Characterization of the Menin-MLL Interaction as Therapeutic Cancer Target

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
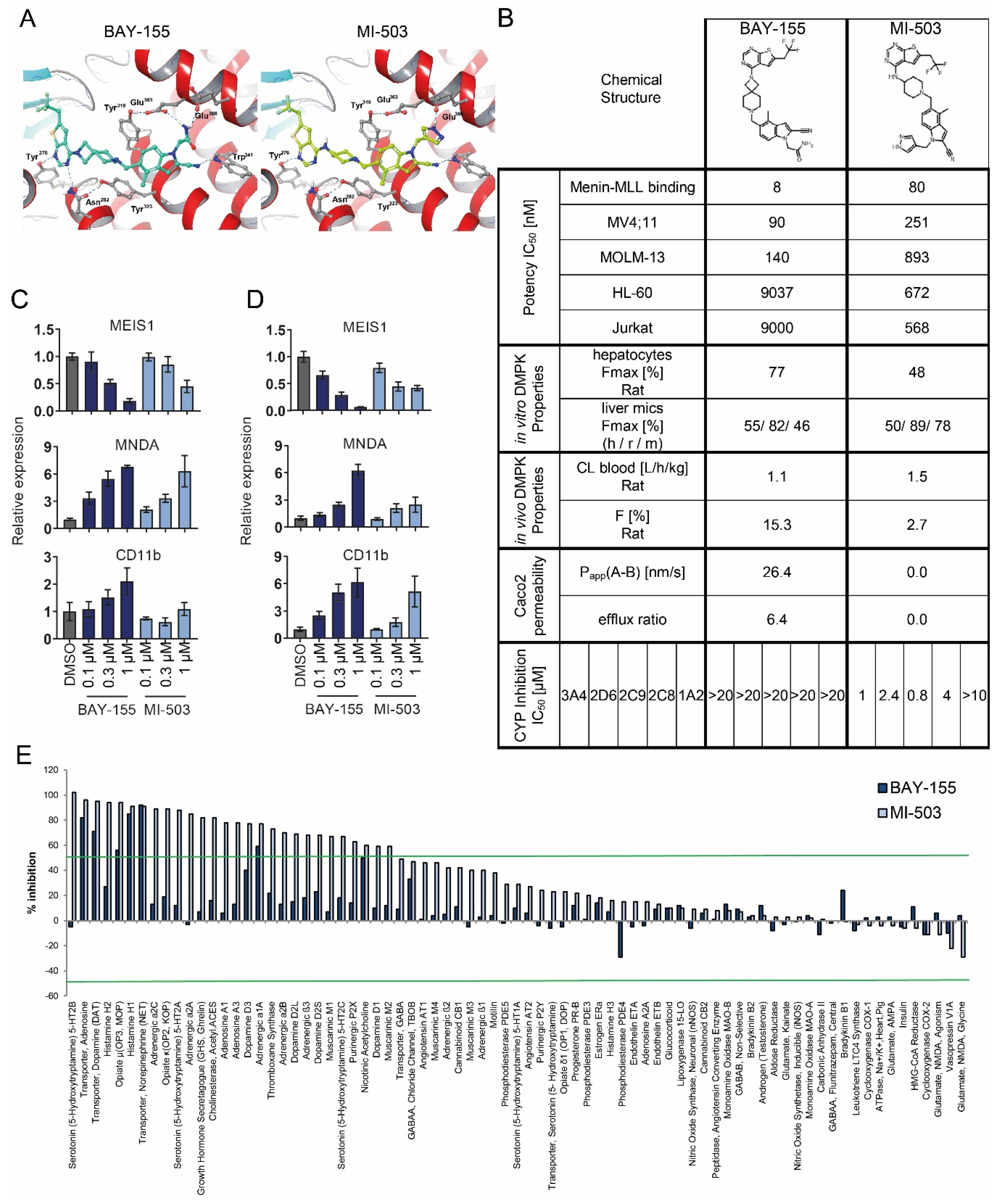
2.1. Characterization of Menin-MLL Tool Inhibitor BAY-155
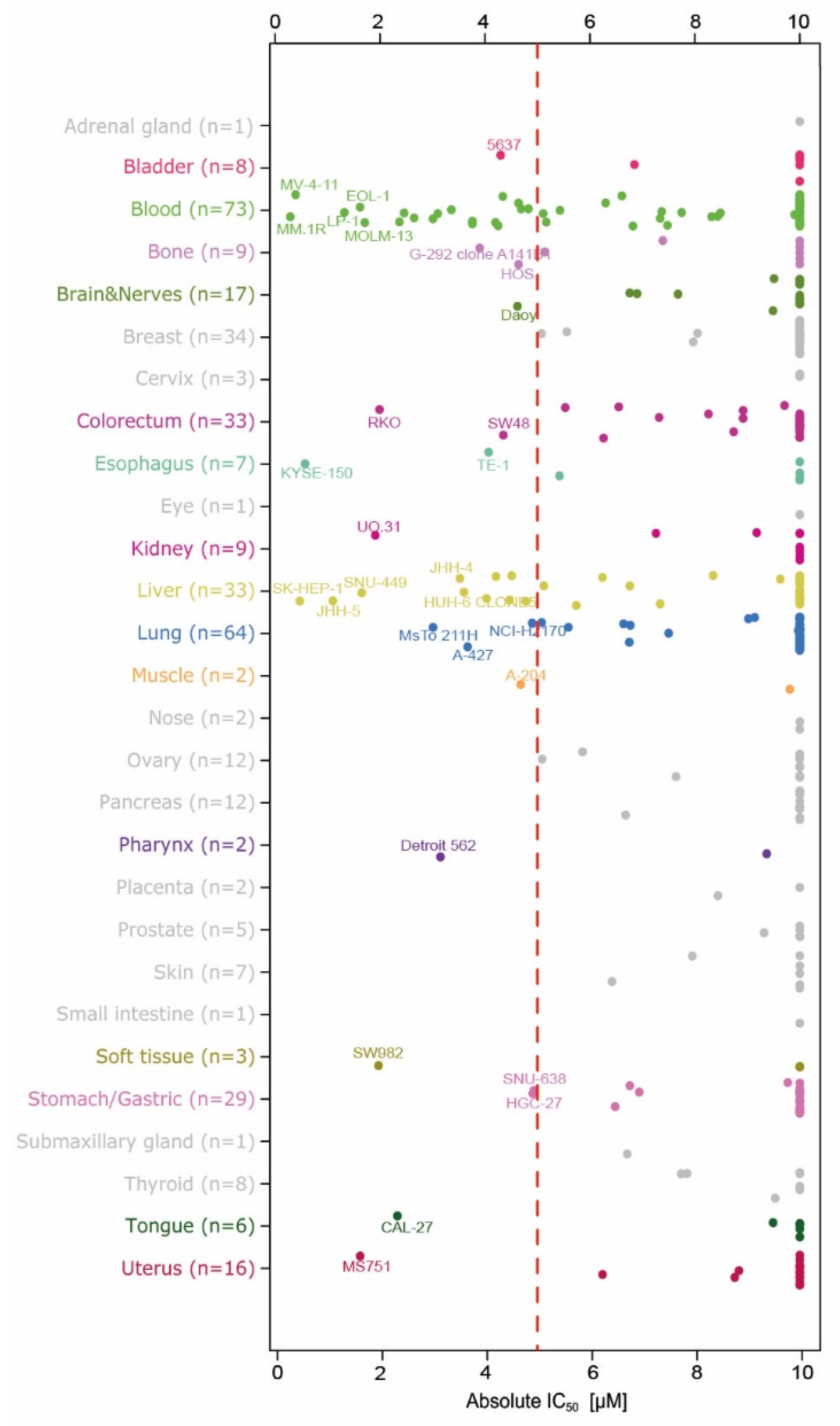
2.2. Evaluation of Anti-Proliferation Effects after Menin-MLL Inhibition in a Large Cancer Cell Line Panel
2.3. Gene Expression Effects after Menin-MLL Inhibition in Cancer Models beyond AML/ALL
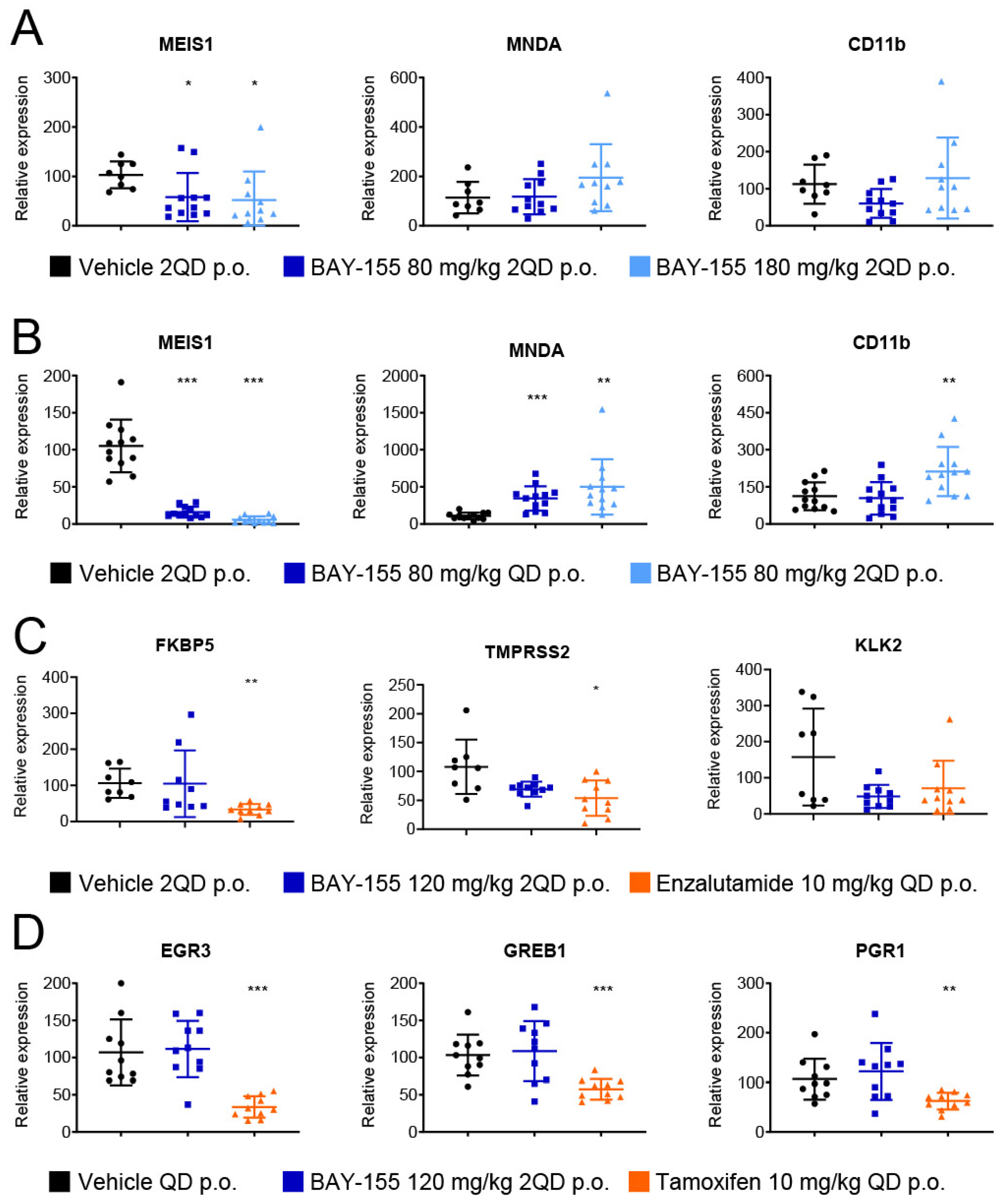
2.4. In Vivo Validation of Menin Inhibition by BAY-155
2.5. Ex Vivo Validation of Cancer Pathway Specific Effects of Menin Inhibition by BAY-155
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Biochemical Assay
4.3. Protein Purification
4.4. Isothermal Titration Calorimetry
4.5. Metabolic Stability in Rat Hepatocytes
4.6. Permeability Assay
4.7. Cell Line Proliferation Panel
4.8. Gene Expression
4.9. Data Analysis and Statistical Methods
4.10. Mouse Studies
4.11. Ex Vivo Analysis
4.12. Availability of Data and Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| ALL | Acute lymphoblastic leukemia |
| AR | androgen receptor |
| ER | estrogen receptor |
| DTT | Dithiothreitol |
| BSA | Bovine Serum Albumin |
| MLL | Mixed-lineage leukemia |
| GPCRs | G-protein coupled receptors |
| GSEA | Gene set enrichment analysis |
| TR-FRET | Time-Resolved Fluorescence Resonance Energy Transfer |
| ITC | Isothermal Titration Calorimetry |
| SCID | severe combined immunodeficiency |
| PCA | principal component analysis |
| NES | normalized enrichment score |
References
- Falchetti, A. Genetics of multiple endocrine neoplasia type 1 syndrome: What’s new and what’s old. F1000Research 2017, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402. [Google Scholar] [CrossRef]
