Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers

 , and
, and 
Abstract
1. Introduction
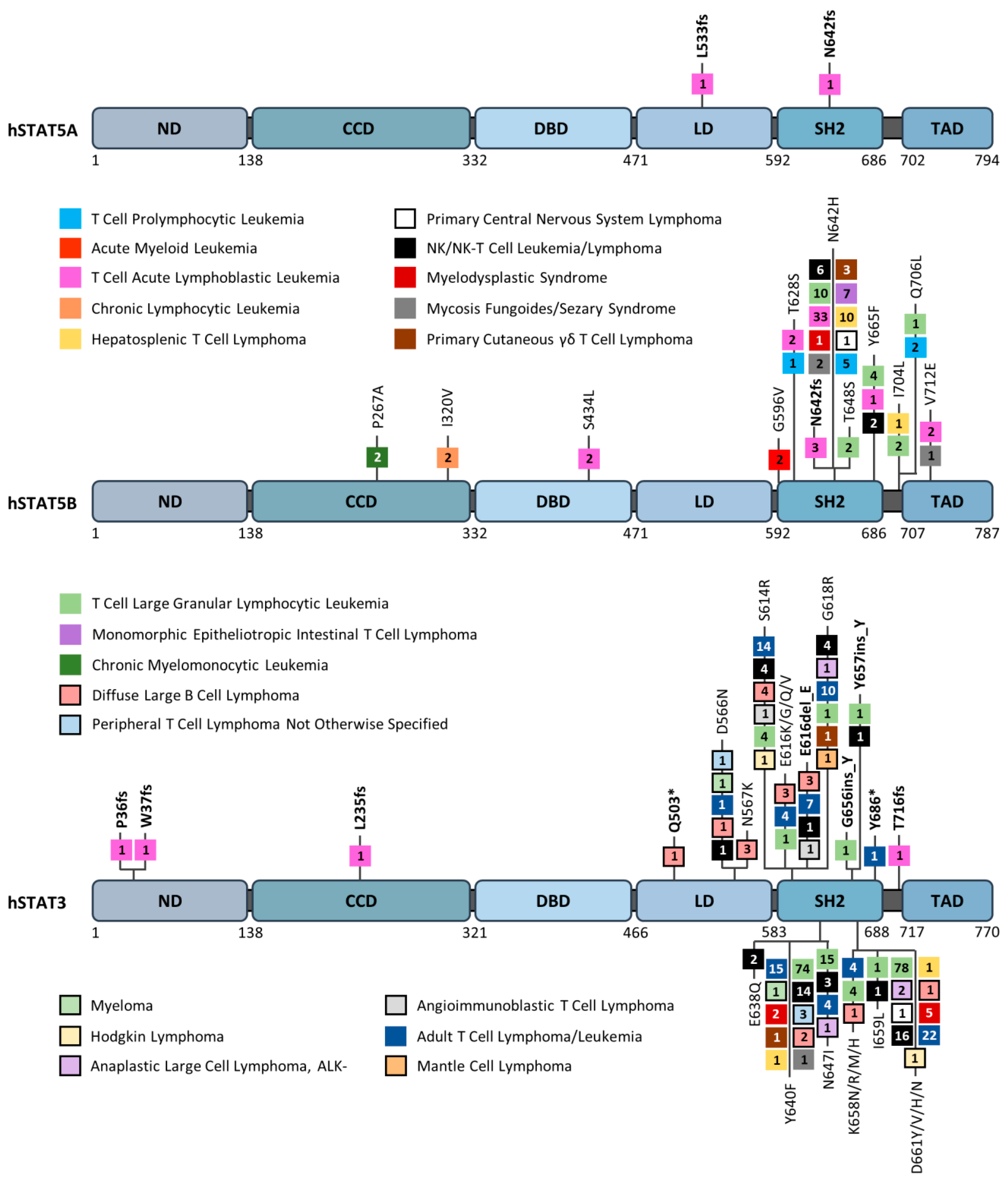
2. STAT3 and STAT5A/5B in Hematopoietic Cancers
2.1. STAT3/STAT5 in Hematopoietic Cancers: An Amazing 23-Year-Old Story
2.2. STAT3/5 in Myeloproliferative Neoplasms (MPNs)
2.3. STAT3/5 in Acute Myeloid Leukemia (AML)
2.4. STAT3/5 in Acute Lymphoblastic Leukemia (ALL)
2.5. STAT3/5 in T Cell Large Granular Lymphocytic (T-LGL) Leukemia
2.6. STAT3/5 in Chronic Lymphocytic Leukemias (CLL)
2.7. STAT3/5 in Lymphomas
2.8. STAT3/5 in Myelomas
3. Canonical and Non-Canonical Roles of STAT3/STAT5 in Hematopoietic Cancers
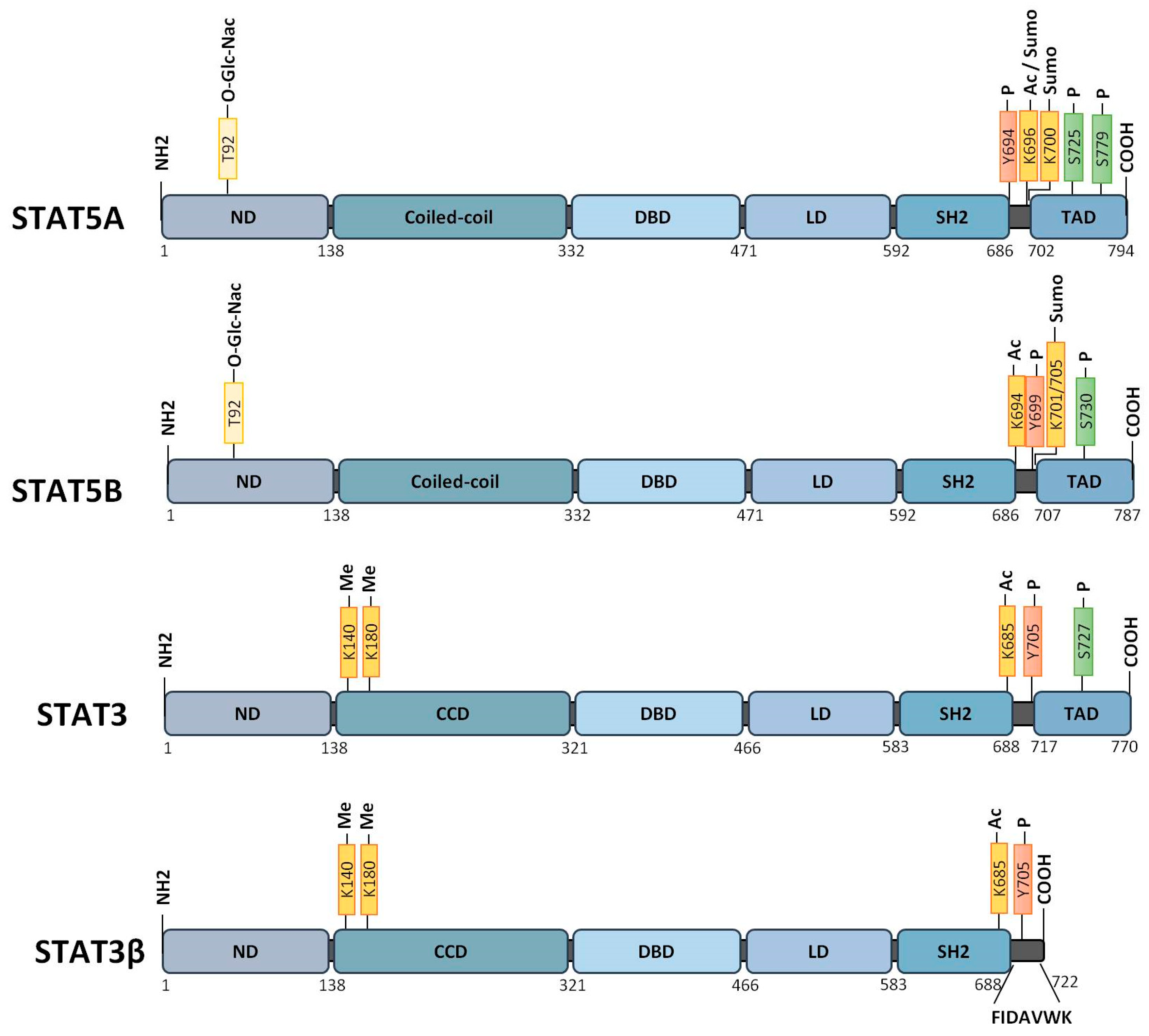
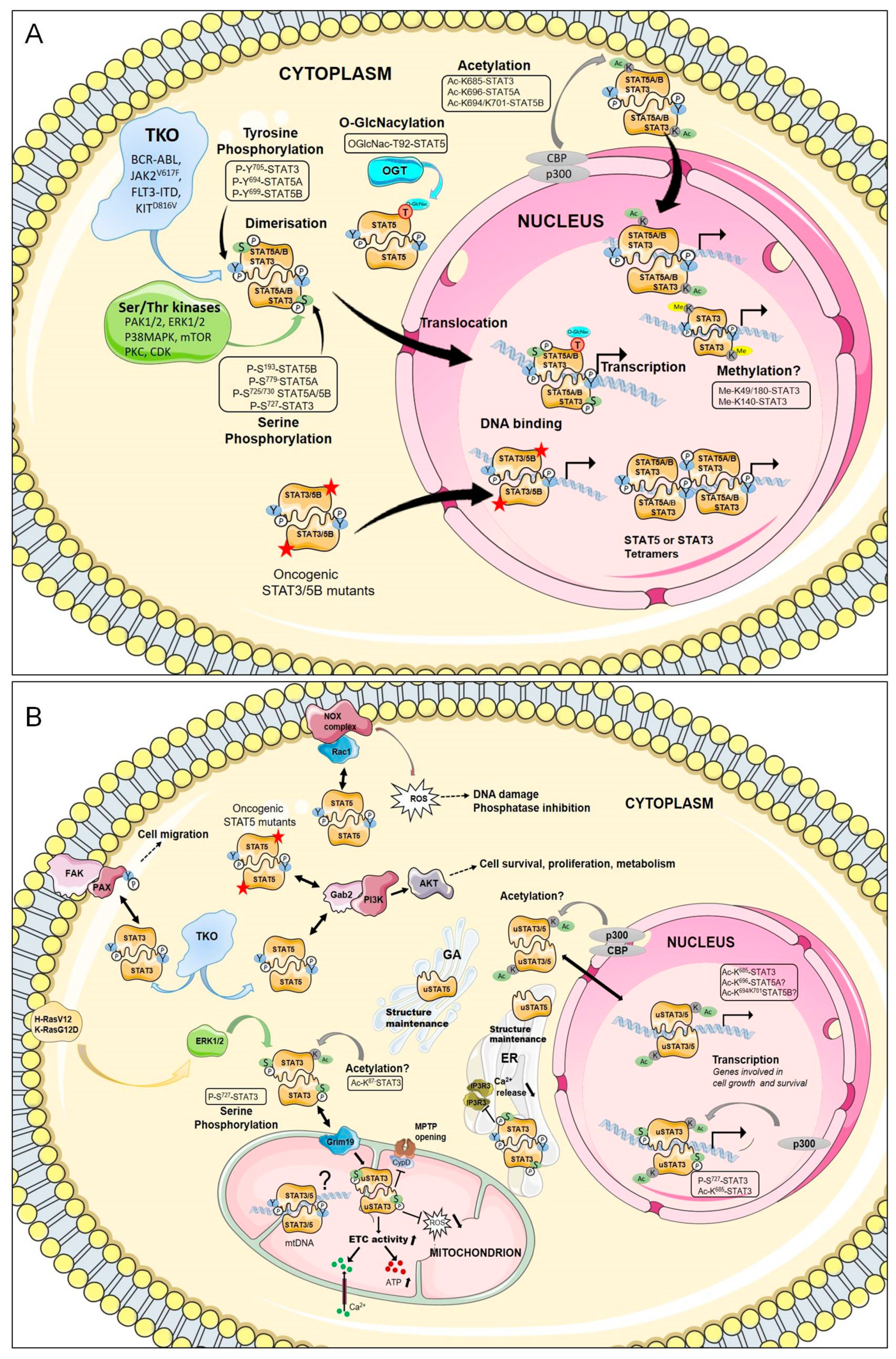
3.1. Canonical Function of STAT3/STAT5
3.2. Non-Canonical Functions of STAT3/STAT5
4. Pharmacological Inhibitors of STAT3 and STAT5
4.1. Introduction
4.2. Indirect Inhibitors of STAT3 and STAT5 Signaling in Hematopoietic Cancers
4.2.1. Targeting Upstream Tyrosine Kinases
4.2.2. Natural and Synthetic Molecules
4.2.3. Drug Repositioning

{kind=link}
{kind=link}
{kind=link}
| Drug | Target(s) and Mode of Inhibition | Hematologic Malignancy | Stage of Clinical Development | References |
|---|---|---|---|---|
| Upstream Kinases | ||||
| Atiprimod | JAK2/JAK3 Inhibits kinase activity | AML JAK2V617F-MPN | Preclinical (cell lines, primary cells) Preclinical (cell lines, primary cells) | [249,250] |
| AZ960 | JAK2/JAK3 Type I inhibitor | T cell leukemia/ lymphoma | Preclinical (cell lines) | [251] |
| AZD1480 | JAK2/JAK1/Aurora/FGFR1/FLT4 Type I inhibitor | MPN B-ALL Lymphomas MM | Phase 1 (terminated) Preclinical (xenografts) Preclinical (cell lines) Preclinical (cell lines, xenografts) | [243,244,245,246] |
| Bosutinib (SKI-606) | BCR-ABL/Src Type I inhibitor | CML | Market authorization (Bosulif®, Pfizer) | [201,202] |
| Crenolanib (CP-868596) | FLT3/PDGFR/KIT Type I inhibitor | FLT3-mutated AML SM, AML | Phase 2 Phase 3 (CT) Preclinical (cell lines, primary cells) | [218,223] |
| Dasatinib (BMS-354825) | BCR-ABL/Src. Inhibits kinase activity Type I inhibitor | CML, Ph+ ALL | Market authorization (Sprycel®, BMS) | [200] |
| Fedratinib (TG101348) | JAK2/FLT3/BRD4 Type I inhibitor | MPN HL, MLBCL | Market authorization (Inrebic®, Celgene) Preclinical (cell lines, xenografts) | [241,242] |
| Gandotinib (LY2784544) | JAK2/JAK1/JAK3 Type I inhibitor | JAK2V617F-MPN | Phase 2 | [240] |
| Gilteritinib (ASP2215) | FLT3/AXL Type I inhibitor | FLT3-mutated AML | Market authorization (Xospata®, Astellas) | [219] |
| Imatinib (STI-571) | BCR-ABL. Type II inhibitor | CML, Ph+ALL | Market authorization (Glivec®, Novartis) | [198,199] |
| INCB20 | Pan JAK | MM | Preclinical (cell lines, primary cells, mouse models) | [247] |
| INCB16562 | JAK1/JAK2. Type I inhibitor | MM | Preclinical (cell lines, primary cells) | [248] |
| Itacitinib (INCB-039110) | JAK1 Type I inhibitor | MF B-cell lymphoma | Phase 2 Phase 1/2 | [252,253] |
| KW-2449 | FLT3/BCR-ABL/ BCR-ABL315I/Aurora Inhibits FLT3 and BCR-ABL phosphorylation | CML, AML MDS, AML, ALL, CML | Preclinical (cell lines, primary cells, xenografts) Phase 1 | [212,213] |
| Lestaurtinib (CEP-701) | JAK2/FLT3/TrkA/ Aurora kinase Type I inhibitor | AML MF PV, ET | Phase 2 Phase 2 Phase 2 | [211,225,226] |
| LS104 (AG490 analog) | JAK2/BCR-ABL Allosteric inhibitor | JAK2V617F-MPN AML Ph+ and Ph− ALL | Preclinical (cell lines, primary cells) Preclinical (cell lines, primary cells) Preclinical (cell lines, primary cells, xenografts) | [262,263,264] |
| Midostaurin (PKC412) | FLT3-ITD/KIT Type I inhibitor | FLT3 mutated AML, SM | Market authorization (Rydapt®, Novartis) | [207,222] |
| Momelotinib (CYT-387) | JAK2/JAK1/JAK3/ALK2 Type I inhibitor | MPN MM | 2 Phase 3 Preclinical (cell lines, primary cells) | [259,260] |
| Nilotinib (AMN107) | BCR-ABL Type II inhibitor | CML, Ph+ ALL | Market authorization (Tasigna®, Novartis) | [203] |
| NS-018 | JAK2/Src Type I inhibitor | MPN MM | Phase 1-2 Preclinical (cell lines, mouse model) | [254,255] |
| NVP-BSK805 | JAK2 (JAK2V617F) Type I inhibitor | JAK2V617F-MPN | Preclinical (cell lines, mouse model) | [256] |
| NVP-BVB808 | JAK2 Type I inhibitor | MPN | Preclinical (cell lines) | [257] |
| NVP-CHZ868 | JAK2/TYK2/KIT/PDGF/VEGFR Type II inhibitor | MPN B-ALL | Preclinical (cell lines, primary cells, mouse models) Preclinical (cell lines, xenografts, PDX) | [238,239] |
| ON044580 | JAK2/BCR-ABL Allosteric inhibitor | JAK2V617F-MPN BCR-ABLT315I-CML | Preclinical (cell lines, primary cells) | [258] |
| Pacritinib (SB1518) | JAK2/FLT3/IRAK1 Type I inhibitor | MF Lymphomas, CLL, LPD | Phase 3 Phase 1 | [233,234] |
| Ponatinib (AP24534) | BCR-ABL and BCR-ABL mutants FGFR/PDGFR/Src/RET/KIT/FLT1 Type II inhibitor | CML, Ph+ ALL | Market authorization (Iclusig®, Ariad) | [204] |
| Ruxolitinib (INCB18424) | JAK2/JAK1/JAK3 Type I Inhibitor | MPN CML HL MM AML, ALL | Market authorization (Jakavi®, Novartis) Phase 1 (CT) Phase 2 Phase 1 (CT) Phase1/2 (terminated) | [227,228,229,230,231,232] |
| Quizartinib (AC220) | FLT3 Type II inhibitor | FLT3-mutated AML | Market authorization (Japan) (Vanflyta®, Daiichi-Sankyo) | [217,218] |
| Sorafenib (BAY43-9006) | FLT3/PDGFR/ VEGFR/c-KIT/RAF Type II inhibitor | FLT3-mutated AML ALL, AML, MDS | Phase 1 Phase ½ (CT) Phase 1 | [208,209,210] |
| Sunitinib (SU11248) | PDGFR/ VEGFR/c-KIT/FLT3 Type I Inhibitor | FLT3-mutated AML | Phase 1 Phase ½ (CT) | [214,215] |
| Tandutinib (MLN518) | FLT3/PDGFR/KIT Inhibits type III receptor kinases | AML/MDS AML SM | Phase ½ Phase ½ (CT) Preclinical (cell lines) | [216,224] |
| TG101209 | JAK2/FLT3 Type I inhibitor | MPN MM T-ALL | Preclinical (cell lines, primary cells, mouse models) Preclinical (cell lines) Preclinical (cell lines, primary cells) | [235,236,237] |
| WP1066 and WP1034 (AG490 analogs) | JAK2 Phosphorylation and/or degradation | JAK2V617F-mutated MPN AML | Preclinical (cell lines, primary cells) Preclinical (cell lines, primary cells) | [265,266,267] |
| XL019 | Pan-JAK. Type I inhibitor | MPN | Phase 1 (terminated) | [268] |
| Natural compounds | ||||
| Avicin D | JAK/STAT3 Inhibits P-Y/JAK1/2 phosphorylation Unknown mechanism | MM CTCL | Preclinical (cell lines) Preclinical (cell lines, primary cells) | [269,270] |
| Butein | STAT3. Inhibits P-Y Activates tyrosine phosphatase SHP1 Unknown mechanism | MM | Preclinical (cell lines) | [271] |
| Capsaicin | JAK1/Src/ STAT3. Inhibits P-Y/ Inhbits kinase activity Unknown mechanism | MM | Preclinical (cell lines, xenografts) | [272] |
| Celastrol | JAK/Src/STAT3. Inhibits P-Y/Kinase activity Unknown mechanism | MM | Preclinical (cell lines) | [273] |
| Curcubitacin I (JSI-124) | STAT3. Inhibits P-Y/P-S of STAT3 Unknown mechanism | B-ALL, CLL, lymphomas | Preclinical (cell lines, primary cells) | [274,275] |
| Curcumin diferuloylmethane | JAK/STAT3/STAT5. Inhibits P-Y/kinase activity Unknown mechanism | CML T-cell leukemia T-cell lymphoma HL MM | Preclinical (cell lines) Preclinical (cell lines) Preclinical (cell lines, primary cells) Preclinical (cell lines) Preclinical (cell lines) | [279,280,281,282,283] |
| Honokiol (HNK) | STAT3. Inhibits P-Y Activate SHP1 Unknown mechanism | AML | Preclinical (cell lines, primary cells) | [276] |
| Piperlongumine | STAT3. Inhibits P-Y and other pathways Binds to cysteine 712 of STAT3 Unknown mechanism | MM | Preclinical (cell lines, xenografts) | [277] |
| Sulforaphane | STAT5. Inhibits transcriptional activity. Deacetylase inhibitor Unknown mechanism | CML | Preclinical (cell lines) | [289] |
| Synthetic molecules | ||||
| CDDO (Imidazolide) | STAT3/ STAT5. Inhibits P-Y Unknown mechanism | MM | Preclinical (cell lines) | [285] |
| E804 (Indirubin derivative) | SFK/STAT5 Inhibits P-Y/ Inhibits kinase activity Unknown mechanism | CML AML | Preclinical (cell lines, primary cells) Preclinical (cell lines, xenografts) | [286,287] |
| FLLL3 (Curcumin derivative) | JAK/STAT3. Inhibits P-Y Unknown mechanism | MM | Preclinical (cell lines) | [284] |
| JQ1 (Thienodiazepine derivative) | STAT5. Inhibits transcriptional activity.BET inhibitor. Unknown mechanism | T-ALL | Preclinical (cell lines, primary cells) | [290] |
| 17f | STAT5. Inhibits P-Y and expression. Unknown Mechanism | AML, CML | Preclinical (cell lines) | [292] |
| Naphtoquinone (NPQ) derivative | STAT5. Inhibits P-Y/ Unknown mechanism | CML | Preclinical (cell lines) | [291] |
| Drug repositionning | ||||
| Niclosamide (Antiparasitic) | STAT3. Inhibits P-Y Unknown mechanism | MM | Preclinical (cell lines, primary cells) | [295] |
| Nifuroxazide (Antidiarrheic) | JAK/STAT3. Inhibits P-Y Unknown mechanism | MM | Preclinical (cell lines, primary cells) | [294] |
| Pimozide (Antipsychotic) | STAT3/STAT5. Inhibits P-Y Unknown mechanism | CML MPN FLT3-mutated AML MM | Preclinical (cell lines, primary cells) Preclinical (cell lines) (CT) Preclinical (cell lines, mouse model) Preclinical (cell lines) | [79,196,298,299] |
| PPARγ ligands (Antidiabetic) (Thiazolinediones) | STAT5. Inhibits STAT5A and STAT5B gene expression STAT3. Inhibits transcriptional activity | CM, Ph+ ALL MM | Preclinical (cell lines, primary cells) Phase 2 (CT) Preclinical (cell lines, primary cells) | [60,301,302,303] |
| Pyrimethamine (Antiparasitic) (Antifolate) | STAT3. Inhibits P-Y Unknown mechanism | AML CLL/SLL MM | Preclinical (cell lines, primary cells, xenografts) Phase ½ Preclinical (cell lines) | [196,296] |
4.3. Direct Inhibitors of STAT3 and STAT5 in Hematopoietic Cancers (Table 3)
4.3.1. Inhibitors Targeting the SH2 Domain
4.3.2. Inhibitors Targeting the DNA Binding Domain (DBD)
