2% | 73% | |
| Anti-PD-L1 | Durvalumab | Wainberg et al. [40] | I/II | 39 | Pre-treated | 13.2 mo | NA | 10.3% | 33% at 6 m |
| Anti-PD-1 + Anti CTLA-4 | Nivolumab + ipilumumab | Yau et al. [41] | II | 148 | Pre-treated | NR | NA | 31%, 5% CR | 49% |
| Durvalumab + tremelimumab | Kelley et al. [42] | I | 40 | Pre-treated and naive | NA | NA | 15% | 57.5% at 4 m | |
| Angiogenesis and immune checkpoints inhibitors | Atezolizumab + bevacizumab | Cheng et al. [43] | III | 501 | Naive | NR | 6.8 mo | 37% | NA |
| Pembrolizumab + lenvatinib | Llovet et al. [44] | Ib | 67 | Naive | NA | NA | 45% | 91% | |
| Camrelizumab + apatinib | Xu et al. [45] | Ib | 16 | Pre-treated | NR | 5.8 mo | 50% | 93.8% | |
| Avelumab + axitinib | Kudo et al. [46] | I | 22 | Naive | NR | 5.5 mo | 31.8% | NA | |
| Cytotoxic agents and Anti-PD-1 | FOLFOX4 or GEMOX + camrelizumab | Qin et al. [38] | Ib | 34 | Naive | NR | 5.5 mo | 26.5% | 79.4% |
| Molecule | Targets | Phase | Population | ClinicalTrial.gov Reference |
|---|---|---|---|---|
| Immunotherapies in Combotherapy | ||||
| Nivolumab plus ipilimumab | PD-1 and CTLA-4 | I-III | Localized HCC Intermediate stage HCC Advanced HCC, L2 Advanced HCC, L1 | NCT03510871 NCT03222076 NCT03203304 NCT01658878 NCT04039607 |
| Durvalumab plus tremelimumab | PD-L1 and CTLA-4 | III | Advanced HCC, L2 | NCT03298451 |
| LY3321367 plus LY3300054 | PD-L1 and TIM-3 | I | Advanced HCC, L2 | NCT03099109 |
| Immunotherapies in association with angiogenesis inhibitors | ||||
| Nivolumab plus bevacizumab | PD-1 | I | Advanced HCC, L2 | NCT03382886 |
| Nivolumab plus lenvatinib | PD-1 | I-II | Advanced HCC, ≥ L1 | NCT03418922 NCT03841201 |
| Nivolumab plus sorafenib | PD-1 | I-III | Advanced HCC, L1 | NCT02576509 NCT01658878 NCT03439891 |
| Nivolumab plus cabozantinib | PD-1 | I-II | Locally advanced HCC Advanced HCC, L1 | NCT03299946 NCT01658878 |
| Nivolumab plus GT90001 | PD-1 | I/II | Advanced HCC | NCT03893695 |
| Pembrolizumab plus lenvatinib | PD-1 | III | Advanced HCC, L1 | NCT03713593 |
| Pembrolizumab plus sorafenib | PD-1 | I/II | Advanced HCC, L1 | NCT03211416 |
| Pembrolizumab plus regorafenib | PD-1 | I | Advanced HCC, L1 | NCT03347292 |
| Atezolizumab plus cabozantinib | PD-L1 | III | Advanced HCC, L1 | NCT03755791 |
| Atezolizumab plus bevacizumab | PD-L1 | III | Advanced HCC, L1 | NCT03434379 |
| Avelumab plus axitinib | PD-L1 | I | Advanced HCC, L1 | NCT03289533 |
| Durvalumab plus bevacizumab | PD-L1 | III | Localized HCC Locally advanced HCC | NCT03847428 NCT03778957 |
| Durvalumab plus tivozanib | PD-L1 | I/II | Advanced HCC, L1 | NCT03970616 |
| Camrelizumab plus apatinib | PD-1 | I III | Advanced HCC, L2 Advanced HCC, L1 | NCT02942329 NCT03764293 |
| Spartalizumab plus sorafenib | PD-1 | I | Advanced HCC, L1 | NCT02988440 |
| Tislelizumab plus sorafenib | PD-1 | III | Advanced HCC, L1 | NCT03412773 |
| Sintilimab plus IBI305 | PD-1 | III | Advanced HCC, L1 | NCT03794440 |
| Immunotherapies in association with loco-regional therapies (radiofrequency ablation, radiotherapy, or intra-arterial treatments) | ||||
| Durvalumab plus tremelimumab | PD-L1 and CTLA-4 | II | Intermediate stage HCC | NCT03638141 |
| Nivolumab ± ipilimumab | PD-1 | I–II | Advanced HCC Intermediate stage HCC | NCT03033446 NCT03572582 NCT03143270 NCT02837029 NCT03203304 NCT03812562 NCT03630640 |
| Pembrolizumab | PD-1 | I–II | Locally advanced | NCT03099564 NCT03397654 NCT03867084 NCT03316872 |
| Immunotherapies in association with another therapy | ||||
| Camrelizumab plus chemotherapy | PD-1 | I | Advanced HCC, L1 | NCT03092895 |
| Nivolumab plus TGF-β inhibitor | PD-1 | I/II | Advanced HCC, L2 | NCT02423343 |
