Heat Shock Protein 90 Chaperone Regulates the E3 Ubiquitin-Ligase Hakai Protein Stability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
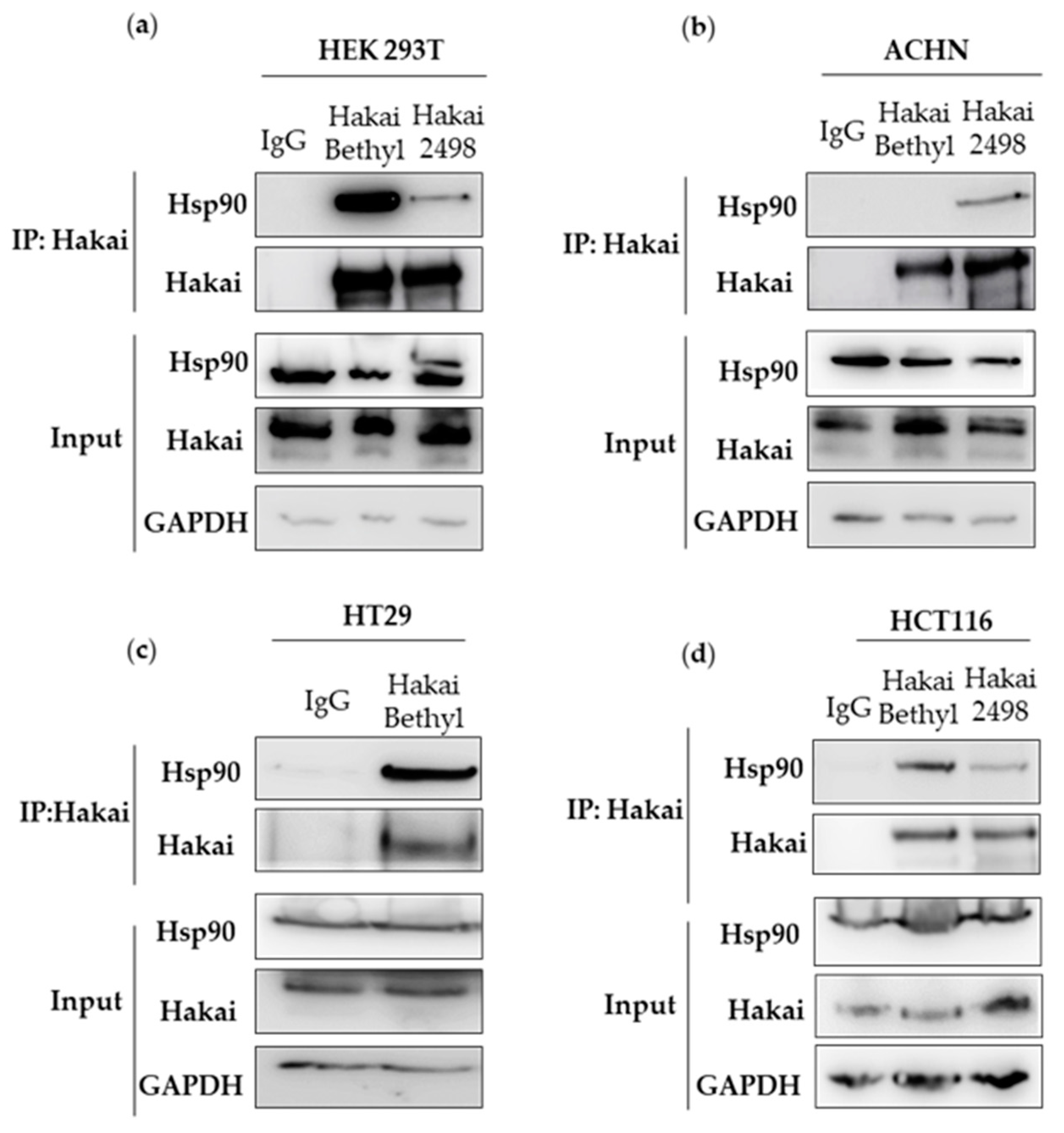
2.1. Hsp90 Chaperone Interacts with Hakai
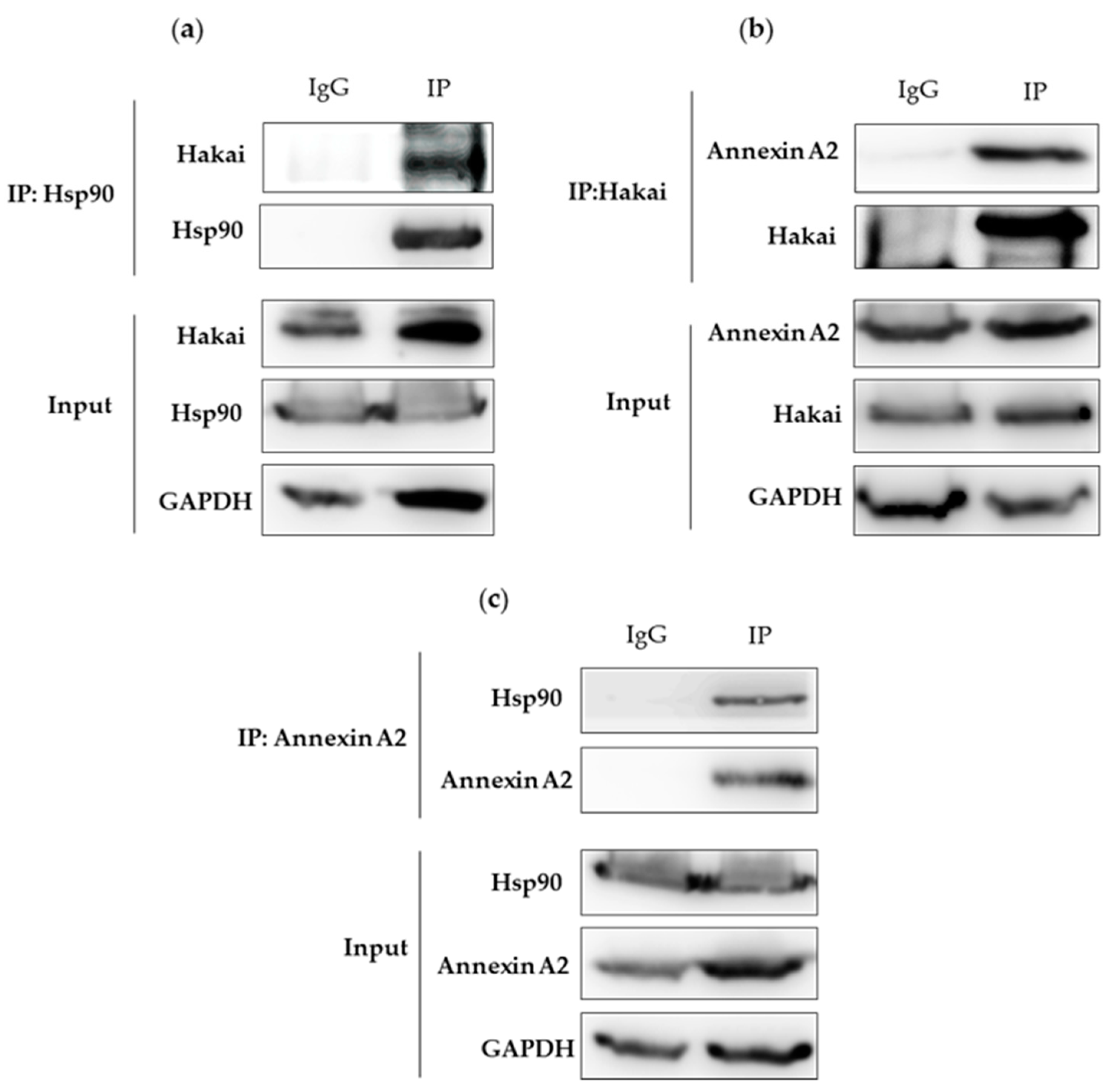
2.2. Interaction between Hsp90, Hakai and Annexin A2
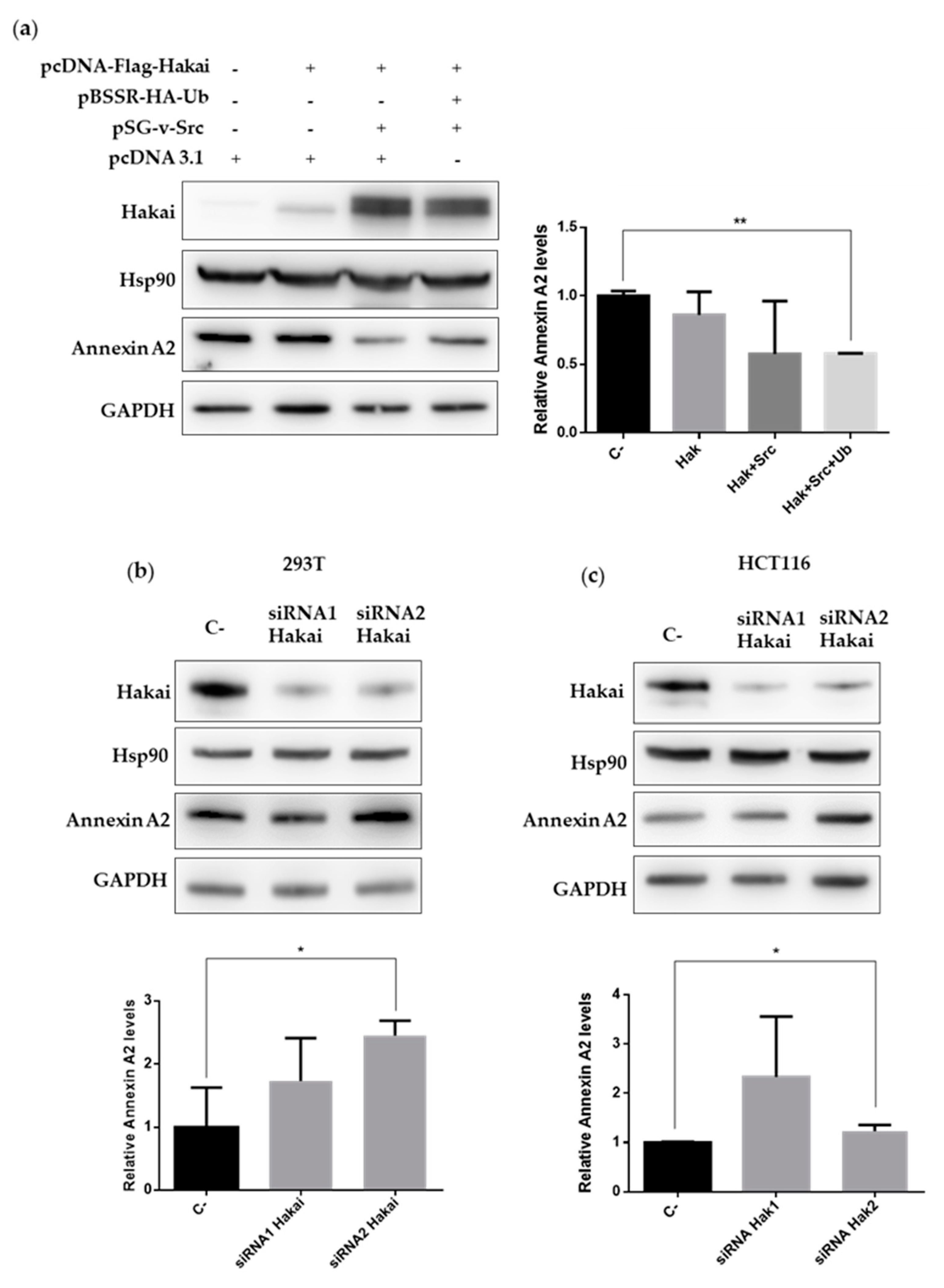
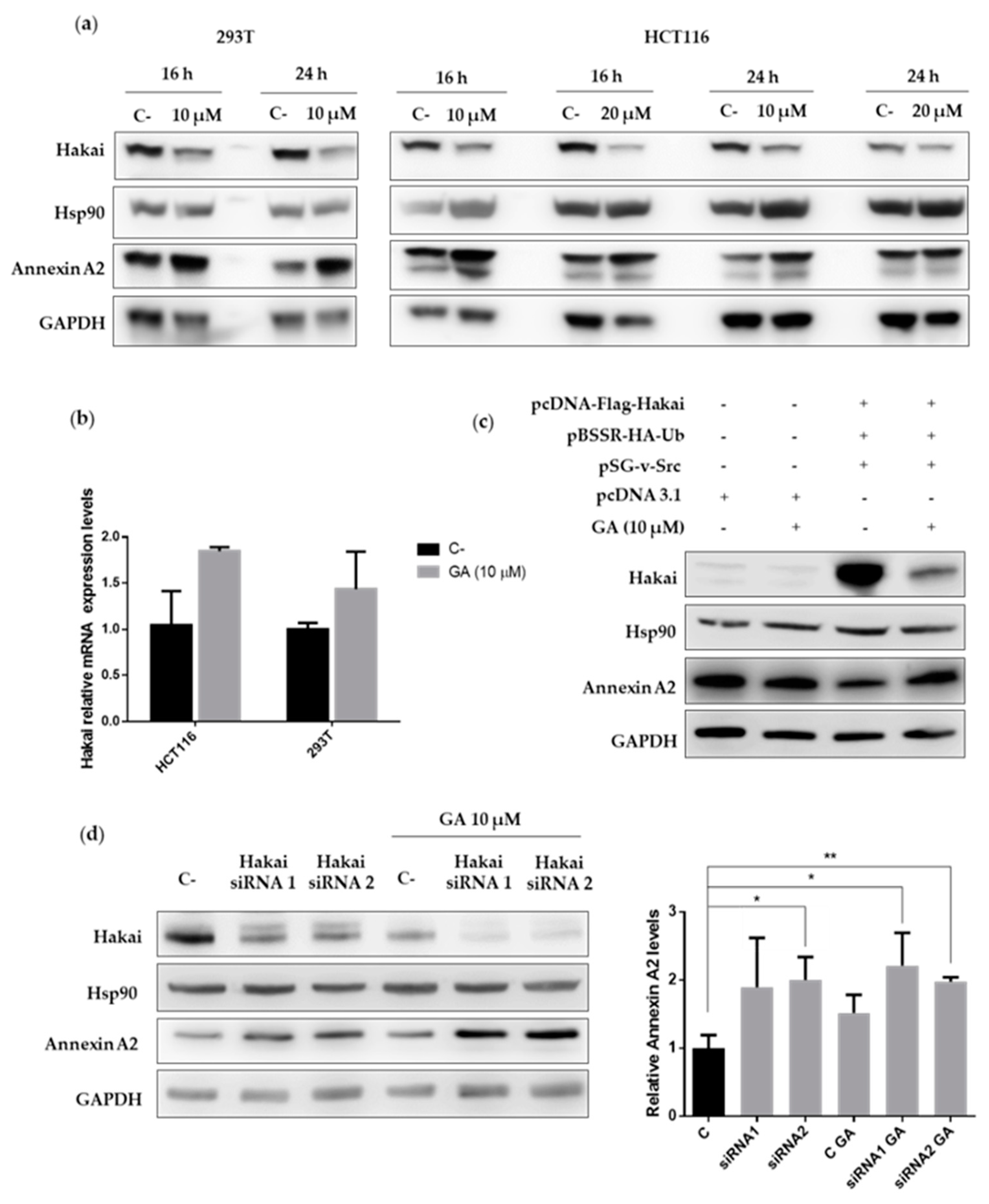
2.3. Hakai Regulates Annexin A2 Protein Expression
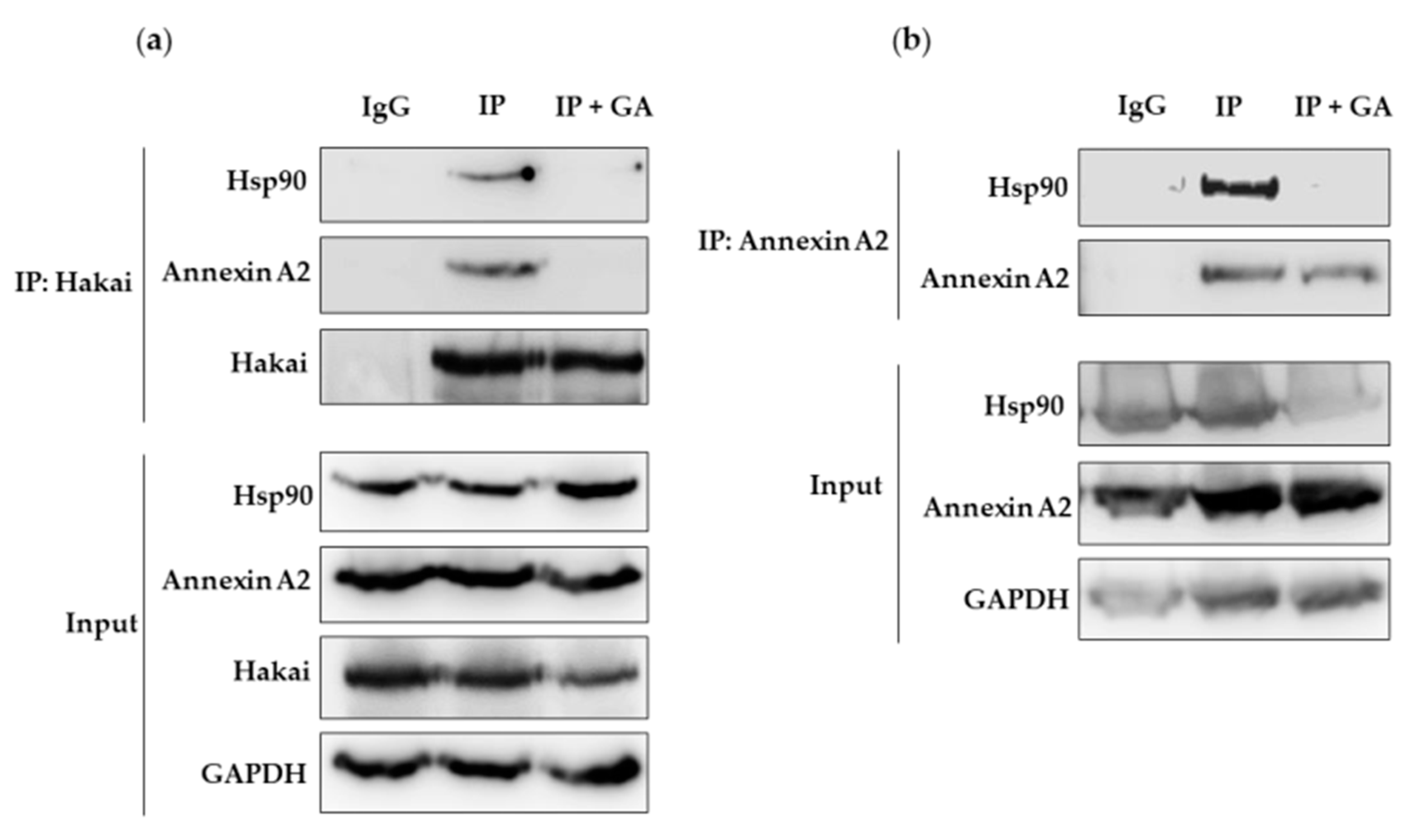
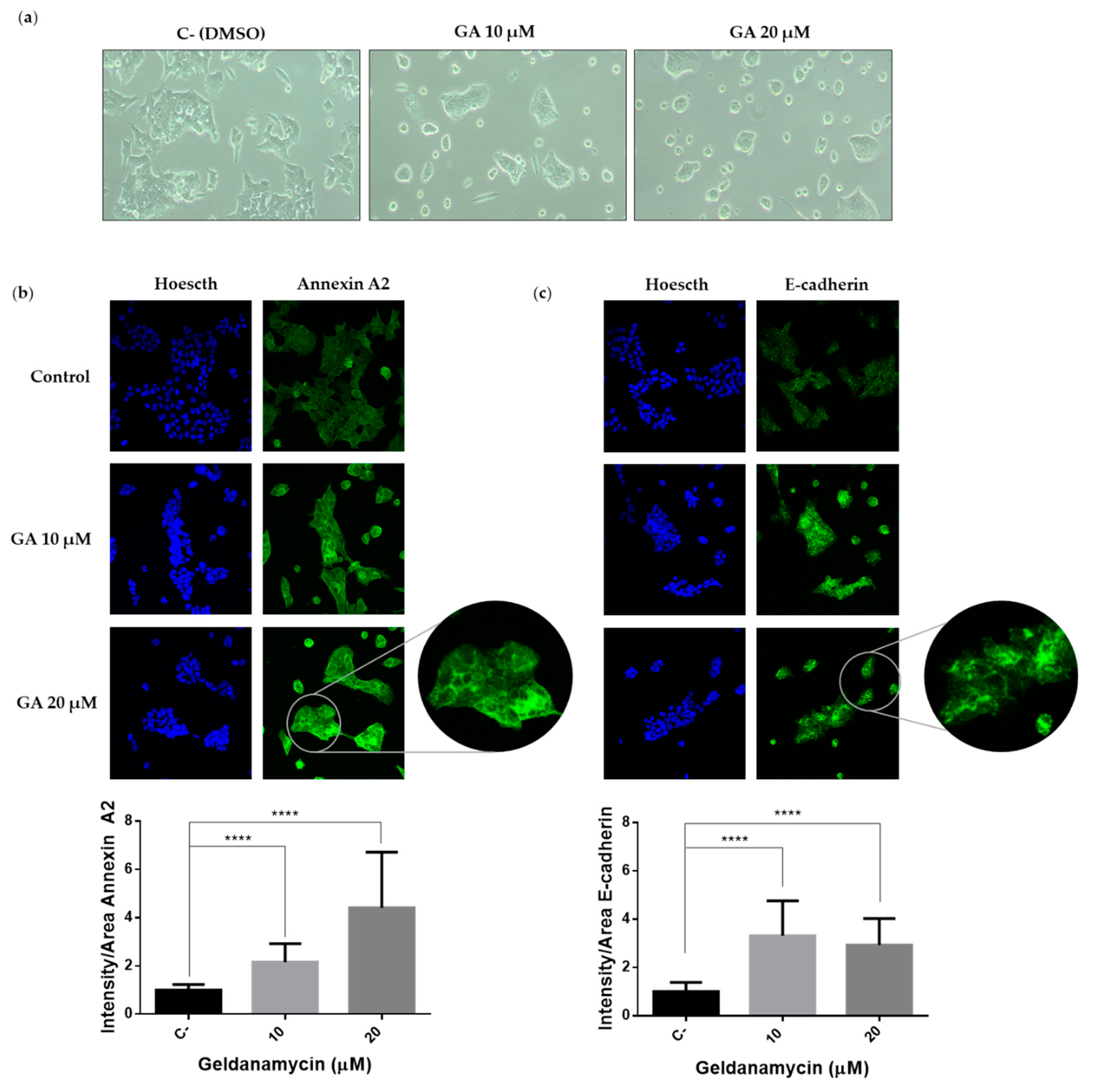
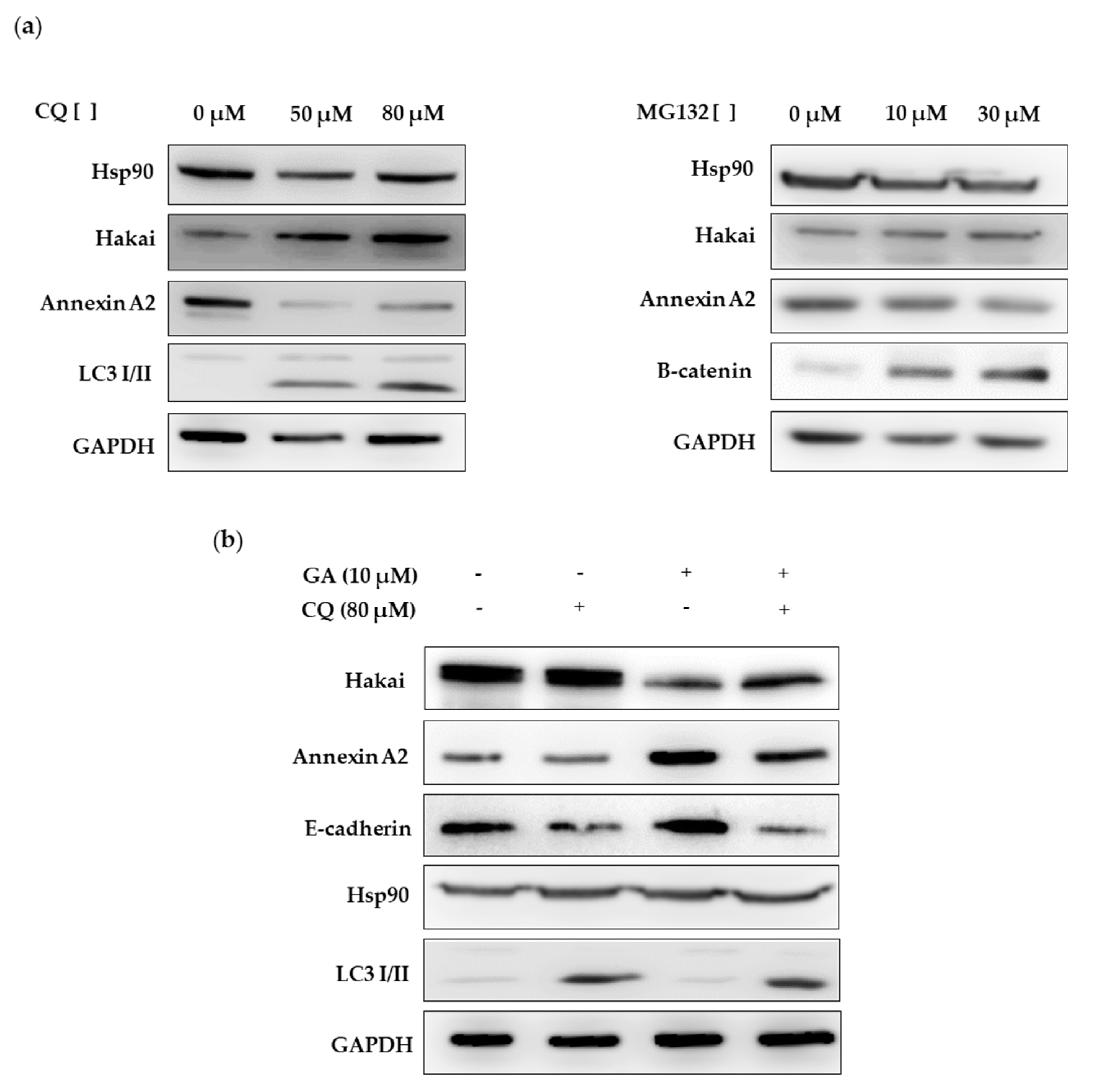
2.4. Hsp90 Inbibitor Geldanamycin Induces Downregulation of Hakai Protein via Lysosome
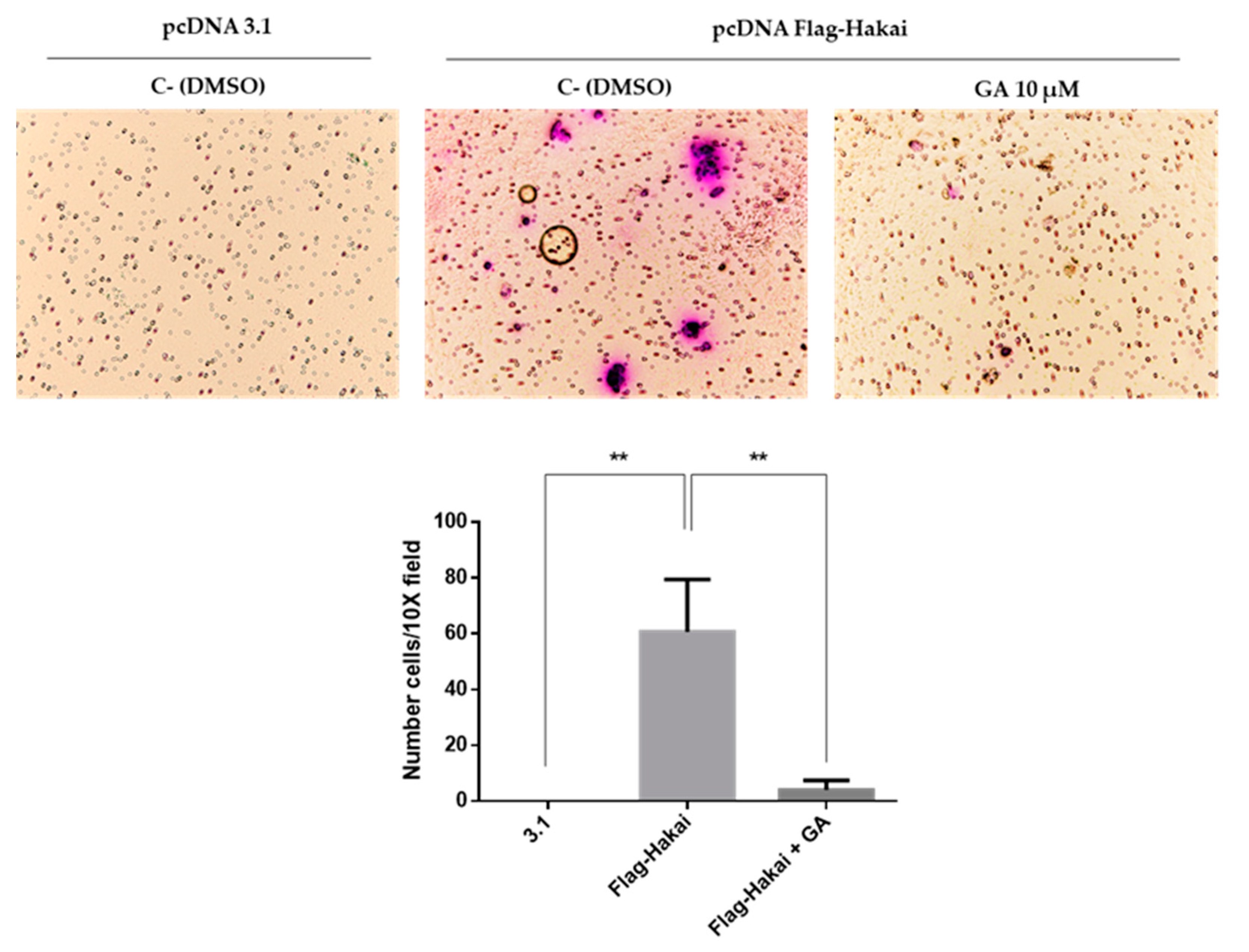
2.5. Downregulation of Hakai May Partially Account for the Pharmacological Anti-Migratory Effect of Geldanamycin Hsp90 Inhibitor
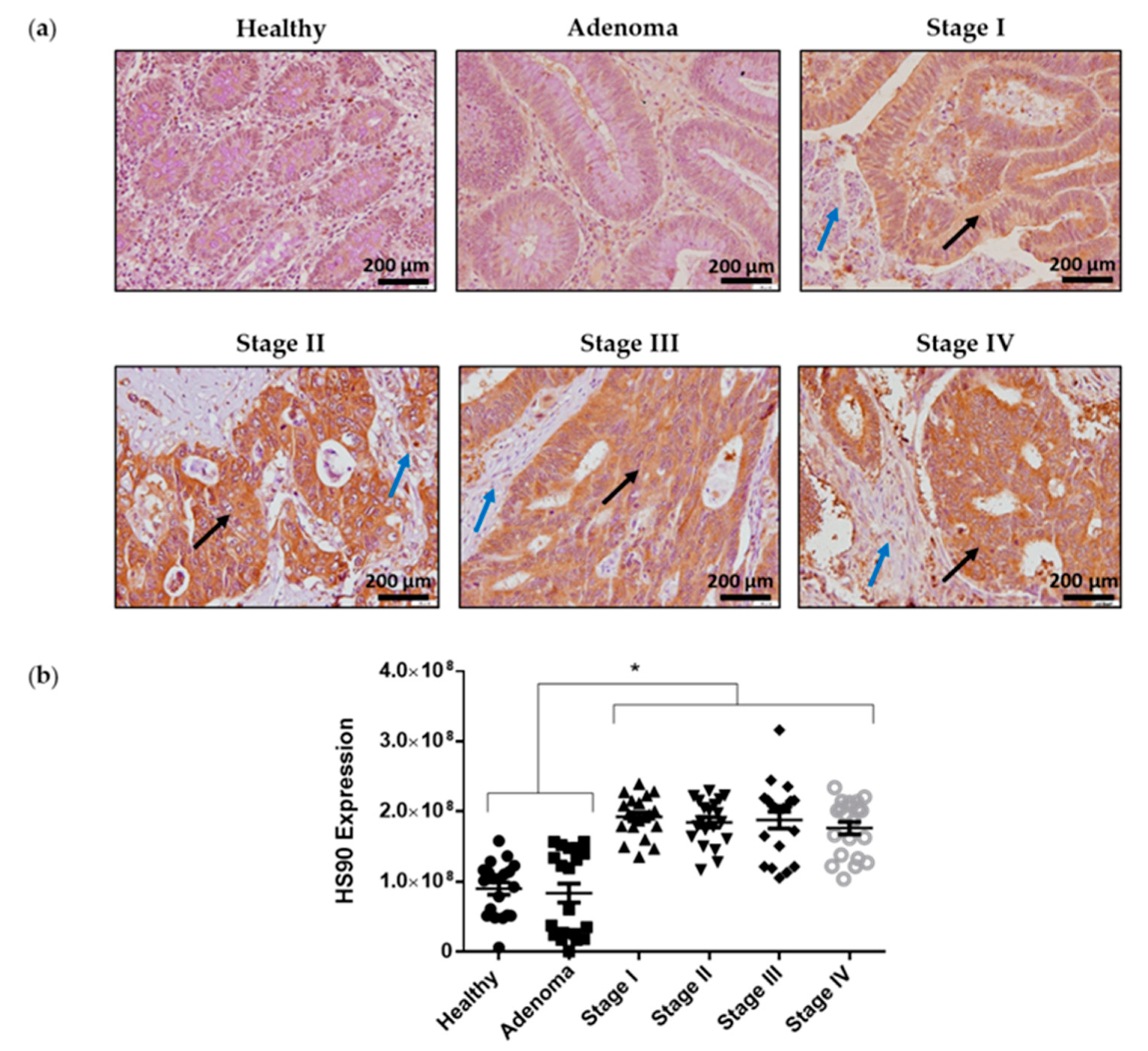
2.6. Hsp90 Is Highly Expressed in Colorectal Cancer Samples Compared to Adjacent Normal Epithelial Tissues
3. Discussion
4. Materials and Methods
4.1. Plasmids, Antibodies, and Inhibitors
4.2. Cell Culture and Transfection
4.3. Western Blot Analysis and Immunoprecipitation
4.4. Human Samples and Immunohistochemistry
4.5. Immunofluorescence Assays
4.6. RNA Interference
4.7. Real-Time Quantitative PCR (RT-qPCR)
