Recent Progress in Mitochondria-Targeted Drug and Drug-Free Agents for Cancer Therapy



Abstract
1. Introduction
Mitochondria-Targeted Cancer Therapeutics
2. Mito–Drug/Toxic Agent Conjugates for Cancer Therapy
2.1. Conventional Drugs Targeting Mitochondria
2.2. Molecules Targeting Mitochondrial Metabolism, Functions, or Proteins
2.3. Mitochondria-Targeted Peptides
2.4. Mitochondria-Targeted Photosensitizers
2.4.1. Mitochondria-Targeted Metal Complexes for PDT
2.4.2. Mitochondria-Targeted Small Molecules for PDT
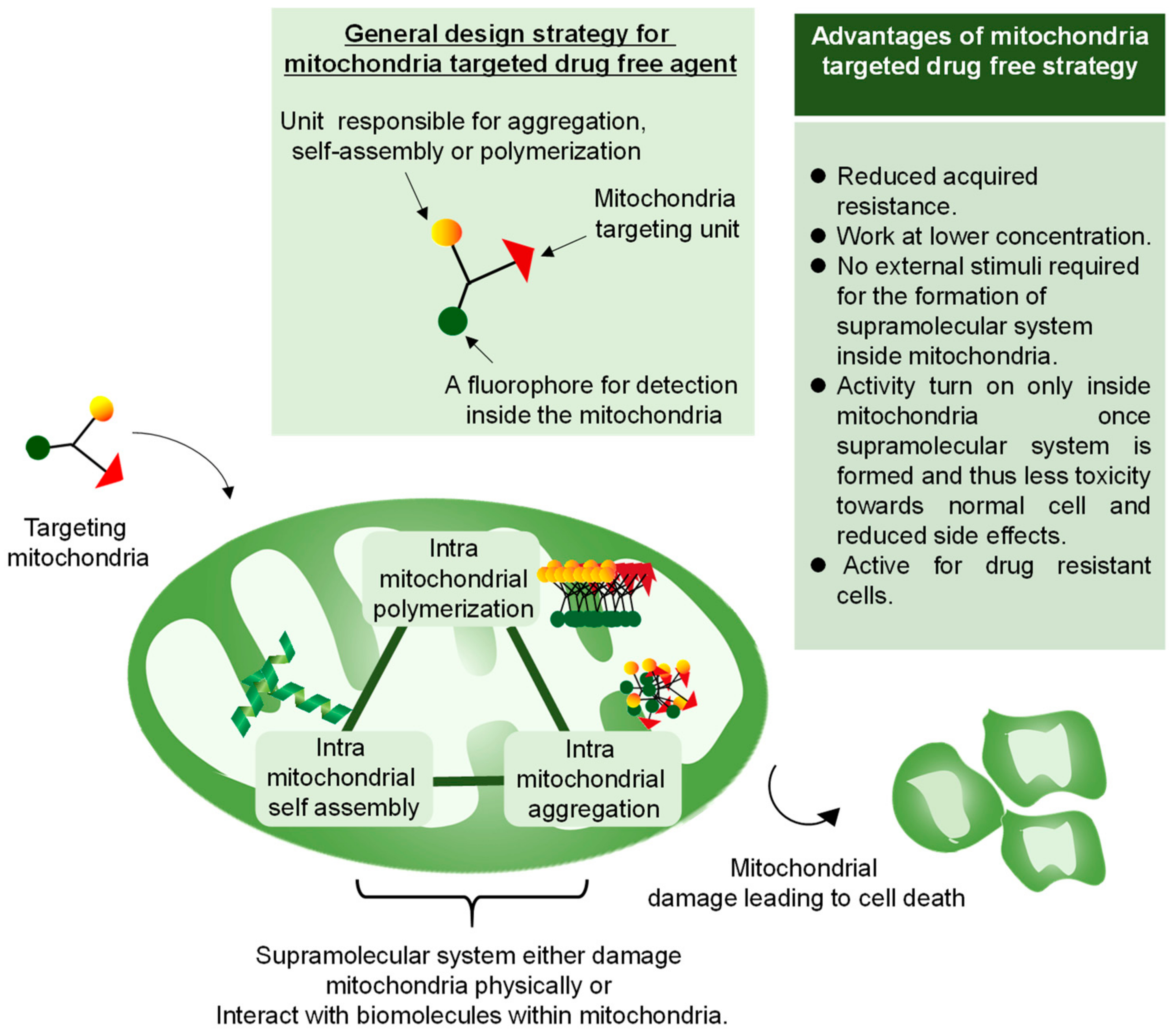
3. Mitochondria-Targeted Drug-Free Agents
3.1. Aggregation of Rationally Designed Molecules Inside the Mitochondria
3.2. In Situ Nanostructures Inside Mitochondria for Cancer Therapy
4. Conclusions and Future Outlook
Funding
Conflicts of Interest
References
- Hoye, A.T.; Davoren, J.E.; Wipf, P.; Fink, M.P.; Kagan, V.E. Taegeting Mitochondria. Acc. Chem. Res. 2008, 41, 87–97. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Yousif, L.F.; Stewart, K.M.; Kelley, S.O. Targeting mitochondria with organelle-specific compounds: Strategies and applications. Chembiochem 2009, 10, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, X.; Zhang, C.; MacKenzie, K.R.; Stossi, F.; Palzkill, T.; Wang, M.C.; Wang, J. Reversible Reaction-Based Fluorescent Probe for Real-Time Imaging of Glutathione Dynamics in Mitochondria. ACS Sens. 2017, 2, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondrial control of apoptosis: An introduction. Biochem. Biophys. Res. Commun. 2003, 304, 433–435. [Google Scholar] [CrossRef]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of apoptosis: The role of cytochrome C. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Nakagawa, T.; Shimizu, S. Mitochondrial membrane permeability transition and cell death. Biochim. Biophys. Acta 2006, 1757, 1297–1300. [Google Scholar] [CrossRef]
- Baines, C.P. Role of the mitochondrion in programmed necrosis. Front. Physiol. 2010, 1, 156. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondria in cancer. Oncogene 2006, 25, 4630–4632. [Google Scholar] [CrossRef] [PubMed]
- Wisnovsky, S.; Lei, E.K.; Jean, S.R.; Kelley, S.O. Mitochondrial Chemical Biology: New Probes Elucidate the Secrets of the Powerhouse of the Cell. Cell Chem. Biol. 2016, 23, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Rin Jean, S.; Tulumello, D.V.; Wisnovsky, S.P.; Lei, E.K.; Pereira, M.P.; Kelley, S.O. Molecular vehicles for mitochondrial chemical biology and drug delivery. ACS Chem. Biol. 2014, 9, 323–333. [Google Scholar] [CrossRef]
- Teixeira, J.; Oliveira, C.; Cagide, F.; Amorim, R.; Garrido, J.; Borges, F.; Oliveira, P.J. Discovery of a new mitochondria permeability transition pore (mPTP) inhibitor based on gallic acid. J. Enzym. Inhib. Med. Chem. 2018, 33, 567–576. [Google Scholar] [CrossRef]