| Nivolumab plus indoleamine dioxygenase inhibitor | PD-1 | I/II | Advanced HCC, L1 | NCT03695250 |
| Nivolumab plus cereblon modulator | PD-1 | I/II | Advanced HCC, L2 | NCT02859324 |
| Nivolumab plus invariant Natural Killer T cell agonist | PD-1 | I/II | Advanced HCC, ≥L2 | NCT03897543 |
| Nivolumab plus Pexa-Vec oncolytic immunotherapy | PD-1 | I/II | Advanced HCC, L1 | NCT03071094 |
| Pembrolizumab plus monoclonal antibody against phosphatidylserine | PD-1 | II | Advanced HCC, L1 | NCT03519997 |
| Spartalizumab plus FGFR4 inhibitor | PD-1 | I/II | Advanced HCC, L2 | NCT02325739 |
| Spartalizumab plus MET inhibitor | PD-1 | I/II | Advanced HCC, L2 | NCT02795429 |
| Molecule | Targets | Phase | Population | ClinicalTrial.gov Reference |
|---|---|---|---|---|
| Immunotherapies in monotherapy or combotherapy | ||||
| Nivolumab plus ipilimumab | PD-1 and CTLA-4 | IIR | Advanced CCA, ≥L1 | NCT03101566 NCT02834013 |
| Pembrolizumab | PD-1 | II | Advanced CCA, L2 | NCT03110328 NCT02628067 |
| Nivolumab | PD-1 | II | Advanced CCA, ≥L2 | NCT02829918 |
| STI-3031 | PD-L1 | II | Advanced CCA, ≥L2 | NCT03999658 |
| Immunotherapies in association with chemotherapy | ||||
| Durvalumab | PD-L1 | III | Advanced CCA, L1 | NCT03875235 |
| KN035 | PD-L1 | III | Advanced CCA, L1 | NCT03478488 |
| Pembrolizumab | PD-1 | II-III | Advanced CCA, ≥L1 | NCT03260712 NCT03111732 NCT04003636 |
| Durvalumab plus tremelimumab | PD-L1 and CTLA-4 | IIR | Advanced CCA, ≥L1 | NCT03473574 NCT03046862 NCT03704480 |
| SHR-1210 | PD-1 | II | Advanced CCA, ≥L1 | NCT03486678 |
| Toripalimab | PD-1 | II | Advanced CCA, L1 | NCT03796429 NCT03982680 NCT04027764 |
| Immunotherapies in association with radiation or ablative therapies | ||||
| Nivolumab ± ipilimumab | PD-1 and CTLA-4 | IIR | Advanced CCA, ≥L2 | NCT02866383 |
| Durvalumab plus tremelimumab | PD-L1 and CTLA-4 | II | Advanced CCA, ≥L2 | NCT03482102 NCT02821754 |
| Camrelizumab | PD-1 | II | Advanced CCA, L1 | NCT03898895 |
| Immunotherapies in association with another therapy | ||||
| SHR-1210 plus apatinib | PD-1 | IIR | Advanced CCA, ≥L2 | NCT03092895 |
| Atezolizumab plus cobimetininb (MEK inhibitor) | PD-L1 | IIR | Metastatic CCA, ≥L2 | NCT03201458 |
| Pembrolizumab plus lenvatinib | PD-1 | II | Advanced CCA, ≥L2 | NCT03797326 NCT03895970 |
| Toripalimab plus axitinib | PD-1 | II | Metastatic CCA, L2 | NCT04010071 |
| Durvalumab plus olaparib | PD-L1 | II | Advanced CCA, IDH1/2 gene mutation, ≥L2 | NCT03991832 |
| Nivolumab plus rucaparib | PD-1 | II | Advanced CCA, ≥L2 | NCT03639935 |
| Atezolizumab plus DKN-01 (DKK1 inhibitor) | PD-L1 | II | Advanced CCA, ≥L2 | NCT03818997 |
| Nivolumab plus DKN-01 (DKK1 inhibitor) | PD-1 | II | Advanced CCA, ≥L2 | NCT04057365 |
| Pembrolizumab plus sargramostim (GM-CSF) | PD-1 | II | Advanced CCA, ≥L1 | NCT02703714 |
| Nivolumab plus entinostat (HDAC inhibitor) | PD-1 | II | Advanced CCA, ≥L2 | NCT03250273 |
| JS001 plus lenvatinib plus GEMOX | PD-1 | II | Advanced iCCA, L1 | NCT03951597 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hilmi, M.; Vienot, A.; Rousseau, B.; Neuzillet, C. Immune Therapy for Liver Cancers. Cancers 2020, 12, 77. https://doi.org/10.3390/cancers12010077
Hilmi M, Vienot A, Rousseau B, Neuzillet C. Immune Therapy for Liver Cancers. Cancers. 2020; 12(1):77. https://doi.org/10.3390/cancers12010077
Chicago/Turabian StyleHilmi, Marc, Angélique Vienot, Benoît Rousseau, and Cindy Neuzillet. 2020. "Immune Therapy for Liver Cancers" Cancers 12, no. 1: 77. https://doi.org/10.3390/cancers12010077
APA StyleHilmi, M., Vienot, A., Rousseau, B., & Neuzillet, C. (2020). Immune Therapy for Liver Cancers. Cancers, 12(1), 77. https://doi.org/10.3390/cancers12010077