4.8. Migration Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Drysdale, M.J.; Brough, P.A.; Massey, A.; Jensen, M.R.; Schoepfer, J. Targeting Hsp90 for the treatment of cancer. Curr. Opin. Drug Discov. Devel. 2006, 9, 483–495. [Google Scholar]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef]
- Schneider, C.; Sepp-Lorenzino, L.; Nimmesgern, E.; Ouerfelli, O.; Danishefsky, S.; Rosen, N.; Hartl, F.U. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc. Natl. Acad. Sci. USA 1996, 93, 14536–14541. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Chow, S.Y.; Yusoff, P.; Seetharaman, J.; Ng, C.; Sinniah, S.; Koh, X.W.; Asgar, N.F.; Li, D.; Yim, D.; et al. Structure of a novel phosphotyrosine-binding domain in Hakai that targets E-cadherin. EMBO J. 2012, 31, 1308–1319. [Google Scholar] [CrossRef]
- Cooper, J.A.; Kaneko, T.; Li, S.S. Cell regulation by phosphotyrosine-targeted ubiquitin ligases. Mol. Cell Biol. 2015, 35, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, L.A.; Valladares, M.; Blanco, M.; Alonso, G.; Figueroa, A. Biological influence of Hakai in cancer: A 10-year review. Cancer Metastasis Rev. 2012, 31, 375–386. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef]
- Aparicio, L.A.; Blanco, M.; Castosa, R.; Concha, Á.; Valladares, M.; Calvo, L.; Figueroa, A. Clinical implications of epithelial cell plasticity in cancer progression. Cancer Lett. 2015, 366, 1–10. [Google Scholar] [CrossRef]