- Dilip, A.; Cheng, G.; Joseph, J.; Kunnimalaiyaan, S.; Kalyanaraman, B.; Kunnimalaiyaan, M.; Gamblin, T.C. Mitochondria-targeted antioxidant and glycolysis inhibition: Synergistic therapy in hepatocellular carcinoma. Anticancer Drugs 2013, 24, 881–888. [Google Scholar] [CrossRef]
- Mannella, C.A. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim. Biophys. Acta 2006, 1763, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Millard, M.; Pathania, D.; Shabaik, Y.; Taheri, L.; Deng, J.; Neamati, N. Preclinical evaluation of novel triphenylphosphonium salts with broad-spectrum activity. PLoS ONE 2010, 5, e13131. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jin, F.; Shu, G.; Xu, X.; Qi, J.; Kang, X.; Yu, H.; Lu, K.; Jiang, S.; Han, F.; et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 2019, 211, 57–67. [Google Scholar] [CrossRef]
- Sweetwyne, M.T.; Pippin, J.W.; Eng, D.G.; Hudkins, K.L.; Chiao, Y.A.; Campbell, M.D.; Marcinek, D.J.; Alpers, C.E.; Szeto, H.H.; Rabinovitch, P.S.; et al. The mitochondrial-targeted peptide, SS-31, improves glomerular architecture in mice of advanced age. Kidney Int. 2017, 91, 1126–1145. [Google Scholar] [CrossRef]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-penetrating peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef]
- Lincoln, R.; Greene, L.E.; Zhang, W.; Louisia, S.; Cosa, G. Mitochondria Alkylation and Cellular Trafficking Mapped with a Lipophilic BODIPY-Acrolein Fluorogenic Probe. J. Am. Chem. Soc. 2017, 139, 16273–16281. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, J.; Wang, L.; Hu, X.; Liu, X.; Liu, M.; Cao, Z.; Shangguan, D.; Tan, W. A Cyanine Dye to Probe Mitophagy: Simultaneous Detection of Mitochondria and Autolysosomes in Live Cells. J. Am. Chem. Soc. 2016, 138, 12368–12374. [Google Scholar] [CrossRef]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Control. Release 2012, 159, 393–402. [Google Scholar] [CrossRef]
- Han, X.; Su, R.; Huang, X.; Wang, Y.; Kuang, X.; Zhou, S.; Liu, H. Triphenylphosphonium-modified mitochondria-targeted paclitaxel nanocrystals for overcoming multidrug resistance. Asian J. Pharm. Sci. 2019, 14, 569–580. [Google Scholar] [CrossRef]
- Han, M.; Vakili, M.R.; Soleymani Abyaneh, H.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol. Pharm. 2014, 11, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Dai, L.; Ji, M.; Wang, H. Mitochondria-targeted triphenylphosphonium conjugated glycyrrhetinic acid derivatives as potent anticancer drugs. Bioorg. Chem. 2019, 85, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.B.; Zhao, Z.L.; Liu, T.; Xie, G.J.; Jin, C.; Deng, T.G.; Sun, Y.; Li, X.; Hu, X.X.; Zhang, X.B.; et al. A Multi-Mitochondrial Anticancer Agent that Selectively Kills Cancer Cells and Overcomes Drug Resistance. ChemMedChem 2017, 12, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Reshetnikov, V.; Daum, S.; Janko, C.; Karawacka, W.; Tietze, R.; Alexiou, C.; Paryzhak, S.; Dumych, T.; Bilyy, R.; Tripal, P.; et al. ROS-Responsive N-Alkylaminoferrocenes for Cancer-Cell-Specific Targeting of Mitochondria. Angew. Chem. 2018, 57, 11943–11946. [Google Scholar] [CrossRef] [PubMed]