| Target | Drugs | Hematologic Malignancy | Stage of Clinical Development | References |
|---|---|---|---|---|
| Direct STAT3 inhibitors | ||||
| mRNA | AZD9150 (IONIS-STAT3Rx) | AML, MDS DLBCL, HL, NHL | Preclinical (cell lines, primary cells PDX) Phase 1 | [347,348,349] |
| CpG-STAT3-siRNA | AML, MM | Preclinical (cell lines, primary cells, mouse model of AML, xenografts) | [73,350] | |
| SH2 domain | BP-5-087, BP-1-102/17o (S3I-201 derivatives) | CML B- ALL, T-ALL MM | Preclinical (cell lines, primary cells) Preclinical (cell lines, xenografts) Preclinical (cell lines) | [94,316,317,318] |
| CD38-S3I-NP (S3I-1757) | MM | Preclinical (cell lines, xenografts) | [313] | |
| C188-9 | AML | Preclinical (cell lines, primary cells) | [329] | |
| LLL-3 | CML | Preclinical (cell lines) | [327] | |
| LLL-12 | MM | Preclinical (cell lines, primary cells, xenografts) | [328] | |
| OPB-51602 | MM, NHL, AML, CML | Phase 1 (terminated) | [336] | |
| SF-1-066 (S3I-201 derivative) | AML, MM | Preclinical (cell lines) | [314,315] | |
| STA-21 | CLPD-NKs T-LGL DLBCL | Preclinical (primary cells) Preclinical (primary cells) Preclinical (cell lines) | [325,326] | |
| STATTIC | MM NK lymphomas T-cell lymphomas | Preclinical (cell lines) Preclinical (primary cells) Preclinical (primary cells) | [331,332] | |
| STX-0119 | NHL | Preclinical (cell lines, xenografts) | [311] | |
| SD-36 | AML, ALCL | Preclinical (cell lines, xenografts) | [337] | |
| YL064 | MM | Preclinical (cell lines, primary cells) | [339] | |
| Withaferin A | MM | Preclinical (cell lines) | [338] | |
| DBD | CpG-STAT3dODN (decoy oligonucleotides) | AML DLBCL | Preclinical (cell lines, xenografts, mouse models) Preclinical (cell lines, xenografts, mouse models) | [74,344] |
| DBD-1-9R (peptide aptamer) | MM | Preclinical (cell lines) | [342] | |
| IS3-295 (Platinum IV) | MM | Preclinical (cell lines) | [341] | |
| T40214 (GQ-ODN) | T-ALL | Preclinical (mouse model) | [346] | |
| Direct STAT5 inhibitors | ||||
| SH2 domain | AC-4-130 | AML, CML | Preclinical (cell lines, primary cells, xenografts) | [81,320] |
| BP-1-107, BP-1-108 | AML, CML | Preclinical (cell lines) | [319] | |
| IST5-002 | CML Ph+ ALL | Preclinical (cell lines, primary cells) Preclinical (cell lines, primary cells, PDX) | [90,321] | |
| Stafib-1 | CML | Preclinical (cell lines) | [323] | |
| Stafib-2 | CML | Preclinical (cell lines) | [324] | |
| DBD | STAT5 dODN | CML | Preclinical (cell lines) | [343] |
| Dual STAT3/STAT5 inhibitors | ||||
| SH2 domain | OPB-31121 | CML, AML, ALL, BL, MM, NHL, MM | Preclinical (cell lines, xenografts, PDX) Phase 1 (terminated) | [334] |
4.3.3. Inhibitors Targeting STAT3/5 mRNAs
5. STAT3/5 Inhibitors in Hematopoietic Cancers: Inherent Limitations and Future Challenges
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leonard, W.J.; O’Shea, J.J. JAKs and STATs: Biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef]
- Leaman, D.W.; Leung, S.; Li, X.; Stark, G.R. Regulation of STAT-dependent pathways by growth factors and cytokines. FASEB J. 1996, 10, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Nevalainen, M.T. Targeting transcription factor Stat5a/b as a therapeutic strategy for prostate cancer. Am. J. Transl. Res. 2011, 3, 133–138. [Google Scholar]
- Benekli, M.; Baer, M.R.; Baumann, H.; Wetzler, M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003, 101, 2940–2954. [Google Scholar] [CrossRef]
- Dorritie, K.A.; McCubrey, J.A.; Johnson, D.E. STAT transcription factors in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2014, 28, 248–257. [Google Scholar] [CrossRef]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 activation in solid cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhance DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar] [CrossRef]
- Lacronique, V.; Boureux, A.; Monni, R.; Dumon, S.; Mauchauffe, M.; Mayeux, P.; Gouilleux, F.; Berger, R.; Gisselbrecht, S.; Ghysdael, J.; et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood 2000, 95, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Harir, N.; Boudot, C.; Friedbichler, K.; Sonneck, K.; Kondo, R.; Martin-Lanneree, S.; Kenner, L.; Kerenyi, M.; Yahiaoui, S.; Gouilleux-Gruart, V.; et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood 2008, 112, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Kanie, T.; Abe, A.; Matsuda, T.; Kuno, Y.; Towatari, M.; Yamamoto, T.; Saito, H.; Emi, N.; Naoe, T. TEL-Syk fusion constitutively activates PI3-K/Akt, MAPK and JAK2-independent STAT5 signal pathways. Leukemia 2004, 18, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Müller, C.; Grüning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [CrossRef]
- Shuai, K.; Halpern, J.; ten Hoeve, J.; Rao, X.; Sawyers, C.L. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 1996, 13, 247–254. [Google Scholar]
- Carlesso, N.; Frank, D.A.; Griffin, J.D. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr-Abl. J. Exp. Med. 1996, 183, 811–820. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Slupianek, A.; Xue, L.; Zhang, Q.; Raghunath, P.N.; Hoser, G.; Wasik, M.A.; Morris, S.W.; Skorski, T. Role of signal transducer and activator of transcription 5 in nucleophosmin/ anaplastic lymphoma kinase-mediated malignant transformation of lymphoid cells. Cancer Res. 2001, 61, 6517–6523. [Google Scholar]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef]
- Wilbanks, A.M.; Mahajan, S.; Frank, D.A.; Druker, B.J.; Gilliland, D.G.; Carroll, M. TEL/PDGFbetaR fusion protein activates STAT1 and STAT5: A common mechanism for transformation by tyrosine kinase fusion proteins. Exp. Hematol. 2000, 28, 584–593. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Schwaller, J.; Parganas, E.; Wang, D.; Cain, D.; Aster, J.C.; Williams, I.R.; Lee, C.K.; Gerthner, R.; Kitamura, T.; Frantsve, J.; et al. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol. Cell 2000, 6, 693–704. [Google Scholar] [CrossRef]
- Onishi, M.; Nosaka, T.; Misawa, K.; Mui, A.L.F.; Gorman, D.; McMahon, M.; Miyajima, A.; Kitamura, T. Identification and Characterization of a Constitutively Active STAT5 Mutant That Promotes Cell Proliferation. Mol. Cell. Biol. 1998, 18, 3871–3879. [Google Scholar] [CrossRef] [PubMed]
- Moriggl, R.; Sexl, V.; Kenner, L.; Duntsch, C.; Stangl, K.; Gingras, S.; Hoffmeyer, A.; Bauer, A.; Piekorz, R.; Wang, D.; et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005, 7, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Kontro, M.; Kuusanmäki, H.; Eldfors, S.; Burmeister, T.; Andersson, E.I.; Bruserud, O.; Brümmendorf, T.H.; Edgren, H.; Gjertsen, B.T.; Itälä-Remes, M.; et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia 2014, 28, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stütz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, 188–192. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef]
- de Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5BN642H driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef]
- Hosui, A.; Kimura, A.; Yamaji, D.; Zhu, B.M.; Na, R.; Hennighausen, L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-beta and STAT3 activation. J. Exp. Med. 2009, 206, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim. Biophys. Acta 2011, 1815, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Lai, R. STAT3 in Cancer—Friend or Foe. Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef] [PubMed]
- Aigner, P.; Mizutani, T.; Horvath, J.; Eder, T.; Heber, S.; Lind, K.; Just, V.; Moll, H.P.; Yeroslaviz, A.; Fischer, M.J.M.; et al. STAT3β is a tumor suppressor in acute myeloid leukemia. Blood Adv. 2019, 3, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, H.Y.; Liu, X.; Wasik, M.A. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat. Med. 2007, 13, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef]
- Gouilleux-Gruart, V.; Gouilleux, F.; Desaint, C.; Claisse, J.F.; Capiod, J.C.; Delobel, J.; Weber-Nordt, R.; Dusanter-Fourt, I.; Dreyfus, F.; Groner, B.; et al. STAT related transcription factors are constitutively activated in peripheral blood cells from acute leukemia patients. Blood 1996, 87, 1692–1697. [Google Scholar] [CrossRef]
- Spivak, J.L. The chronic myeloproliferative disorders: Clonality and clinical heterogeneity. Semin. Hematol. 2004, 41, 1–5. [Google Scholar] [CrossRef]
- Van Etten, R.A.; Koschmieder, S.; Delhommeau, F.; Perrotti, D.; Holyoake, T.; Pardanani, A.; Mesa, R.; Green, T.; Ibrahim, A.R.; Mughal, T.; et al. The Ph-positive and Ph-negative myeloproliferative neoplasms: Some topical pre-clinical and clinical issues. Haematologica 2011, 96, 590–601. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Busers, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Horny, H.P.; Escribano, L.; Longley, B.J.; Li, C.Y.; Schwartz, L.B.; Marone, G.; Nuñez, R.; Akin, C.; Sotlar, K.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef]
- Lim, K.H.; Pardanani, A.; Tefferi, A. KIT and mastocytosis. Acta Haematol. 2008, 119, 194–198. [Google Scholar] [CrossRef]
- Goemans, B.F.; Zwaan, C.M.; Miller, M.; Zimmermann, M.; Harlow, A.; Meshinchi, S.; Loonen, A.H.; Hählen, K.; Reinhardt, D.; Creutzig, U.; et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 2005, 19, 1536–1542. [Google Scholar] [CrossRef]
- Fritsche-Polanz, R.; Fritz, M.; Huber, A.; Sotlar, K.; Sperr, W.R.; Mannhalter, C.; Födinger, M.; Valent, P. High frequency of concomitant mastocytosis in patients with acute myeloid leukemia exhibiting the transforming KIT mutation D816V. Mol. Oncol. 2010, 4, 335–346. [Google Scholar] [CrossRef]