- Figueroa, A.; Kotani, H.; Toda, Y.; Mazan-Mamczarz, K.; Mueller, E.; Otto, A.; Disch, L.; Norman, M.; Ramdasi, R.; Keshtgar, M.; et al. Novel roles of hakai in cell proliferation and oncogenesis. Mol. Biol. Cell 2009, 20, 3533–3542. [Google Scholar] [CrossRef]
- Rodríguez-Rigueiro, T.; Valladares-Ayerbes, M.; Haz-Conde, M.; Aparicio, L.A.; Figueroa, A. Hakai reduces cell-substratum adhesion and increases epithelial cell invasion. BMC Cancer 2011, 11, 474. [Google Scholar] [CrossRef]
- Abella, V.; Valladares, M.; Rodriguez, T.; Haz, M.; Blanco, M.; Tarrío, N.; Iglesias, P.; Aparicio, L.A.; Figueroa, A. miR-203 Regulates Cell Proliferation through Its Influence on Hakai Expression. PLoS ONE 2012, 7, e52568. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, S.; Wudu, M.; Ren, H.; Xu, Y.; Zhang, Q.; Qiu, X. CBLL1 is highly expressed in non-small cell lung cancer and promotes cell proliferation and invasion. Thorac. Cancer 2019, 10, 1479–1488. [Google Scholar] [CrossRef]
- Castosa, R.; Martinez-Iglesias, O.; Roca-Lema, D.; Casas-Pais, A.; Díaz-Díaz, A.; Iglesias, P.; Santamarina, I.; Graña, B.; Calvo, L.; Valladares-Ayerbes, M.; et al. Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci. Rep. 2018, 8, 3466. [Google Scholar] [CrossRef]