- Balogh, K.; Racz, K.; Patocs, A.; Hunyady, L. Menin and its interacting proteins: Elucidation of menin function. Trends Endocrinol. Metab. 2006, 17, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; Klossowski, S.; Pollock, J.; Miao, H.; Linhares, B.M.; Kempinska, K.; Jin, Z.; Purohit, T.; Wen, B.; He, M.; et al. Complexity of blocking bivalent protein-protein interactions: Development of a highly potent inhibitor of the menin-mixed-lineage leukemia interaction. J. Med. Chem. 2018, 61, 4832–4850. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both mll and jund but has opposite effects on transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Del Rizzo, P.A.; Trievel, R.C. Substrate and product specificities of set domain methyltransferases. Epigenetics 2011, 6, 1059–1067. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for mll-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Dreijerink, K.M.A.; Groner, A.C.; Vos, E.S.M.; Font-Tello, A.; Gu, L.; Chi, D.; Reyes, J.; Cook, J.; Lim, E.; Lin, C.Y.; et al. Enhancer-mediated oncogenic function of the menin tumor suppressor in breast cancer. Cell Rep. 2017, 18, 2359–2372. [Google Scholar] [CrossRef]
- Malik, R.; Khan, A.P.; Asangani, I.A.; Cieslik, M.; Prensner, J.R.; Wang, X.; Iyer, M.K.; Jiang, X.; Borkin, D.; Escara-Wilke, J.; et al. Targeting the mll complex in castration-resistant prostate cancer. Nat. Med. 2015, 21, 344–352. [Google Scholar] [CrossRef]
- Kempinska, K.; Malik, B.; Borkin, D.; Klossowski, S.; Shukla, S.; Miao, H.; Wang, J.; Cierpicki, T.; Grembecka, J. Pharmacologic inhibition of the menin-mll interaction leads to transcriptional repression of peg10 and blocks hepatocellular carcinoma. Mol. Cancer Ther. 2018, 17, 26–38. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Bailey, N.; Van Noord, R.A.; Krook, M.A.; Harris, A.; Cramer, C.; Jasman, B.; Patel, R.M.; Thomas, D.; Borkin, D.; et al. Tumorigenicity of ewing sarcoma is critically dependent on the trithorax proteins mll1 and menin. Oncotarget 2017, 8, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Dreijerink, K.M.; Mulder, K.W.; Winkler, G.S.; Hoppener, J.W.; Lips, C.J.; Timmers, H.T. Menin links estrogen receptor activation to histone h3k4 trimethylation. Cancer Res. 2006, 66, 4929–4935. [Google Scholar] [CrossRef] [PubMed]