- Coppo, P.; Flamant, S.; De Mas, V.; Jarrier, P.; Guillier, M.; Bonnet, M.L.; Lacout, C.; Guilhot, F.; Vainchenker, W.; Turhan, A.G. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of Bcr/abl-positive leukemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Chung, Y.J.; Park, B.B.; Kang, Y.J.; Kim, T.M.; Eaves, C.J.; Oh, I.H. Unique effects of Stat3 on the early phase of hematopoietic stem cell regeneration. Blood 2006, 108, 1208–1215. [Google Scholar] [CrossRef][Green Version]
- Schepers, H.; van Gosliga, D.; Wierenga, A.T.; Eggen, B.J.; Schuringa, J.J.; Vellenga, E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood 2007, 110, 2880–2888. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Iwama, A.; Tadokoro, Y.; Shimoda, K.; Minoguchi, M.; Akira, S.; Tanaka, M.; Miyajima, A.; Kitamura, T.; Nakauchi, H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J. Exp. Med. 2005, 202, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, S.; Friedbichler, K.; Boudot, C.; Bourgeais, J.; Gouilleux-Gruart, V.; Régnier, A.; Herault, O.; Moriggl, R.; Gouilleux, F. STAT5A/5B-specific expansion and transformation of hematopoietic stem cells. Blood Cancer J. 2017, 7, 514. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B-but not STAT5A-has a key role in BCR/ABL-induced leukemia. Leukemia 2019, 33, 1583–1597. [Google Scholar] [CrossRef] [PubMed]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.H.; Ame, S.; et al. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.B.; Gunby, R.H.; Piazza, R.; Galietta, A.; Rostagno, R.; Scapozza, L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003, 4, 75–85. [Google Scholar] [CrossRef]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Warsch, W.; Grundschober, E.; Berger, A.; Gille, L.; Cerny-Reiterer, S.; Tigan, A.S.; Hoelbl-Kovacic, A.; Valent, P.; Moriggl, R.; Sexl, V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukemia. Oncotarget 2012, 3, 1669–1687. [Google Scholar] [CrossRef]
- Bourgeais, J.; Ishac, N.; Medrzycki, M.; Brachet-Botineau, M.; Desbourdes, L.; Gouilleux-Gruart, V.; Pecnard, E.; Rouleux-Bonnin, F.; Gyan, E.; Domenech, J.; et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget 2017, 8, 41876–41889. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Casetti, L.; Martin-Lannerée, S.; Najjar, I.; Plo, I.; Augé, S.; Roy, L.; Chomel, J.C.; Lauret, E.; Turhan, A.G.; Dusanter-Fourt, I. Differential contributions of STAT5A and STAT5B to stress protection and tyrosine kinase inhibitor resistance of chronic myeloid leukemia stem/progenitor cells. Cancer Res. 2013, 73, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Bewry, N.N.; Nair, R.R.; Emmons, M.; Boulware, D.; Pinilla-Ibarz, J.; Hazlehurst, L.A. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol. Cancer Ther. 2008, 7, 3169–3175. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Schneeweiss, M.; Eisenwort, G.; Berger, D.; Herrmann, H.; Blatt, K.; Greiner, G.; Byrgazov, K.; Hoermann, G.; Konopleva, M.; et al. Combined targeting of STAT3 and STAT5: A novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica 2017, 102, 1519–1529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; Van Etten, R.A. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef]
- Yan, D.; Hutchison, R.E.; Mohi, G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood 2012, 119, 3539–3549. [Google Scholar] [CrossRef]
- Yan, D.; Jobe, F.; Hutchison, R.E.; Mohi, G. Deletion of Stat3 enhances myeloid cell expansion and increases the severity of myeloproliferative neoplasms in Jak2V617F knock-in mice. Leukemia 2015, 29, 2050–2061. [Google Scholar] [CrossRef]
- Grisouard, J.; Shimizu, T.; Duek, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Deletion of Stat3 in hematopoietic cells enhances thrombocytosis and shortens survival in a JAK2-V617F mouse model of MPN. Blood 2015, 125, 2131–2140. [Google Scholar] [CrossRef]
- Chaix, A.; Lopez, S.; Voisset, E.; Gros, L.; Dubreuil, P.; De Sepulveda, P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011, 286, 5956–5966. [Google Scholar] [CrossRef]
- Gilliland, D.G. Molecular genetics of human leukemias: New insights into therapy. Semin. Hematol. 2002, 39, 6–11. [Google Scholar] [CrossRef]
- Martignoles, J.A.; Delhommeau, F.; Hirsch, P. Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. Int. J. Mol. Sci. 2018, 19, 3850. [Google Scholar] [CrossRef]
- Benekli, M.; Xia, Z.; Donohue, K.A.; Ford, L.A.; Pixley, L.A.; Baer, M.R.; Baumann, H.; Wetzler, M. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood 2002, 99, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Heydt, Q.; Saland, E.; Boutzen, H.; Kaoma, T.; Sarry, J.E.; Joffre, C.; Récher, C. Oncogenic KIT mutations induce STAT3-dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis 2019, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.; Dos Santos, C.; Zhang, Q.; Kozlowska, A.; Liu, H.; Gao, C.; Moreira, D.; Swiderski, P.; Jozwiak, A.; Kline, J.; et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood 2014, 123, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hossain, D.M.; Duttagupta, P.; Moreira, D.; Zhao, X.; Won, H.; Buettner, R.; Nechaev, S.; Majka, M.; Zhang, B.; et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood 2016, 127, 1687–1700. [Google Scholar] [CrossRef]
- Kiyoi, H.; Naoe, T. FLT3 in human hematologic malignancies. Leuk. Lymphoma 2002, 43, 1541–1547. [Google Scholar] [CrossRef]
- Spiekermann, K.; Bagrintseva, K.; Schwab, R.; Schmieja, K.; Hiddemann, W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin. Cancer Res. 2003, 9, 2140–2150. [Google Scholar]
- Obermann, E.C.; Arber, C.; Jotterand, M.; Tichelli, A.; Hirschmann, P.; Tzankov, A. Expression of pSTAT5 predicts FLT3 internal tandem duplications in acute myeloid leukemia. Ann. Hematol. 2010, 89, 663–669. [Google Scholar] [CrossRef][Green Version]
- Rocnik, J.L.; Okabe, R.; Yu, J.C.; Lee, B.H.; Giese, N.; Schenkein, D.P.; Gilliland, D.G. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood 2006, 108, 339–345. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Xiang, M.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Liu, S.; Kharbanda, S.; Christie, A.L.; Nicolais, M.; et al. The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer 2012, 3, 503–511. [Google Scholar] [CrossRef]
- Müller, T.A.; Grundler, R.; Istvanffy, R.; Rudelius, M.; Hennighausen, L.; Illert, A.L.; Duyster, J. Lineage-specific STAT5 target gene activation in hematopoietic progenitor cells predicts the FLT3(+) -mediated leukemic phenotype. Leukemia 2016, 30, 1725–1733. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.F.; Naughton, R.; Stanicka, J.; Gough, D.R.; Bhatt, L.; Dickinson, B.C.; Chang, C.J.; Cotter, T.G. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS ONE 2012, 7, e34050. [Google Scholar] [CrossRef] [PubMed]
- Dumas, P.Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossée, M.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Arnould, C.; Philippe, C.; Bourdon, V.; Gregoire, M.J.; Berger, R.; Jonveaux, P. The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor alpha in acute promyelocytic-like leukaemia. Hum. Mol. Genet. 1999, 8, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Tweardy, D.J. Interactions of STAT5b-RARalpha, a novel acute promyelocytic leukemia fusion protein, with retinoic acid receptor and STAT3 signaling pathways. Blood 2002, 99, 2637–2646. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Gajjar, A.J.; Kane, J.R.; Qaddoumi, I.A.; Pappo, A.S. Challenging issues in pediatric oncology. Nat. Rev. Clin. Oncol. 2011, 8, 540–549. [Google Scholar] [CrossRef]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef]
- Joliot, V.; Cormier, F.; Medyouf, H.; Alcalde, H.; Ghysdael, J. Constitutive STAT5 activation specifically cooperates with the loss of p53 function in B-cell lymphomagenesis. Oncogene 2006, 25, 4573–4584. [Google Scholar] [CrossRef][Green Version]
- Minieri, V.; De Dominici, M.; Porazzi, P.; Mariani, S.A.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; Porcu, P.; Nevalainen, M.T.; Calabretta, B. Targeting STAT5 or STAT5-Regulated Pathways Suppresses Leukemogenesis of Ph+ Acute Lymphoblastic Leukemia. Cancer Res. 2018, 78, 5793–5807. [Google Scholar] [CrossRef]
- Katerndahl, C.D.S.; Heltemes-Harris, L.M.; Willette, M.J.L.; Henzler, C.M.; Frietze, S.; Yang, R.; Schjerven, H.; Silverstein, K.A.T.; Ramsey, L.B.; Hubbard, G.; et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 2017, 18, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Heltemes-Harris, L.M.; Willette, M.J.; Ramsey, L.B.; Qiu, Y.H.; Neeley, E.S.; Zhang, N.; Thomas, D.A.; Koeuth, T.; Baechler, E.C.; Kornblau, S.M.; et al. Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Medrzycki, M.; Bunting, S.T.; Bunting, K.D. Stat5-deficient hematopoiesis is permissive for Myc-induced B-cell leukemogenesis. Oncotarget 2015, 6, 28961–28972. [Google Scholar] [CrossRef] [PubMed]
- Belton, A.; Xian, L.; Huso, T.; Koo, M.; Luo, L.Z.; Turkson, J.; Page, B.D.; Gunning, P.T.; Liu, G.; Huso, D.L.; et al. STAT3 inhibitor has potent antitumor activity in B-lineage acute lymphoblastic leukemia cells overexpressing the high mobility group A1 (HMGA1)-STAT3 pathway. Leuk. Lymphoma 2016, 57, 2681–2684. [Google Scholar] [CrossRef]
- Fasan, A.; Kern, W.; Grossmann, V.; Haferlach, C.; Haferlach, T.; Schnittger, S. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia 2013, 27, 1598–1600. [Google Scholar] [CrossRef]
- Rajala, H.L.; Porkka, K.; Maciejewski, J.P.; Loughran, T.P., Jr.; Mustjoki, S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann. Med. 2014, 46, 114–122. [Google Scholar] [CrossRef]
- Rajala, H.L.; Eldfors, S.; Kuusanmäki, H.; van Adrichem, A.J.; Olson, T.; Lagström, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef]