- Díaz-Díaz, A.; Casas-Pais, A.; Calamia, V.; Castosa, R.; Martinez-Iglesias, O.; Roca-Lema, D.; Santamarina, I.; Valladares-Ayerbes, M.; Calvo, L.; Chantada, V.; et al. Proteomic Analysis of the E3 Ubiquitin-Ligase Hakai Highlights a Role in Plasticity of the Cytoskeleton Dynamics and in the Proteasome System. J. Proteome Res. 2017, 16, 2773–2788. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, E.S.; Wang, T.; Luo, K.; Xiao, Z.; Niewiadomska, A.M.; Martinez, T.; Xu, W.; Neckers, L.; Yu, X.F. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 20330–20335. [Google Scholar] [CrossRef] [PubMed]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Marcu, M.; Yuan, X.; Mimnaugh, E.; Patterson, C.; Neckers, L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. USA 2002, 99, 12847–12852. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Romeo, G.; Kazlauskas, A. Heat shock protein 90alpha-dependent translocation of annexin II to the surface of endothelial cells modulates plasmin activity in the diabetic rat aorta. Circ Res. 2004, 94, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.; Bydoun, M.; Holloway, R.; Waisman, D. Annexin A2 heterotetramer: Structure and function. Int. J. Mol. Sci. 2013, 14, 6259–6305. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Garg, G.; Khandelwal, A.; Blagg, B.S. Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects. Adv. Cancer Res. 2016, 129, 51–88. [Google Scholar]