- Imachi, H.; Murao, K.; Dobashi, H.; Bhuyan, M.M.; Cao, X.; Kontani, K.; Niki, S.; Murazawa, C.; Nakajima, H.; Kohno, N.; et al. Menin, a product of the meni gene, binds to estrogen receptor to enhance its activity in breast cancer cells: Possibility of a novel predictive factor for tamoxifen resistance. Breast Cancer Res. Treat. 2010, 122, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ozawa, A.; Zaman, S.; Prasad, N.B.; Chandrasekharappa, S.C.; Agarwal, S.K.; Marx, S.J. The tumor suppressor protein menin inhibits akt activation by regulating its cellular localization. Cancer Res. 2011, 71, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Wuescher, L.; Angevine, K.; Hinds, T.; Ramakrishnan, S.; Najjar, S.M.; Mensah-Osman, E.J. Insulin regulates menin expression, cytoplasmic localization, and interaction with foxo1. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E474–E483. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Egido, J.; Cunningham, J.; Berg, M.; Oberg, K.; Bongcam-Rudloff, E.; Gobl, A. Menin’s interaction with glial fibrillary acidic protein and vimentin suggests a role for the intermediate filament network in regulating menin activity. Exp. Cell Res. 2002, 278, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yuan, M.; Shen, S.; Ma, X.; Fang, J.; Zhu, L.; Sun, L.; Liu, Z.; He, X.; Huang, D.; et al. Menin enhances c-myc-mediated transcription to promote cancer progression. Nat. Commun. 2017, 8, 15278. [Google Scholar] [CrossRef]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-mll inhibitors reverse oncogenic activity of mll fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef]
- He, S.; Senter, T.J.; Pollock, J.; Han, C.; Upadhyay, S.K.; Purohit, T.; Gogliotti, R.D.; Lindsley, C.W.; Cierpicki, T.; Stauffer, S.R.; et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (mll) interaction closely mimic a natural protein-protein interaction. J. Med. Chem. 2014, 57, 1543–1556. [Google