- Dutta, A.; Yan, D.; Hutchison, R.E.; Mohi, G. STAT3 mutations are not sufficient to induce large granular lymphocytic leukaemia in mice. Br. J. Haematol. 2018, 180, 911–915. [Google Scholar] [CrossRef]
- Klein, K.; Witalisz-Siepracka, A.; Maurer, B.; Prinz, D.; Heller, G.; Leidenfrost, N.; Prchal-Murphy, M.; Suske, T.; Moriggl, R.; Sexl, V. STAT5BN642H drives transformation of NKT cells: A novel mouse model for CD56+ T-LGL leukemia. Leukemia 2019, 33, 2336–2340. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Capron, C.; Jondeau, K.; Casetti, L.; Jalbert, V.; Costa, C.; Verhoeyen, E.; Massé, J.M.; Coppo, P.; Béné, M.C.; Bourdoncle, P.; et al. Viability and stress protection of chronic lymphoid leukemia cells involves overactivation of mitochondrial phosphoSTAT3Ser727. Cell Death Dis. 2014, 5, e1451. [Google Scholar] [CrossRef] [PubMed]
- de Totero, D.; Meazza, R.; Capaia, M.; Fabbi, M.; Azzarone, B.; Balleari, E.; Gobbi, M.; Cutrona, G.; Ferrarini, M.; Ferrini, S. The opposite effects of IL-15 and IL-21 on CLL B cells correlate with differential activation of the JAK/STAT and ERK1/2 pathways. Blood 2008, 111, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Holtick, U.; Vockerodt, M.; Ahmadi, T.; Haier, B.; Behrmann, I.; Heinrich, P.C.; Diehl, V.; Tesch, H. STAT3 is constitutively activated in Hodgkin cell lines. Blood 2001, 98, 762–770. [Google Scholar] [CrossRef]
- Holtick, U.; Vockerodt, M.; Pinkert, D.; Schoof, N.; Stürzenhofecker, B.; Kussebi, N.; Lauber, K.; Wesselborg, S.; Löffler, D.; Horn, F.; et al. STAT3 is essential for Hodgkin lymphoma cell proliferation and is a target of tyrphostin AG17 which confers sensitization for apoptosis. Leukemia 2005, 19, 936–944. [Google Scholar] [CrossRef]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.; et al. IL-21 is expressed in Hodgkin lymphoma and activates STAT5: Evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood 2008, 111, 4706–4715. [Google Scholar] [CrossRef]
- Ding, B.B.; Yu, J.J.; Yu, R.Y.; Mendez, L.M.; Shaknovich, R.; Zhang, Y.; Cattoretti, G.; Ye, B.H. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008, 111, 1515–1523. [Google Scholar] [CrossRef]
- Lam, L.T.; Wright, G.; Davis, R.E.; Lenz, G.; Farinha, P.; Dang, L.; Chan, J.W.; Rosenwald, A.; Gascoyne, R.D.; Staudt, L.M. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008, 111, 3701–3713. [Google Scholar] [CrossRef]
- Pan, Y.R.; Chen, C.C.; Chan, Y.T.; Wang, H.J.; Chien, F.T.; Chen, Y.L.; Liu, J.L.; Yang, M.H. STAT3-coordinated migration facilitates the dissemination of diffuse large B-cell lymphomas. Nat. Commun. 2018, 9, 3696. [Google Scholar] [CrossRef]
- Skarbnik, A.P.; Burki, M.; Pro, B. Peripheral T-cell lymphomas: A review of current approaches and hopes for the future. Front Oncol. 2013, 3, 138. [Google Scholar] [CrossRef]
- Netchiporouk, E.; Litvinov, I.V.; Moreau, L.; Gilbert, M.; Sasseville, D.; Duvic, M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle 2014, 13, 3331–3335. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Song, T.L.; Nairismägi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Yu, H. Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 2008, 20, 228–233. [Google Scholar] [CrossRef]
- Roodman, G.D. Pathogenesis of myeloma bone disease. Leukemia 2009, 23, 435–441. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Núñez, G.; et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Bharti, A.C.; Shishodia, S.; Reuben, J.M.; Weber, D.; Alexanian, R.; Raj-Vadhan, S.; Estrov, Z.; Talpaz, M.; Aggarwal, B.B. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004, 103, 3175–3184. [Google Scholar] [CrossRef]
- Jung, S.H.; Ahn, S.Y.; Choi, H.W.; Shin, M.G.; Lee, S.S.; Yang, D.H.; Ahn, J.S.; Kim, Y.K.; Kim, H.J.; Lee, J.J. STAT3 expression is associated with poor survival in non-elderly adult patients with newly diagnosed multiple myeloma. Blood Res. 2017, 52, 293–299. [Google Scholar] [CrossRef]
- Chong, P.S.Y.; Chng, W.J.; de Mel, S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers 2019, 11, 731. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito Maria, A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Beldi-Ferchiou, A.; Skouri, N.; Ben Ali, C.; Safra, I.; Abdelkefi, A.; Ladeb, S.; Mrad, K.; Ben Othman, T.; Ben Ahmed, M. Abnormal repression of SHP-1, SHP-2 and SOCS-1 transcription sustains the activation of the JAK/STAT3 pathway and the progression of the disease in multiple myeloma. PLoS ONE 2017, 12, e0174835. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Fung, T.K.; Cheung, W.C.; Liang, R.; Kwong, Y.L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef] [PubMed]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Hösler, N.; Riedel, D.; Zipfel, I.; Buschmann, T.; Kämpf, C.; Reiche, K.; Burger, R.; Gramatzki, M.; Hackermüller, J.; et al. STAT3-induced long noncoding RNAs in multiple myeloma cells display different properties in cancer. Sci. Rep. 2017, 7, 7976. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Yang, S.; Weatherburn, C.; Gibson, J.; Ho, P.J.; Suen, H.; Hart, D.; Joshua, D. Phospho-flow detection of constitutive and cytokine-induced pSTAT3/5, pAKT and pERK expression highlights novel prognostic biomarkers for patients with multiple myeloma. Leukemia 2015, 29, 483–490. [Google Scholar] [CrossRef]
- Kloth, M.T.; Catling, A.D.; Silva, C.M. Novel activation of STAT5b in response to epidermal growth factor. J. Biol. Chem. 2002, 277, 8693–8701. [Google Scholar] [CrossRef]
- Schaller-Schönitz, M.; Barzan, D.; Williamson, A.J.; Griffiths, J.R.; Dallmann, I.; Battmer, K.; Ganser, A.; Whetton, A.D.; Scherr, M.; Eder, M. BCR-ABL affects STAT5A and STAT5B differentially. PLoS ONE 2014, 9, e97243. [Google Scholar] [CrossRef]
- Reich, N.C. STATs get their move on. JAK-STAT 2013, 2, e27080. [Google Scholar] [CrossRef]
- Kawashima, T.; Bao, Y.C.; Nomura, Y.; Moon, Y.; Tonozuka, Y.; Minoshima, Y.; Hatori, T.; Tsuchiya, A.; Kiyono, M.; Nosaka, T.; et al. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J. Cell Biol. 2006, 175, 937–946. [Google Scholar] [CrossRef]
- Kawashima, T.; Bao, Y.C.; Minoshima, Y.; Nomura, Y.; Hatori, T.; Hori, T.; Fukagawa, T.; Fukada, T.; Takahashi, N.; Nosaka, T.; et al. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol. Cell. Biol. 2009, 29, 1796–1813. [Google Scholar] [CrossRef] [PubMed]
- Tonozuka, Y.; Minoshima, Y.; Bao, Y.C.; Moon, Y.; Tsubono, Y.; Hatori, T.; Nakajima, H.; Nosaka, T.; Kawashima, T.; Kitamura, T. A GTPase-activating protein binds STAT3 and is required for IL-6-induced STAT3 activation and for differentiation of a leukemic cell line. Blood 2004, 104, 3550–3557. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Hoelbl-Kovacic, A.; Bourgeais, J.; Hoefling, L.; Warsch, W.; Grundschober, E.; Uras, I.Z.; Menzl, I.; Putz, E.M.; Hoermann, G.; et al. Pak-dependent stat5 serine phosphorylation is required for bcr-abl-induced leukemogenesis. Leukemia 2014, 28, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Ghosh, J.; Ramdas, B.; Mali, R.S.; Martin, H.; Kobayashi, M.; Vemula, S.; Canela, V.H.; Waskow, E.R.; Visconte, V.; et al. Regulation of Stat5 by FAK and PAK1 in Oncogenic FLT3- and KIT-Driven Leukemogenesis. Cell Rep. 2014, 9, 1333–1348. [Google Scholar] [CrossRef]
- Harir, N.; Pecquet, C.; Kerenyi, M.; Sonneck, K.; Kovacic, B.; Nyga, R.; Brevet, M.; Dhennin, I.; Gouilleux-Gruart, V.; Beug, H.; et al. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood 2007, 109, 1678–1686. [Google Scholar] [CrossRef]
- Chatain, N.; Ziegler, P.; Fahrenkamp, D.; Jost, E.; Moriggl, R.; Schmitz-Van de Leur, H.; Muller-Newen, G. Src family kinases mediate cytoplasmic retention of activated stat5 in bcr-abl-positive cells. Oncogene 2013, 32, 3587–3597. [Google Scholar] [CrossRef]
- Meyer, W.K.; Reichenbach, P.; Schindler, U.; Soldaini, E.; Nabholz, M. Interaction of STAT5 dimers on two low affinity binding sites mediates interleukin 2 (IL-2) stimulation of IL-2 receptor alpha gene transcription. J. Biol. Chem. 1997, 272, 31821–31828. [Google Scholar] [CrossRef]
- Soldaini, E.; John, S.; Moro, S.; Bollenbacher, J.; Schindler, U.; Leonard, W.J. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol. Cell. Biol. 2000, 20, 389–401. [Google Scholar] [CrossRef]
- Hu, T.; Yeh, J.E.; Pinello, L.; Jacob, J.; Chakravarthy, S.; Yuan, G.C.; Chopra, R.; Frank, D.A. Impact of the N-Terminal Domain of STAT3 in STAT3-Dependent Transcriptional Activity. Mol. Cell. Biol. 2015, 35, 3284–3300. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. Maximal STAT5-induced proliferation and self-renewal at intermediate STAT5 activity levels. Mol. Cell. Biol. 2008, 28, 6668–6680. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Grgurevic, S.; Faderl, S.; Ferrajoli, A.; Wierda, W.G.; Martinez, M.; et al. At High Levels, Constitutively Activated STAT3 Induces Apoptosis of Chronic Lymphocytic Leukemia Cells. J. Immunol. 2016, 196, 4400–4409. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, T.; Kawashima, T.; Misawa, K.; Ikuta, K.; Mui, A.L.; Kitamura, T. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 1999, 18, 4754–4765. [Google Scholar] [CrossRef] [PubMed]
- Friedbichler, K.; Kerenyi, M.A.; Kovacic, B.; Li, G.; Hoelbl, A.; Yahiaoui, S.; Sexl, V.; Mullner, E.W.; Fajmann, S.; Cerny-Reiterer, S.; et al. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood 2010, 116, 1548–1558. [Google Scholar] [CrossRef]
- Mitra, A.; Ross, J.A.; Rodriguez, G.; Nagy, Z.S.; Wilson, H.L.; Kirken, R.A. Signal transducer and activator of transcription 5b (Stat5b) serine 193 is a novel cytokine-induced phospho-regulatory site that is constitutively activated in primary hematopoietic malignancies. J. Biol. Chem. 2012, 287, 16596–16608. [Google Scholar] [CrossRef]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132–2142. [Google Scholar] [CrossRef]