- Stapelberg, M.; Gellert, N.; Swettenham, E.; Tomasetti, M.; Witting, P.K.; Procopio, A.; Neuzil, J. Alpha-tocopheryl succinate inhibits malignant mesothelioma by disrupting the fibroblast growth factor autocrine loop: Mechanism and the role of oxidative stress. J. Biol. Chem. 2005, 280, 25369–25376. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Low, P.; Dyason, J.C.; Wang, X.F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J.; et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4335. [Google Scholar] [CrossRef] [PubMed]
- Juan, M.E.; Wenzel, U.; Daniel, H.; Planas, J.M. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J. Agric. Food Chem. 2008, 56, 4813–4818. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Singh, K.K. Mitochondria as targets for detection and treatment of cancer. Expert Rev. Mol. Med. 2002, 4, 1–19. [Google Scholar] [CrossRef]
- Lehenkari, P.P.; Kellinsalmi, M.; Napankangas, J.P.; Ylitalo, K.V.; Monkkonen, J.; Rogers, M.J.; Azhayev, A.; Vaananen, H.K.; Hassinen, I.E. Further insight into mechanism of clodronate: Inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adnine-containing metabolite. Mol. Pharmacol. 2002, 62, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, H.K.; Jeong, H.; Lim, J.; Lee, A.J.; Cheon, K.Y.; Kim, C.S.; Thomas, A.P.; Bae, B.; Kim, N.D.; et al. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J. Am. Chem. Soc. 2015, 137, 4358–4367. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, C.; He, Y.; Zhang, Z.; Zhou, W.; Muhammad, N.; Guo, Y.; Wang, X.; Guo, Z. Restraining Cancer Cells by Dual Metabolic Inhibition with a Mitochondrion-Targeted Platinum(II) Complex. Angew. Chem. 2019, 58, 4638–4643. [Google Scholar] [CrossRef]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Self-assembly of peptides to nanostructures. Org. Biomol. Chem. 2014, 12, 3544–3561. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, F.; Xu, H.; Yaseen, M.; Shan, H.; Hauser, C.A.; Zhang, S.; Lu, J.R. Molecular self-assembly and applications of designer peptide amphiphiles. Chem. Soc. Rev. 2010, 39, 3480–3498. [Google Scholar] [CrossRef]
- Lowik, D.W.; van Hest, J.C. Peptide based amphiphiles. Chem. Soc. Rev. 2004, 33, 234–245. [Google Scholar] [CrossRef]
- Lei, E.K.; Kelley, S.O. Delivery and Release of Small-Molecule Probes in Mitochondria Using Traceless Linkers. J. Am. Chem. Soc. 2017, 139, 9455–9458. [Google Scholar] [CrossRef]
- Chamberlain, G.R.; Tulumello, D.V.; Kelley, S.O. Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 2013, 8, 1389–1395. [Google Scholar] [CrossRef]