Scholar] [CrossRef]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-mll interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-mll interaction blocks progression of mll leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Aguilar, A.; Xu, T.; Zheng, K.; Huang, L.; Stuckey, J.; Chinnaswamy, K.; Bernard, D.; Fernandez-Salas, E.; Liu, L.; et al. Design of the first-in-class, highly potent irreversible inhibitor targeting the menin-mll protein-protein interaction. Angew. Chem. 2018, 57, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, L.K.; Teh, S.S.K.; Sud, S.; Kerk, S.; Zebolsky, A.; Treichel, S.; Thomas, D.; Halbrook, C.J.; Lee, H.J.; Kremer, D.; et al. Menin regulates the serine biosynthetic pathway in ewing sarcoma. J. Pathol. 2018, 245, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.; Long, K.; Yeo, D.J.; Miles, J.A.; Azzarito, V.; Burslem, G.M.; Prabhakaran, P.; Edwards, T.A.; Wilson, A.J. Orthogonal functionalisation of alpha-helix mimetics. Org. Biomol. Chem. 2014, 12, 6794–6799. [Google Scholar] [CrossRef] [PubMed]
- Brzezinka, K.; Nevedomskaya, E.; Lesche, R.; Steckel, M.; Eheim, A.L.; Haegebarth, A.; Stresemann, C. Functional diversity of inhibitors tackling the differentiation blockage of mll-rearranged leukemia. J. Hematol. Oncol. 2019, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.; Haendler, B.; Stresemann, C.; Fernandez-Montalvan, A.E.; Ter Laak, A.; Stoeckigt, D. Spiro Condensed Azetidine Derivatives as Inhibitors of the Menin-Mml1 Interaction. Patent WO/2017/207387, 7 December 2017. [Google Scholar]
- Siegel, S.; Haendler, B.; Stresemann, C.; Fernandez-Montalvan, A.E.; Ter Laak, A.; Stoeckigt, D.; Harb, H.Y.; Kosemund, D.; Eheim, A.L.; Monning, U. 2,7-Diazaspiro[4.4]Nonanes. Patent WO/2018/024602, 8 February 2018. [Google Scholar]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of crispr-cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global analysis of h3k4 methylation defines mll family member targets and points to a role for mll1-mediated h3k4 methylation in the regulation of transcriptional initiation by rna polymerase ii. Mol. Cell. Biol. 2009, 29, 6074–6085. [Google Scholar] [CrossRef]