- Zhuang, S. Regulation of STAT Signaling by Acetylation. Cell Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [PubMed]
- Kosan, C.; Ginter, T.; Heinzel, T.; Krämer, O.H. STAT5 acetylation: Mechanisms and consequences for immunological control and leukemogenesis. JAKSTAT 2013, 2, e26102. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Unal, H.; Willard, B.; Yang, J.; Karnik, S.S.; Stark, G.R. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J. Biol. Chem. 2014, 289, 30763–30771. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound stat3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of ezh2 activates stat3 signaling via stat3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 2011, 12, 1212–1220. [Google Scholar] [CrossRef]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef]
- Li, L.; Cheung, S.H.; Evans, E.L.; Shaw, P.E. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010, 70, 8222–8232. [Google Scholar] [CrossRef]
- Sebastián, C.; Herrero, C.; Serra, M.; Lloberas, J.; Blasco, M.A.; Celada, A. Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation. J. Immunol. 2009, 183, 2356–2364. [Google Scholar] [CrossRef]
- Krämer, O.H.; Moriggl, R. Acetylation and sumoylation control STAT5 activation antagonistically. JAKSTAT 2012, 1, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, M.; Li, J.; Xiao, M.; Chin, Y.E.; Cheng, J.; Yeh, E.T.; Yang, J.; Yi, J. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene 2016, 35, 5826–5838. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Xiang, M.; Frank, D.A. Distinct roles of STAT3 and STAT5 in the pathogenesis and targeted therapy of breast cancer. Mol. Cell. Endocrinol. 2014, 382, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Nelson, E.A.; Yeh, J.E.; Pinello, L.; Yuan, G.C.; Frank, D.A. STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol. Cell. Biol. 2013, 33, 2879–2890. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wie, L.; Vahedi, G.; et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells. Cancers 2015, 7, 503–537. [Google Scholar] [CrossRef]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Tse, W.; Gouilleux, F.; Moriggl, R.; Bunting, K.D. Effective targeting of stat5-mediated survival in myeloproliferative neoplasms using abt-737 combined with rapamycin. Leukemia 2010, 24, 1397–1405. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (stat) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef]
- Avalle, L.; Camporeale, A.; Morciano, G.; Caroccia, N.; Ghetti, E.; Orecchia, V.; Viavattene, D.; Giorgi, C.; Pinton, P.; Poli, V. Stat3 localizes to the ER, acting as a gatekeeper for er-mitochondrion ca (2+) fluxes and apoptotic responses. Cell Death Differ. 2019, 26, 932–942. [Google Scholar] [CrossRef]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L. Mitochondrial Ca2⁺ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife 2015, 4, e06376. [Google Scholar] [CrossRef] [PubMed]
- Carbognin, E.; Betto, R.M.; Soriano, M.E.; Smith, A.G.; Martello, G. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J. 2016, 35, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Chueh, F.Y.; Leong, K.F.; Yu, C.L. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem. Biophys. Res. Commun. 2010, 402, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Ferrajoli, A.; Burger, J.; Thompson, P.; Jain, N.; Wierda, W.; et al. STAT3 is constitutively acetylated on lysine 685 residues in chronic lymphocytic leukemia cells. Oncotarget 2018, 9, 33710–33718. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Gough, D.J.; Marié, I.J.; Lobry, C.; Aifantis, I.; Levy, D.E. STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood 2014, 124, 2252–2261. [Google Scholar] [CrossRef]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor stat3 into mitochondria depends on grim-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Mantel, C.; Messina-Graham, S.; Moh, A.; Cooper, S.; Hangoc, G.; Fu, X.Y.; Broxmeyer, H.E. Mouse hematopoietic cell-targeted STAT3 deletion: Stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood 2012, 120, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Yang, Y.M.; Liang, F.X.; Gough, D.J.; Levy, D.E.; Sehgal, P.B. Nongenomic stat5-dependent effects on golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 2012, 302, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Cholez, E.; Debuysscher, V.; Bourgeais, J.; Boudot, C.; Leprince, J.; Tron, F.; Brassart, B.; Regnier, A.; Bissac, E.; Pecnard, E.; et al. Evidence for a protective role of the STAT5 transcription factor against oxidative stress in human leukemic pre-B cells. Leukemia 2012, 26, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Domoszlai, T.; Kleshchanok, D.; Lehmann, S.; Schmitt, A.; Poli, V.; Richtering, W.; Muller-Newen, G. The role of the N-terminal domain in dimerization and nucleocytoplasmic shuttling of latent STAT3. J. Cell Sci. 2011, 124, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Göttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016, 35, 580–594. [Google Scholar] [CrossRef]
- Nagy, Z.S.; Rui, H.; Stepkowski, S.M.; Karras, J.; Kirken, R.A. A preferential role for STAT5, not constitutively active STAT3, in promoting survival of a human lymphoid tumor. J. Immunol. 2006, 177, 5032–5040. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the jak/stat pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. Socs: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000, 113, 2813–2819. [Google Scholar]
- Shuai, K. Modulation of stat signaling by stat-interacting proteins. Oncogene 2000, 19, 2638–2644. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Kim, J.S.; Zhang, S.; Yuan, J.; Huang, M.; Glenn, M.; Haura, E.; Sebti, S.; Hamilton, A.D.; Jove, R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 2004, 3, 261–269. [Google Scholar] [PubMed]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef]
- Sen, M.; Grandis, J.R. Nucleic acid-based approaches to STAT inhibition. JAKSTAT 2012, 1, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Sharma, S.V.; Settleman, J.; Frank, D.A. A chemical biology approach to developing STAT inhibitors: Molecular strategies for accelerating clinical translation. Oncotarget 2011, 2, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Frank, D.A. Screening approaches to generating STAT inhibitors: Allowing the hits to identify the targets. JAKSTAT 2012, 1, 292–299. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63, 375–381. [Google Scholar] [PubMed]
- Konig, H.; Holyoake, T.L.; Bhatia, R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood 2008, 111, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q.; et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef]
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sawyers, C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef]
- Crump, M.; Hedley, D.; Kamel-Reid, S.; Leber, B.; Wells, R.; Brandwein, J.; Buckstein, R.; Kassis, J.; Minden, M.; Matthews, J.; et al. A randomized phase I clinical and biologic study of two schedules of sorafenib in patients with myelodysplastic syndrome or acute myeloid leukemia: A NCIC (National Cancer Institute of Canada) Clinical Trials Group Study. Leuk. Lymphoma 2010, 51, 252–260. [Google Scholar] [CrossRef]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; O’Brien, S.; Estrov, Z.; Borthakur, G.; Thomas, D.; et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856–1862. [Google Scholar] [CrossRef]
- Knapper, S.; Burnett, A.K.; Littlewood, T.; Kell, W.J.; Agrawal, S.; Chopra, R.; Clark, R.; Levis, M.J.; Small, D. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 2006, 108, 3262–3270. [Google Scholar] [CrossRef] [PubMed]
- Shiotsu, Y.; Kiyoi, H.; Ishikawa, Y.; Tanizaki, R.; Shimizu, M.; Umehara, H.; Ishii, K.; Mori, Y.; Ozeki, K.; Minami, Y.; et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood 2009, 114, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Cortes, J.; Roboz, G.J.; Rao, N.; Arowojolu, O.; Stine, A.; Shiotsu, Y.; Shudo, A.; Akinaga, S.; Small, D.; et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood 2009, 113, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, A.M.; Foran, J.M.; Fiedler, W.; Serve, H.; Paquette, R.L.; Cooper, M.A.; Yuen, H.A.; Louie, S.G.; Kim, H.; Nicholas, S.; et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin. Cancer Res. 2003, 9, 5465–5476. [Google Scholar] [PubMed]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Götze, K.; Weber, D.; Göhring, G.; Teleanu, V.; et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Paz, K. Tandutinib, an oral, small-molecule inhibitor of FLT3 for the treatment of AML and other cancer indications. IDrugs 2008, 11, 46–56. [Google Scholar] [PubMed]
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A promising option for acute myeloid leukemia. Drug Des. Dev. Ther. 2019, 13, 1117–1125. [Google Scholar] [CrossRef]