- Rodina, A.; Vilenchik, M.; Moulick, K.; Aguirre, J.; Kim, J.; Chiang, A.; Litz, J.; Clement, C.C.; Kang, Y.; She, Y.; et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat. Chem. Biol. 2007, 3, 498–507. [Google Scholar] [CrossRef]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Rao, R.; Balusu, R.; Venkannagari, S.; Rao, N.N.; Ha, K.; Smith, J.E.; Hembruff, S.L.; et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin. Cancer Res. 2011, 17, 7347–7358. [Google Scholar] [CrossRef]
- Ha, K.; Fiskus, W.; Rao, R.; Balusu, R.; Venkannagari, S.; Nalabothula, N.R.; Bhalla, K.N. Hsp90 inhibitor-mediated disruption of chaperone association of ATR with hsp90 sensitizes cancer cells to DNA damage. Mol. Cancer 2011, 10, 1194–1206. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; Fang, J.M.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fernandes, N.; Dodge, I.L.; Reddi, A.L.; Rao, N.; Safran, H.; DiPetrillo, T.A.; Wazer, D.E.; Band, V.; Band, H. ErbB2 degradation mediated by the co-chaperone protein CHIP. J. Biol. Chem. 2003, 278, 13829–13837. [Google Scholar] [CrossRef]