- De Laat, J.M.; van Leeuwaarde, R.S.; Valk, G.D. The importance of an early and accurate men1 diagnosis. Front. Endocrinol. 2018, 9, 533. [Google Scholar] [CrossRef]
- Xu, B.; Li, S.H.; Zheng, R.; Gao, S.B.; Ding, L.H.; Yin, Z.Y.; Lin, X.; Feng, Z.J.; Zhang, S.; Wang, X.M.; et al. Menin promotes hepatocellular carcinogenesis and epigenetically up-regulates yap1 transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 17480–17485. [Google Scholar] [CrossRef]
- Bunnage, M.E.; Chekler, E.L.; Jones, L.H. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Dreijerink, K.M.A.; Timmers, H.T.M.; Brown, M. Twenty years of menin: Emerging opportunities for restoration of transcriptional regulation in men1. Endocr. Related Cancer 2017, 24, T135–T145. [Google Scholar] [CrossRef] [PubMed]
- Van Nuland, R.; Smits, A.H.; Pallaki, P.; Jansen, P.W.; Vermeulen, M.; Timmers, H.T. Quantitative dissection and stoichiometry determination of the human set1/mll histone methyltransferase complexes. Mol. Cell. Biol. 2013, 33, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Song, T.Y.; Park, J.; Lee, J.; Lim, J.; Jang, H.; Kim, Y.N.; Yang, J.H.; Song, Y.; Choi, A.; et al. Menin mediates epigenetic regulation via histone h3 lysine 9 methylation. Cell Death Dis. 2013, 4, e583. [Google Scholar] [CrossRef]
- Katona, B.W.; Glynn, R.A.; Paulosky, K.E.; Feng, Z.; Davis, C.I.; Ma, J.; Berry, C.T.; Szigety, K.M.; Matkar, S.; Liu, Y.; et al. Combined menin and egfr inhibitors synergize to suppress colorectal cancer via egfr-independent and calcium-mediated repression of skp2 transcription. Cancer Res. 2019, 79, 2195–2207. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Safarian, S.; Carroll, J.S.; Sheibani, N.; Bidkhori, G. Tamoxifen-induced apoptosis of mcf-7 cells via gpr30/pi3k/mapks interactions: Verification by ode modeling and rna sequencing. Front. Physiol. 2018, 9, 907. [Google Scholar] [CrossRef]