- Sutamtewagul, G.; Vigil, C.E. Clinical use of FLT3 inhibitors in acute myeloid leukemia. Onco Targets Ther. 2018, 11, 7041–7052. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark Res. 2019, 7, 19. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol. Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.H.; Gotlib, J. Clinical Validation of KIT Inhibition in Advanced Systemic Mastocytosis. Curr. Hematol. Malig. Rep. 2018, 13, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Kampa-Schittenhelm, K.M.; Frey, J.; Haeusser, L.A.; Illing, B.; Pavlovsky, A.A.; Blumenstock, G.; Schittenhelm, M.M. Crenolanib is a type I tyrosine kinase inhibitor that inhibits mutant KIT D816 isoforms prevalent in systemic mastocytosis and core binding factor leukemia. Oncotarget 2017, 8, 82897–82909. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Griswold, I.J.; La Rosée, P.; Yee, K.W.; Heinrich, M.C.; Reimer, C.L.; Druker, B.J.; Deininger, M.W. Sensitivity of oncogenic KIT mutants to the kinase inhibitors MLN518 and PD180970. Blood 2004, 104, 3754–3757. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mascarenhas, J.; Baer, M.R.; Kessler, C.; Hexner, E.; Tremblay, D.; Price, L.; Sandy, L.; Weinberg, R.; Pahl, H.; Silverman, L.R.; et al. Phase II trial of Lestaurtinib, a JAK2 inhibitor, in patients with myelofibrosis. Leuk. Lymphoma 2019, 60, 1343–1345. [Google Scholar] [CrossRef] [PubMed]
- Hexner, E.; Roboz, G.; Hoffman, R.; Luger, S.; Mascarenhas, J.; Carroll, M.; Clementi, R.; Bensen-Kennedy, D.; Moliterno, A. Open-label study of oral CEP-701 (lestaurtinib) in patients with polycythaemia vera or essential thrombocythaemia with JAK2-V617F mutation. Br. J. Haematol. 2014, 164, 83–93. [Google Scholar] [CrossRef]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000 Res. 2018, 7, 82. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef]
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jørgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood 2014, 124, 1492–1501. [Google Scholar] [CrossRef]
- Van Den Neste, E.; André, M.; Gastinne, T.; Stamatoullas, A.; Haioun, C.; Belhabri, A.; Reman, O.; Casasnovas, O.; Ghesquieres, H.; Verhoef, G.; et al. A phase II study of the oral JAK1/JAK2 inhibitor ruxolitinib in advanced relapsed/refractory Hodgkin lymphoma. Haematologica 2018, 103, 840–848. [Google Scholar] [CrossRef]
- Ghermezi, M.; Spektor, T.M.; Berenson, J.R. The role of JAK inhibitors in multiple myeloma. Clin. Adv. Hematol. Oncol. 2019, 17, 500–505. [Google Scholar] [PubMed]
- Pemmaraju, N.; Kantarjian, H.; Kadia, T.; Cortes, J.; Borthakur, G.; Newberry, K.; Garcia-Manero, G.; Ravandi, F.; Jabbour, E.; Dellasala, S.; et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.E.; Mesa, R.A. Pacritinib and its use in the treatment of patients with myelofibrosis who have thrombocytopenia. Future Oncol. 2018, 14, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Romaguera, J.; Fanale, M.; McLaughlin, P.; Hagemeister, F.; Copeland, A.; Neelapu, S.; Kwak, L.; Shah, J.; de Castro Faria, S.; et al. Phase I study of a novel oral Janus kinase 2 inhibitor, SB1518, in patients with relapsed lymphoma: Evidence of clinical and biologic activity in multiple lymphoma subtypes. J. Clin. Oncol. 2012, 30, 4161–4167. [Google Scholar] [CrossRef]
- Pardanani, A.; Hood, J.; Lasho, T.; Levine, R.L.; Martin, M.B.; Noronha, G.; Finke, C.; Mak, C.C.; Mesa, R.; Zhu, H.; et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007, 21, 1658–1668. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Kimlinger, T.; Haug, J.; Timm, M.; Wellik, L.; Halling, T.; Pardanani, A.; Tefferi, A.; Rajkumar, S.V.; Kumar, S. TG101209, a novel JAK2 inhibitor, has significant in vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. Am. J. Hematol. 2010, 85, 675–686. [Google Scholar] [CrossRef]
- Cheng, Z.; Yi, Y.; Xie, S.; Yu, H.; Peng, H.; Zhang, G. The effect of the JAK2 inhibitor TG101209 against T cell acute lymphoblastic leukemia (T-ALL) is mediated by inhibition of JAK-STAT signaling and activation of the crosstalk between apoptosis and autophagy signaling. Oncotarget 2017, 8, 106753–106763. [Google Scholar] [CrossRef]
- Meyer, S.C.; Keller, M.D.; Chiu, S.; Koppikar, P.; Guryanova, O.A.; Rapaport, F.; Xu, K.; Manova, K.; Pankov, D.; O’Reilly, R.J.; et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2015, 28, 15–28. [Google Scholar] [CrossRef]
- Wu, S.C.; Li, L.S.; Kopp, N.; Montero, J.; Chapuy, B.; Yoda, A.; Christie, A.L.; Liu, H.; Christodoulou, A.; van Bodegom, D.; et al. Activity of the Type II JAK2 Inhibitor CHZ868 in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 29–41. [Google Scholar] [CrossRef]
- Berdeja, J.; Palandri, F.; Baer, M.R.; Quick, D.; Kiladjian, J.J.; Martinelli, G.; Verma, A.; Hamid, O.; Walgren, R.; Pitou, C.; et al. Phase 2 study of gandotinib (LY2784544) in patients with myeloproliferative neoplasms. Leuk. Res. 2018, 71, 82–88. [Google Scholar] [CrossRef]
- Blair, H.A. Fedratinib: First Approval. Drugs 2019, 79, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chapuy, B.; Monti, S.; Sun, H.H.; Rodig, S.J.; Shipp, M.A. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin. Cancer Res. 2014, 20, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Hoffman, R.; Mascarenhas, J.; Soria, J.C.; Bahleda, R.; McCoon, P.; Tang, W.; Cortes, J.; Kantarjian, H.; Ribrag, V. A phase I, open-label, multi-center study of the JAK2 inhibitor AZD1480 in patients with myelofibrosis. Leuk. Res. 2015, 39, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Suryani, S.; Bracken, L.S.; Harvey, R.C.; Sia, K.C.; Carol, H.; Chen, I.M.; Evans, K.; Dietrich, P.A.; Roberts, K.G.; Kurmasheva, R.T.; et al. Evaluation of the in vitro and in vivo efficacy of the JAK inhibitor AZD1480 against JAK-mutated acute lymphoblastic leukemia. Mol. Cancer Ther. 2015, 14, 364–374. [Google Scholar] [CrossRef]
- Derenzini, E.; Lemoine, M.; Buglio, D.; Katayama, H.; Ji, Y.; Davis, R.E.; Sen, S.; Younes, A. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. 2011, 1, e46. [Google Scholar] [CrossRef]
- Scuto, A.; Krejci, P.; Popplewell, L.; Wu, J.; Wang, Y.; Kujawski, M.; Kowolik, C.; Xin, H.; Chen, L.; Wang, Y.; et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia 2011, 25, 538–550. [Google Scholar] [CrossRef]
- Burger, R.; Le Gouill, S.; Tai, Y.T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef]
- Li, J.; Favata, M.; Kelley, J.A.; Caulder, E.; Thomas, B.; Wen, X.; Sparks, R.B.; Arvanitis, A.; Rogers, J.D.; Combs, A.P.; et al. INCB16562, a JAK1/2 selective inhibitor, is efficacious against multiple myeloma cells and reverses the protective effects of cytokine and stromal cell support. Neoplasia 2010, 12, 28–38. [Google Scholar] [CrossRef]
- Faderl, S.; Ferrajoli, A.; Harris, D.; Van, Q.; Kantarjian, H.M.; Estrov, Z. Atiprimod blocks phosphorylation of JAK-STAT and inhibits proliferation of acute myeloid leukemia (AML) cells. Leuk. Res. 2007, 31, 91–95. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Manshouri, T.; Estrov, Z.; Harris, D.; Zhang, Y.; Gaikwad, A.; Kantarjian, H.M.; Verstovsek, S. Preclinical characterization of atiprimod, a novel JAK2 AND JAK3 inhibitor. Investig. New Drugs 2011, 29, 818–826. [Google Scholar] [CrossRef]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Furihata, M.; Yokoyama, A. AZ960, a novel Jak2 inhibitor, induces growth arrest and apoptosis in adult T-cell leukemia cells. Mol. Cancer Ther. 2010, 9, 3386–3395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mascarenhas, J.O.; Talpaz, M.; Gupta, V.; Foltz, L.M.; Savona, M.R.; Paquette, R.; Turner, A.R.; Coughlin, P.; Winton, E.; Burn, T.C.; et al. Primary analysis of a phase II open-label trial of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Haematologica 2017, 102, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.J.; Forero-Torres, A.; Sher, T.; Diefenbach, C.S.; Johnston, P.; Talpaz, M.; Pulini, J.; Zhou, L.; Scherle, P.; Chen, X.; et al. Phase 1 study of the PI3Kδ inhibitor INCB040093 ± JAK1 inhibitor itacitinib in relapsed/refractory B-cell lymphoma. Blood 2018, 132, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Talpaz, M.; Ritchie, E.; Wadleigh, M.; Odenike, O.; Jamieson, C.; Stein, B.; Uno, T.; Mesa, R.A. A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis. Leukemia 2017, 31, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Kuramoto, K.; Niwa, T.; Naito, H. NS-018 reduces myeloma cell proliferation and suppresses osteolysis through inhibition of the JAK2 and Src signaling pathways. Blood Cancer J. 2018, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Baffert, F.; Régnier, C.H.; De Pover, A.; Pissot-Soldermann, C.; Tavares, G.A.; Blasco, F.; Brueggen, J.; Chène, P.; Drueckes, P.; Erdmann, D.; et al. Potent and selective inhibition of polycythemia by the quinoxaline JAK2 inhibitor NVP-BSK805. Mol. Cancer Ther. 2010, 9, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Ringel, F.; Kaeda, J.; Schwarz, M.; Oberender, C.; Grille, P.; Dörken, B.; Marque, F.; Manley, P.W.; Radimerski, T.; le Coutre, P. Effects of Jak2 type 1 inhibitors NVP-BSK805 and NVP-BVB808 on Jak2 mutation-positive and Bcr-Abl-positive cell lines. Acta Haematol. 2014, 132, 75–86. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Cosenza, S.C.; Reddy, M.V.; Ha, J.H.; Baker, S.J.; Samanta, A.K.; Olnes, M.J.; Pfannes, L.; Sloand, E.M.; Arlinghaus, R.B.; et al. A Non-ATP-Competitive Dual Inhibitor of JAK2 and BCR-ABL Kinases: Elucidation of a Novel Therapeutic Spectrum Based on Substrate Competitive Inhibition. Genes Cancer. 2010, 1, 331–345. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; McLornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor-Naïve Patients With Myelofibrosis. J. Clin. Oncol. 2017, 35, 3844–3850. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Monaghan, K.A.; Khong, T.; Burns, C.J.; Spencer, A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia 2011, 25, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Lipka, D.B.; Hoffmann, L.S.; Heidel, F.; Markova, B.; Blum, M.C.; Breitenbuecher, F.; Kasper, S.; Kindler, T.; Levine, R.L.; Huber, C.; et al. LS104, a non-ATP-competitive small-molecule inhibitor of JAK2, is potently inducing apoptosis in JAK2V617F-positive cells. Mol. Cancer Ther. 2008, 7, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Breitenbuecher, F.; Hoehn, Y.; Heidel, F.; Lipka, D.B.; Markova, B.; Huber, C.; Kindler, T.; Fischer, T. The kinase inhibitor LS104 induces apoptosis, enhances cytotoxic effects of chemotherapeutic drugs and is targeting the receptor tyrosine kinase FLT3 in acute myeloid leukemia. Leuk. Res. 2008, 32, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Grunberger, T.; Demin, P.; Rounova, O.; Sharfe, N.; Cimpean, L.; Dadi, H.; Freywald, A.; Estrov, Z.; Roifman, C.M. Inhibition of acute lymphoblastic and myeloid leukemias by a novel kinase inhibitor. Blood 2003, 102, 4153–4158. [Google Scholar] [CrossRef][Green Version]