- Ding, G.; Chen, P.; Zhang, H.; Huang, X.; Zang, Y.; Li, J.; Wong, J. Regulation of Ubiquitin-like with Plant Homeodomain and RING Finger Domain 1 (UHRF1) Protein Stability by Heat Shock Protein 90 Chaperone Machinery. J. Biol. Chem. 2016, 291, 20125–20135. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, J.K.; Katz, A.A.; Schäfer, G. Dynamic reciprocity: The role of annexin A2 in tissue integrity. J. Cell Commun. Signal. 2014, 8, 125–133. [Google Scholar] [CrossRef]
- Hayes, M.J.; Moss, S.E. Annexin 2 has a dual role as regulator and effector of v-Src in cell transformation. J. Biol. Chem. 2009, 284, 10202–10210. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Long, T.E.; Park, W.; Landry, J.C.; Taliaferro-Smith, L.; Farris, A.B.; Diaz, R.; El-Rayes, B.F. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol. Carcinog. 2015, 54, 1147–1158. [Google Scholar] [CrossRef]
- Wei, W.S.; Chen, X.; Guo, L.Y.; Li, X.D.; Deng, M.H.; Yuan, G.J.; He, L.Y.; Li, Y.H.; Zhang, Z.L.; Jiang, L.J.; et al. TRIM65 supports bladder urothelial carcinoma cell aggressiveness by promoting ANXA2 ubiquitination and degradation. Cancer Lett. 2018, 435, 10–22. [Google Scholar] [CrossRef]