- Jean, S.R.; Tulumello, D.V.; Riganti, C.; Liyanage, S.U.; Schimmer, A.D.; Kelley, S.O. Mitochondrial Targeting of Doxorubicin Eliminates Nuclear Effects Associated with Cardiotoxicity. ACS Chem. Biol. 2015, 10, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Wisnovsky, S.P.; Wilson, J.J.; Radford, R.J.; Pereira, M.P.; Chan, M.R.; Laposa, R.R.; Lippard, S.J.; Kelley, S.O. Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem. Biol. 2013, 20, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hu, R.; Lu, F.; Guo, X.; Wang, S.; Zeng, Y.; Li, Y.; Yang, G. Traceable cancer cell photoablation with a new mitochondria-responsive and -activatable red-emissive photosensitizer. Chem. Commun. 2019, 55, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Hilf, R. Mitochondria are targets of photodynamic therapy. J. Bioenerg. Biomembr. 2007, 39, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gao, P.; Huang, Y.; Lu, X.; Chang, Q.; Pan, W.; Li, N.; Tang, B. Boosting the photodynamic therapy efficiency with a mitochondria-targeted nanophotosensitizer. Chin. Chem. Lett. 2019, 30, 1293–1296. [Google Scholar] [CrossRef]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef]
- Xia, D.; Xu, P.; Luo, X.; Zhu, J.; Gu, H.; Huo, D.; Hu, Y. Overcoming Hypoxia by Multistage Nanoparticle Delivery System to Inhibit Mitochondrial Respiration for Photodynamic Therapy. Adv. Funct. Mater. 2019, 29, 1807294. [Google Scholar] [CrossRef]
- Lv, W.; Zhang, Z.; Zhang, K.Y.; Yang, H.; Liu, S.; Xu, A.; Guo, S.; Zhao, Q.; Huang, W. A Mitochondria-Targeted Photosensitizer Showing Improved Photodynamic Therapy Effects Under Hypoxia. Angew. Chem. 2016, 55, 9947–9951. [Google Scholar] [CrossRef]
- Yi, S.; Lu, Z.; Zhang, J.; Wang, J.; Xie, Z.; Hou, L. Amphiphilic Gemini Iridium(III) Complex as a Mitochondria-Targeted Theranostic Agent for Tumor Imaging and Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 15276–15289. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, J.; Rees, T.W.; Wang, H.; Li, X.; Chao, H.; Stang, P.J. Heterometallic Ru-Pt metallacycle for two-photon photodynamic therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 5664–5669. [Google Scholar] [CrossRef]
- Tian, N.; Sun, W.; Guo, X.; Lu, J.; Li, C.; Hou, Y.; Wang, X.; Zhou, Q. Mitochondria targeted and NADH triggered photodynamic activity of chloromethyl modified Ru(ii) complexes under hypoxic conditions. Chem. Commun. 2019, 55, 2676–2679. [Google Scholar] [CrossRef] [PubMed]
- Chakrabortty, S.; Agrawalla, B.K.; Stumper, A.; Vegi, N.M.; Fischer, S.; Reichardt, C.; Kogler, M.; Dietzek, B.; Feuring-Buske, M.; Buske, C.; et al. Mitochondria Targeted Protein-Ruthenium Photosensitizer for Efficient Photodynamic Applications. J. Am. Chem. Soc. 2017, 139, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, X.; Cao, Z.; Wu, Y.; Chen, J.A.; Gao, J.; Wang, Z.; Guo, W.; Gu, X. Mitochondria-targeting BODIPY-loaded micelles as novel class of photosensitizer for photodynamic therapy. Eur. J. Med. Chem. 2018, 157, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Noh, I.; Lee, D.; Kim, H.; Jeong, C.U.; Lee, Y.; Ahn, J.O.; Hyun, H.; Park, J.H.; Kim, Y.C. Enhanced Photodynamic Cancer Treatment by Mitochondria-Targeting and Brominated Near-Infrared Fluorophores. Adv. Sci. 2018, 5, 1700481. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.P.; Palanikumar, L.; Jeena, M.T.; Kim, K.; Ryu, J.H. Cancer-mitochondria-targeted photodynamic therapy with supramolecular assembly of HA and a water soluble NIR cyanine dye. Chem. Sci. 2017, 8, 8351–8356. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.-M.; Zong, Y.-N.; Cao, S.-M. Current cancer situation in China: Good or bad news from the 2018 global cancer statics? Cancer Commun. 2019, 39, 22. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resisitance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Zhao, R.; Wang, B.; Yang, X.; Xiao, Y.; Wang, X.; Shao, C.; Tang, R. A drug-free tumor therapy strategy: Cancer-cell-targeting calcification. Angew. Chem. 2016, 55, 5225–5229. [Google Scholar] [CrossRef]
- Hu, Q.; Gao, M.; Feng, G.; Liu, B. Mitochondria-targeted cancer therapy using a light-up probe with aggregation-induced-emission characteristics. Angew. Chem. 2014, 53, 14225–14229. [Google Scholar] [CrossRef]
- Kim, S.; Palanikumar, L.; Choi, H.; Jeena, M.T.; Kim, C.; Ryu, J.H. Intra-mitochondrial biomineralization for inducing apoptosis of cancer cells. Chem. Sci. 2018, 9, 2474–2479. [Google Scholar] [CrossRef]
- Kuang, Y.; Shi, J.; Li, J.; Yuan, D.; Alberti, K.A.; Xu, Q.; Xu, B. Pericellular hydrogel/nanonets inhibit cancer cells. Angew. Chem. 2014, 53, 8104–8107. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, Z.; Wang, Y.; Zhou, R.; Yang, Z.; Xu, B. Integrating Enzymatic Self-Assembly and Mitochondria Targeting for Selectively Killing Cancer Cells without Acquired Drug Resistance. J. Am. Chem. Soc. 2016, 138, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Jeena, M.T.; Palanikumar, L.; Go, E.M.; Kim, I.; Kang, M.G.; Lee, S.; Park, S.; Choi, H.; Kim, C.; Jin, S.M.; et al. Mitochondria localization induced self-assembly of peptide amphiphiles for cellular dysfunction. Nat. Commun. 2017, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Jeena, M.T.; Jeong, K.; Go, E.M.; Cho, Y.; Lee, S.; Jin, S.; Hwang, S.-K.; Jang, J.H.; Kang, C.S.; Bang, W.-Y.; et al. Heterochiral Assembly of Amphiphilic Peptides Inside the Mitochondria for Supramolecular Cancer Therapeutics. ACS Nano 2019, 13, 11022–11033. [Google Scholar] [CrossRef]
- Cheng, D.B.; Zhang, X.H.; Gao, Y.J.; Ji, L.; Hou, D.; Wang, Z.; Xu, W.; Qiao, Z.Y.; Wang, H. Endogenous Reactive Oxygen Species-Triggered Morphology Transformation for Enhanced Cooperative Interaction with Mitochondria. J. Am. Chem. Soc. 2019, 141, 7235–7239. [Google Scholar] [CrossRef]
- He, H.; Wang, J.; Wang, H.; Zhou, N.; Yang, D.; Green, D.R.; Xu, B. Enzymatic Cleavage of Branched Peptides for Targeting Mitochondria. J. Am. Chem. Soc. 2018, 140, 1215–1218. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Mitochondrial Protein/Metabolism Targeted Drug | Target of Interest | Mode of Cell Death | Reference |
|---|---|---|---|---|
| 1 | alpha-tocopheryl succinate (α-TOS) | inhibits succinate dehydrogenase (SDH) | ROS accumulation apoptosis. | [36] |