- Nazarali, S.A.; Narod, S.A. Tamoxifen for women at high risk of breast cancer. Breast Cancer 2014, 6, 29–36. [Google Scholar]
- Linder, S.; van der Poel, H.G.; Bergman, A.M.; Zwart, W.; Prekovic, S. Enzalutamide therapy for advanced prostate cancer: Efficacy, resistance and beyond. Endocr. Related Cancer 2018, 26, R31–R52. [Google Scholar] [CrossRef]
- Grembecka, J.; Belcher, A.M.; Hartley, T.; Cierpicki, T. Molecular basis of the mixed lineage leukemia-menin interaction: Implications for targeting mixed lineage leukemias. J. Biol. Chem. 2010, 285, 40690–40698. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzezinka, K.; Nevedomskaya, E.; Lesche, R.; Haegebarth, A.; ter Laak, A.; Fernández-Montalván, A.E.; Eberspaecher, U.; Werbeck, N.D.; Moenning, U.; Siegel, S.; et al. Characterization of the Menin-MLL Interaction as Therapeutic Cancer Target. Cancers 2020, 12, 201. https://doi.org/10.3390/cancers12010201
Brzezinka K, Nevedomskaya E, Lesche R, Haegebarth A, ter Laak A, Fernández-Montalván AE, Eberspaecher U, Werbeck ND, Moenning U, Siegel S, et al. Characterization of the Menin-MLL Interaction as Therapeutic Cancer Target. Cancers. 2020; 12(1):201. https://doi.org/10.3390/cancers12010201
Chicago/Turabian StyleBrzezinka, Krzysztof, Ekaterina Nevedomskaya, Ralf Lesche, Andrea Haegebarth, Antonius ter Laak, Amaury E. Fernández-Montalván, Uwe Eberspaecher, Nicolas D. Werbeck, Ursula Moenning, Stephan Siegel, and et al. 2020. "Characterization of the Menin-MLL Interaction as Therapeutic Cancer Target" Cancers 12, no. 1: 201. https://doi.org/10.3390/cancers12010201
APA StyleBrzezinka, K., Nevedomskaya, E., Lesche, R., Haegebarth, A., ter Laak, A., Fernández-Montalván, A. E., Eberspaecher, U., Werbeck, N. D., Moenning, U., Siegel, S., Haendler, B., Eheim, A. L., & Stresemann, C. (2020). Characterization of the Menin-MLL Interaction as Therapeutic Cancer Target. Cancers, 12(1), 201. https://doi.org/10.3390/cancers12010201