- Chen, G.; Chen, W.; Ye, M.; Tan, W.; Jia, B. TRIM59 knockdown inhibits cell proliferation by down-regulating the Wnt/β-catenin signaling pathway in neuroblastoma. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Yamada, A.; Irie, K.; Hirota, T.; Ooshio, T.; Fukuhara, A.; Takai, Y. Involvement of the annexin II-S100A10 complex in the formation of E-cadherin-based adherens junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 2005, 280, 6016–6027. [Google Scholar] [CrossRef]
- Bonvini, P.; An, W.G.; Rosolen, A.; Nguyen, P.; Trepel, J.; Garcia de Herreros, A.; Dunach, M.; Neckers, L.M. Geldanamycin abrogates ErbB2 association with proteasome-resistant beta-catenin in melanoma cells, increases beta-catenin-E-cadherin association, and decreases beta-catenin-sensitive transcription. Cancer Res. 2001, 61, 1671–1677. [Google Scholar] [PubMed]
- Barrott, J.J.; Haystead, T.A. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013, 280, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kryeziu, K.; Bruun, J.; Guren, T.K.; Sveen, A.; Lothe, R.A. Combination therapies with HSP90 inhibitors against colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 240–247. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, S.; Li, Z.; Li, D.; Zhan, Q. High expression of HSP90 is associated with poor prognosis in patients with colorectal cancer. PeerJ 2019, 7, e7946. [Google Scholar] [CrossRef]