- Verstovsek, S.; Manshouri, T.; Quintás-Cardama, A.; Harris, D.; Cortes, J.; Giles, F.J.; Kantarjian, H.; Priebe, W.; Estrov, Z. WP1066, a novel JAK2 inhibitor, suppresses proliferation and induces apoptosis in erythroid human cells carrying the JAK2 V617F mutation. Clin. Cancer Res. 2008, 14, 788–796. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Faderl, S.; Van, Q.; Koch, P.; Harris, D.; Liu, Z.; Hazan-Halevy, I.; Wang, Y.; Kantarjian, H.M.; Priebe, W.; et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007, 67, 11291–11299. [Google Scholar] [CrossRef]
- Faderl, S.; Ferrajoli, A.; Harris, D.; Van, Q.; Priebe, W.; Estrov, Z. WP-1034, A Novel JAK-STAT Inhibitor, with Proapoptotic and Antileukemic Activity in Acute Myeloid Leukemia (AML). Anticancer Res. 2005, 25, 1841–1850. [Google Scholar]
- Verstovsek, S.; Tam, C.S.; Wadleigh, M.; Sokol, L.; Smith, C.C.; Bui, L.A.; Song, C.; Clary, D.O.; Olszynski, P.; Cortes, J.; et al. Phase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitor. Leuk. Res. 2014, 38, 316–322. [Google Scholar] [CrossRef][Green Version]
- Haridas, V.; Nishimura, G.; Xu, Z.X.; Connolly, F.; Hanausek, M.; Walaszek, Z.; Zoltaszek, R.; Gutterman, J.U. Avicin D: A protein reactive plant isoprenoid dephosphorylates Stat 3 by regulating both kinase and phosphatase activities. PLoS ONE 2009, 4, e5578. [Google Scholar] [CrossRef]
- Zhang, C.; Li, B.; Gaikwad, A.S.; Haridas, V.; Xu, Z.; Gutterman, J.U.; Duvic, M. Avicin D selectively induces apoptosis and downregulates p-STAT-3, bcl-2, and survivin in cutaneous T-cell lymphoma cells. J. Investig. Dermatol. 2008, 128, 2728–2735. [Google Scholar] [CrossRef]
- Pandey, M.K.; Sung, B.; Ahn, K.S.; Aggarwal, B.B. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol. Pharmacol. 2009, 75, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Pathak, A.K.; Nair, A.S.; Kunnumakkara, A.B.; Guha, S.; Sethi, G.; Aggarwal, B.B. Capsaicin is a novel blocker of constitutive and interleukin-6-inducible STAT3 activation. Clin. Cancer Res. 2007, 13, 3024–3032. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Hay, H.S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-κB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Ishdorj, G.; Johnston, J.B.; Gibson, S.B. Inhibition of constitutive activation of STAT3 by curcurbitacin-I (JSI-124) sensitized human B-leukemia cells to apoptosis. Mol. Cancer Ther. 2010, 9, 3302–3314. [Google Scholar] [CrossRef]
- van Kester, M.S.; Out-Luiting, J.J.; von dem Borne, P.A.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Cucurbitacin I inhibits Stat3 and induces apoptosis in Sézary cells. J. Investig. Dermatol. 2008, 128, 1691–1695. [Google Scholar] [CrossRef]
- Bi, L.; Yu, Z.; Wu, J.; Yu, K.; Hong, G.; Lu, Z.; Gao, S. Honokiol Inhibits Constitutive and Inducible STAT3 Signaling via PU.1-Induced SHP1 Expression in Acute Myeloid Leukemia Cells. Tohoku J. Exp. Med. 2015, 237, 163–172. [Google Scholar] [CrossRef]
- Yao, Y.; Sun, Y.; Shi, M.; Xia, D.; Zhao, K.; Zeng, L.; Yao, R.; Zhang, Y.; Li, Z.; Niu, M.; et al. Piperlongumine induces apoptosis and reduces bortezomib resistance by inhibiting STAT3 in multiple myeloma cells. Oncotarget 2016, 7, 73497–73508. [Google Scholar] [CrossRef]
- Kuttan, G.; Kumar, K.B.; Guruvayoorappan, C.; Kuttan, R. Antitumor, anti-invasion, and antimetastatic effects of curcumin. Adv. Exp. Med. Biol. 2007, 595, 173–184. [Google Scholar]
- Blasius, R.; Reuter, S.; Henry, E.; Dicato, M.; Diederich, M. Curcumin regulates signal transducer and activator of transcription (STAT) expression in K562 cells. Biochem. Pharmacol. 2006, 72, 1547–1554. [Google Scholar] [CrossRef]
- Rajasingh, J.; Raikwar, H.P.; Muthian, G.; Johnson, C.; Bright, J.J. Curcumin induces growth-arrest and apoptosis in association with the inhibition of constitutively active JAK-STAT pathway in T cell leukemia. Biochem. Biophys. Res. Commun. 2006, 340, 359–368. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.R.; Manogaran, P.S.; Al-Hussein, K.; Platanias, L.C.; Gutierrez, M.I.; Bhatia, K.G. Curcumin suppresses growth and induces apoptosis in primary effusion lymphoma. Oncogene 2005, 24, 7022–7030. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Queisser, N.; Wolfson, M.L.; Fraga, C.G.; Adamo, A.M.; Oteiza, P.I. Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin’s lymphoma cells. Int. J. Cancer. 2008, 123, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Donato, N.; Aggarwal, B.B. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J. Immunol. 2003, 171, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Deangelis, S.; Foust, E.; Fuchs, J.; Li, C.; Li, P.K.; Schwartz, E.B.; Lesinski, G.B.; Benson, D.; Lü, J.; et al. A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol. Cancer. 2010, 9, 217. [Google Scholar] [CrossRef]
- Liby, K.; Voong, N.; Williams, C.R.; Risingsong, R.; Royce, D.B.; Honda, T.; Gribble, G.W.; Sporn, M.B.; Letterio, J.J. The synthetic triterpenoid CDDO-Imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin. Cancer Res. 2006, 12, 4288–4293. [Google Scholar] [CrossRef]
- Nam, S.; Scuto, A.; Yang, F.; Chen, W.; Park, S.; Yoo, H.S.; Konig, H.; Bhatia, R.; Cheng, X.; Merz, K.H.; et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol. Oncol. 2012, 6, 276–283. [Google Scholar] [CrossRef]
- Zhou, J.; Bi, C.; Janakakumara, J.V.; Liu, S.C.; Chng, W.J.; Tay, K.G.; Poon, L.F.; Xie, Z.; Palaniyandi, S.; Yu, H.; et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood 2009, 113, 4052–4062. [Google Scholar] [CrossRef]
- Nam, S.; Buettner, R.; Turkson, J.; Kim, D.; Cheng, J.Q.; Muehlbeyer, S.; Hippe, F.; Vatter, S.; Merz, K.H.; Eisenbrand, G.; et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5998–6003. [Google Scholar] [CrossRef]
- Pinz, S.; Unser, S.; Rascle, A. The natural chemopreventive agent sulforaphane inhibits STAT5 activity. PLoS ONE 2014, 9, e99391. [Google Scholar] [CrossRef]
- Liu, S.; Walker, S.R.; Nelson, E.A.; Cerulli, R.; Xiang, M.; Toniolo, P.A.; Qi, J.; Stone, R.M.; Wadleigh, M.; Bradner, J.E.; et al. Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Mol. Cancer Ther. 2014, 13, 1194–1205. [Google Scholar] [CrossRef]
- Guerra, B.; Martín-Rodríguez, P.; Díaz-Chico, J.C.; McNaughton-Smith, G.; Jiménez-Alonso, S.; Hueso-Falcón, I.; Montero, J.C.; Blanco, R.; León, J.; Rodríguez-González, G.; et al. CM363, a novel naphthoquinone derivative which acts as multikinase modulator and overcomes imatinib resistance in chronic myelogenous leukemia. Oncotarget 2017, 8, 29679–29698. [Google Scholar] [CrossRef] [PubMed]
- Juen, L.; Brachet-Botineau, M.; Parmenon, C.; Bourgeais, J.; Hérault, O.; Gouilleux, F.; Viaud-Massuard, M.C.; Prié, G. New inhibitor targeting signal transducer and activator of transcription 5 (STAT5) signaling in myeloid leukemias. J. Med. Chem. 2017, 60, 6119–6136. [Google Scholar] [CrossRef] [PubMed]
- Brachet-Botineau, M.; Deynoux, M.; Vallet, N.; Polomski, M.; Juen, L.; Hérault, O.; Mazurier, F.; Viaud-Massuard, M.C.; Prié, G.; Gouilleux, F. A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells. Cancers 2019, 11, 2043. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Walker, S.R.; Kepich, A.; Gashin, L.B.; Hideshima, T.; Ikeda, H.; Chauhan, D.; Anderson, K.C.; Frank, D.A. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 2008, 112, 5095–5102. [Google Scholar] [CrossRef]
- Khanim, F.L.; Merrick, B.A.; Giles, H.V.; Jankute, M.; Jackson, J.B.; Giles, L.J.; Birtwistle, J.; Bunce, C.M.; Drayson, M.T. Redeployment-based drug screening identifies the anti-helminthic niclosamide as anti-myeloma therapy that also reduces free light chain production. Blood Cancer J. 2011, 1, e39. [Google Scholar] [CrossRef]
- Sharma, A.; Jyotsana, N.; Lai, C.K.; Chaturvedi, A.; Gabdoulline, R.; Görlich, K.; Murphy, C.; Blanchard, J.E.; Ganser, A.; Brown, E.; et al. Pyrimethamine as a Potent and Selective Inhibitor of Acute Myeloid Leukemia Identified by High-throughput Drug Screening. Curr. Cancer Drug Targets 2016, 16, 818–828. [Google Scholar] [CrossRef]
- Liu, H.; Qin, Y.; Zhai, D.; Zhang, Q.; Gu, J.; Tang, Y.; Yang, J.; Li, K.; Yang, L.; Chen, S.; et al. Antimalarial Drug Pyrimethamine Plays a Dual Role in Antitumor Proliferation and Metastasis through Targeting DHFR and TP. Mol. Cancer Ther. 2019, 18, 541–555. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Walker, S.R.; Kuang, Y.; Distel, R.J.; Frank, D.A. Dual inhibition of Jak2 and STAT5 enhances killing of myeloproliferative neoplasia cells. Leukemia 2012, 26, 1407–1410. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M.; Andreeff, M.; Ohsaka, A. Effects of PPARγ Ligands on Leukemia. PPAR Res. 2012, 2012. [Google Scholar] [CrossRef]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zang, C.; Fenner, M.H.; Liu, D.; Possinger, K.; Koeffler, H.P.; Elstner, E. Growth inhibition and apoptosis in human Philadelphia chromosome-positive lymphoblastic leukemia cell lines by treatment with the dual PPARα/γ ligand TZD18. Blood 2006, 107, 3683–3692. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Yang, X.Y.; Zhang, X.; Huang, J.; Hou, J.; Li, J.; Xiong, H.; Mihalic, K.; Zhu, H.; Xiao, W.; et al. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Ren, Z.; Cabell, L.A.; Schaefer, T.S.; McMurray, J.S. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003, 13, 633–636. [Google Scholar] [CrossRef]