| 2 | resveratrol | inhibits complex III, induce the expression of mitochondrial superoxide dismutase (SOD2) | apoptosis | [38] |
| 3 | clodronate | inhibit the activity of adenine nucleotide transporter (ANT), inhibit mitochondrial oxygen consumption | apoptosis | [40] |
| 4 | AppCCl2P | ADP/ATP translocation | apoptosis | [40] |
| 5 | ABT-737 | inhibits anti-apoptotic proteins, disturb mitochondrial membrane potential, increase intra cellular ROS | apoptosis | [43] |
| 6 | SMTIN-P01 | inhibits mitochondrial TRAP1 | apoptosis | [44] |
| 7 | TPP conjugated terpyridine-Pt | inhibits thioredoxin (TrxR) | apoptosis | [45] |
| No. | Molecular Design | Advantages | Disadvantages |
|---|---|---|---|
| 1 | Conventional drug conjugate with mitochondria targeting ligand | Enhanced cell cytotoxicity | Induces toxicity towards normal cell as well |
| 2 | Photosensitizer conjugate with mitochondria targeting ligand | Quick action, higher toxicity | Induces toxicity towards normal cell as well, requires an external aid of laser for PS activation |
| 3 | Protein inhibitor conjugate with mitochondria targeting ligand | High cancer selectivity | Induces acquired resistance as a result of targeted protein mutation upon repeated administration |
| 4 | Mitochondria penetrating peptide conjugate with cargo | High mitochondria penetrating ability | Could penetrate normal cells as well, cannot be used for higher molecular weight cargo, complicated design and synthesis. |
| No. | Molecular Design | Barrier for Practical Application | Advantages for Practical Application |
|---|---|---|---|
| 1 | Conventional drug conjugate with mitochondria targeting ligand | Fetal toxicity and side effects | - |
| 2 | Protein inhibitor conjugate with mitochondria targeting ligand | Complicated synthetic procedure, acquired resistance | High tumor selectivity |
| 3 | Mitochondria penetrating peptide conjugate with cargo | Complicated molecular design, needs to carry a cargo. | - |
| 4 | Mitochondria-targeted drug-free agents | - | Work at small concertation, ease of production, high selectivity towards the tumor, fewer side effects. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeena, M.T.; Kim, S.; Jin, S.; Ryu, J.-H. Recent Progress in Mitochondria-Targeted Drug and Drug-Free Agents for Cancer Therapy. Cancers 2020, 12, 4. https://doi.org/10.3390/cancers12010004
Jeena MT, Kim S, Jin S, Ryu J-H. Recent Progress in Mitochondria-Targeted Drug and Drug-Free Agents for Cancer Therapy. Cancers. 2020; 12(1):4. https://doi.org/10.3390/cancers12010004
Chicago/Turabian StyleJeena, M.T., Sangpil Kim, Seongeon Jin, and Ja-Hyoung Ryu. 2020. "Recent Progress in Mitochondria-Targeted Drug and Drug-Free Agents for Cancer Therapy" Cancers 12, no. 1: 4. https://doi.org/10.3390/cancers12010004
APA StyleJeena, M. T., Kim, S., Jin, S., & Ryu, J.-H. (2020). Recent Progress in Mitochondria-Targeted Drug and Drug-Free Agents for Cancer Therapy. Cancers, 12(1), 4. https://doi.org/10.3390/cancers12010004