- Aparicio, L.A.; Castosa, R.; Haz-Conde, M.; Rodríguez, M.; Blanco, M.; Valladares, M.; Figueroa, A. Role of the microtubule-targeting drug vinflunine on cell-cell adhesions in bladder epithelial tumour cells. BMC Cancer 2014, 14, 507. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Díaz, A.; Roca-Lema, D.; Casas-Pais, A.; Romay, G.; Colombo, G.; Concha, Á.; Graña, B.; Figueroa, A. Heat Shock Protein 90 Chaperone Regulates the E3 Ubiquitin-Ligase Hakai Protein Stability. Cancers 2020, 12, 215. https://doi.org/10.3390/cancers12010215
Díaz-Díaz A, Roca-Lema D, Casas-Pais A, Romay G, Colombo G, Concha Á, Graña B, Figueroa A. Heat Shock Protein 90 Chaperone Regulates the E3 Ubiquitin-Ligase Hakai Protein Stability. Cancers. 2020; 12(1):215. https://doi.org/10.3390/cancers12010215
Chicago/Turabian StyleDíaz-Díaz, Andrea, Daniel Roca-Lema, Alba Casas-Pais, Gabriela Romay, Giovanni Colombo, Ángel Concha, Begoña Graña, and Angélica Figueroa. 2020. "Heat Shock Protein 90 Chaperone Regulates the E3 Ubiquitin-Ligase Hakai Protein Stability" Cancers 12, no. 1: 215. https://doi.org/10.3390/cancers12010215
APA StyleDíaz-Díaz, A., Roca-Lema, D., Casas-Pais, A., Romay, G., Colombo, G., Concha, Á., Graña, B., & Figueroa, A. (2020). Heat Shock Protein 90 Chaperone Regulates the E3 Ubiquitin-Ligase Hakai Protein Stability. Cancers, 12(1), 215. https://doi.org/10.3390/cancers12010215