- Coleman, D.R., IV; Ren, Z.; Mandal, P.K.; Cameron, A.G.; Dyer, G.A.; Muranjan, S.; Campbell, M.; Chen, X.; McMurray, J.S. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J. Med. Chem. 2005, 48, 6661–6670. [Google Scholar] [CrossRef]
- Siddiquee, K.A.; Gunning, P.T.; Glenn, M.; Katt, W.P.; Zhang, S.; Schrock, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef]
- Becker, S.; Groner, B.; Müller, C.W. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998, 394, 145–151. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef]
- Xu, X.; Kasembeli, M.; Jiang, X.; Tweardy, B.J.; Tweardy, D.J. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS ONE 2009, 4, e4783. [Google Scholar] [CrossRef]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; et al. Identification of a New Series of STAT3 Inhibitors by Virtual Screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers 2019, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Singh, J.; Zhang, X.; Yue, P.; Page, B.D.; Sharmeen, S.; Shahani, V.M.; Zhao, W.; Schimmer, A.D.; Turkson, J.; et al. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: Potent in vitro and tumor cell activities. Chembiochem 2009, 10, 1959–1964. [Google Scholar] [CrossRef]
- Fletcher, S.; Page, B.D.; Zhang, X.; Yue, P.; Li, Z.H.; Sharmeen, S.; Singh, J.; Zhao, W.; Schimmer, A.D.; Trudel, S.; et al. Antagonism of the Stat3-Stat3 protein dimer with salicylic acid based small molecules. Chem. Med. Chem. 2011, 6, 1459–1470. [Google Scholar] [CrossRef]
- Page, B.D.; Fletcher, S.; Yue, P.; Li, Z.; Zhang, X.; Sharmeen, S.; Datti, A.; Wrana, J.L.; Trudel, S.; Schimmer, A.D.; et al. Identification of a non-phosphorylated, cell permeable, small molecule ligand for the Stat3 SH2 domain. Bioorg. Med. Chem. Lett. 2011, 21, 5605–5609. [Google Scholar] [CrossRef]
- Page, B.D.; Croucher, D.C.; Li, Z.H.; Haftchenary, S.; Jimenez-Zepeda, V.H.; Atkinson, J.; Spagnuolo, P.A.; Wong, Y.L.; Colaguori, R.; Lewis, A.M.; et al. Inhibiting aberrant signal transducer and activator of transcription protein activation with tetrapodal, small molecule Src homology 2 domain binders: Promising agents against multiple myeloma. J. Med. Chem. 2013, 56, 7190–7200. [Google Scholar] [CrossRef]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586–597. [Google Scholar] [CrossRef]
- Page, B.D.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-potency small molecule inhibitor of STAT5 protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/b with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Sperl, B.; Reindl, W.; Kiessling, A.; Berg, T. Discovery of chromone-based inhibitors of the transcription factor STAT5. Chembiochem 2008, 9, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, N.; Berg, A.; Natarajan, K.; Scharow, A.; Berg, T. Nanomolar inhibitors of the transcription factor STAT5b with high selectivity over STAT5a. Angew. Chem. Int. Ed. Engl. 2015, 54, 4758–4763. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Kaneko, N.; Kita, A.; Yamanaka, K.; Mori, M. Combination of YM155, a survivin suppressant with a STAT3 inhibitor: A new strategy to treat diffuse large B-cell lymphoma. Leuk. Res. 2013, 37, 1156–1161. [Google Scholar] [CrossRef]
- Mencalha, A.L.; Du Rocher, B.; Salles, D.; Binato, R.; Abdelhay, E. LLL-3, a STAT3 inhibitor, represses BCR-ABL-positive cell proliferation, activates apoptosis and improves the effects of Imatinib mesylate. Cancer Chemother. Pharmacol. 2010, 65, 1039–1046. [Google Scholar] [CrossRef]
- Lin, L.; Benson, D.M., Jr.; DeAngelis, S.; Bakan, C.E.; Li, P.K.; Li, C.; Lin, J. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int. J. Cancer 2012, 130, 1459–1469. [Google Scholar] [CrossRef]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. Stat3 signaling in acute myeloid leukemia: Ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood 2011, 117, 5701–5709. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Huang, Y.H.; Molavi, O.; Alshareef, A.; Haque, M.; Wang, Q.; Chu, M.P.; Venner, C.P.; Sandhu, I.; Peters, A.C.; Lavasanifar, A.; et al. Constitutive Activation of STAT3 in Myeloma Cells Cultured in a Three-Dimensional, Reconstructed Bone Marrow Model. Cancers 2018, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, L.; Li, D.; Nong, L.; Zheng, Y.; Huang, S.; Zhang, B.; Li, T. JAK3/STAT3 oncogenic pathway and PRDM1 expression stratify clinicopathologic features of extranodal NK/T-cell lymphoma, nasal type. Oncol. Rep. 2019, 41, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.D.; Liu, J.Y.; Zhang, J.T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 2018, 191, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, 3, e166. [Google Scholar] [CrossRef]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W.; et al. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511. [Google Scholar] [CrossRef]
- Yco, L.P.; Mocz, G.; Opoku-Ansah, J.; Bachmann, A.S. Withaferin A Inhibits STAT3 and Induces Tumor Cell Death in Neuroblastoma and Multiple Myeloma. Biochem. Insights 2014, 7, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, L.; Cai, H.; Lei, H.; Ma, C.M.; Yang, L.; Xu, H.; Zhu, Q.; Yao, Z.; Wu, Y. Sinomenine derivative YL064: A novel STAT3 inhibitor with promising anti-myeloma activity. Cell Death Dis. 2018, 9, 1093. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Buerger, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol. Cancer Res. 2004, 2, 170–182. [Google Scholar] [PubMed]
- Wang, X.; Zeng, J.; Shi, M.; Zhao, S.; Bai, W.; Cao, W.; Tu, Z.; Huang, Z.; Feng, W. Targeted blockage of signal transducer and activator of transcription 5 signaling pathway with decoy oligodeoxynucleotides suppresses leukemic k562 cell growth. DNA Cell Biol. 2011, 30, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Z.; Moreira, D.; Su, Y.L.; Won, H.; Adamus, T.; Dong, Z.; Liang, Y.; Yin, H.H.; Swiderski, P.; et al. B Cell Lymphoma Immunotherapy Using TLR9-Targeted Oligonucleotide STAT3 Inhibitors. Mol. Ther. 2018, 26, 695–707. [Google Scholar] [CrossRef]
- Jing, N.; Li, Y.; Xiong, W.; Sha, W.; Jing, L.; Tweardy, D.J. G-quartet oligonucleotides: A new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004, 64, 6603–6609. [Google Scholar] [CrossRef]
- Hillion, J.; Belton, A.M.; Shah, S.N.; Turkson, J.; Jing, N.; Tweardy, D.J.; di Cello, F.; Huso, D.L.; Resar, L.M. Nanoparticle delivery of inhibitory signal transducer and activator of transcription 3 G-quartet oligonucleotides blocks tumor growth in HMGA1 transgenic model of T-cell leukemia. Leuk. Lymphoma 2014, 55, 1194–1197. [Google Scholar] [CrossRef]
- Shastri, A.; Choudhary, G.; Teixeira, M.; Gordon-Mitchell, S.; Ramachandra, N.; Bernard, L.; Bhattacharyya, S.; Lopez, R.; Pradhan, K.; Giricz, O.; et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J. Clin. Investig. 2018, 128, 5479–5488. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer. 2018, 6, 119. [Google Scholar] [CrossRef]
- Zhang, Q.; Hossain, D.M.; Nechaev, S.; Kozlowska, A.; Zhang, W.; Liu, Y.; Kowolik, C.M.; Swiderski, P.; Rossi, J.J.; Forman, S.; et al. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood 2013, 121, 1304–1315. [Google Scholar] [CrossRef]
- Von Manstein, V.; Yang, C.M.; Richter, D.; Delis, N.; Vafaizadeh, V.; Groner, B. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr. Signal. Transduct. Ther. 2013, 8, 193–202. [Google Scholar] [CrossRef] [PubMed]



| Hematopoietic Lineage | Hematologic Malignancies | Contribution of STAT3 and/or STAT5A/5B | |||
|---|---|---|---|---|---|
| Myeloid compartment | Ph+ MPN | CML (BCR-ABL) | STAT3 | STAT5A | STAT5B |
| Ph− MPN | PV JAK2V617F | STAT3 | STAT5 | ||
| ET JAK2V617F | |||||
| PMF JAK2V617F | |||||
| SM KITD816V | |||||
| AML | Flt3-ITD | STAT5 | |||
| CBF-AML KitD816V | STAT3 | STAT5 | |||
| APL | STAT3 | STAT5 | |||
| t (15;17) | |||||
| Lymphoid compartment | ALL | Pre-B-ALL | STAT3 | STAT5 | |
| B-ALL | |||||
| T-ALL | STAT3 | STAT5A | STAT5B | ||
| T-LGL Leukemias | STAT3 | STAT5A | STAT5B | ||
| CLL | STAT3 | ||||
| Lymphomas | HL | STAT3 | STAT5 | ||
| DLBCL | STAT3 | STAT5 | |||
| ALCL | STAT3 | STAT5B | |||
| γδ-T cell Lymphomas | STAT3 | STAT5B | |||
| NK-T cell Lymphomas | STAT3 | STAT5B | |||
| Multiple Myelomas | STAT3 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brachet-Botineau, M.; Polomski, M.; Neubauer, H.A.; Juen, L.; Hédou, D.; Viaud-Massuard, M.-C.; Prié, G.; Gouilleux, F. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers 2020, 12, 240. https://doi.org/10.3390/cancers12010240
Brachet-Botineau M, Polomski M, Neubauer HA, Juen L, Hédou D, Viaud-Massuard M-C, Prié G, Gouilleux F. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers. 2020; 12(1):240. https://doi.org/10.3390/cancers12010240
Chicago/Turabian StyleBrachet-Botineau, Marie, Marion Polomski, Heidi A. Neubauer, Ludovic Juen, Damien Hédou, Marie-Claude Viaud-Massuard, Gildas Prié, and Fabrice Gouilleux. 2020. "Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers" Cancers 12, no. 1: 240. https://doi.org/10.3390/cancers12010240
APA StyleBrachet-Botineau, M., Polomski, M., Neubauer, H. A., Juen, L., Hédou, D., Viaud-Massuard, M.-C., Prié, G., & Gouilleux, F. (2020). Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers, 12(1), 240. https://doi.org/10.3390/cancers12010240