A Novel Prostate Cell Type-Specific Gene Signature to Interrogate Prostate Tumor Differentiation Status and Monitor Therapeutic Response

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
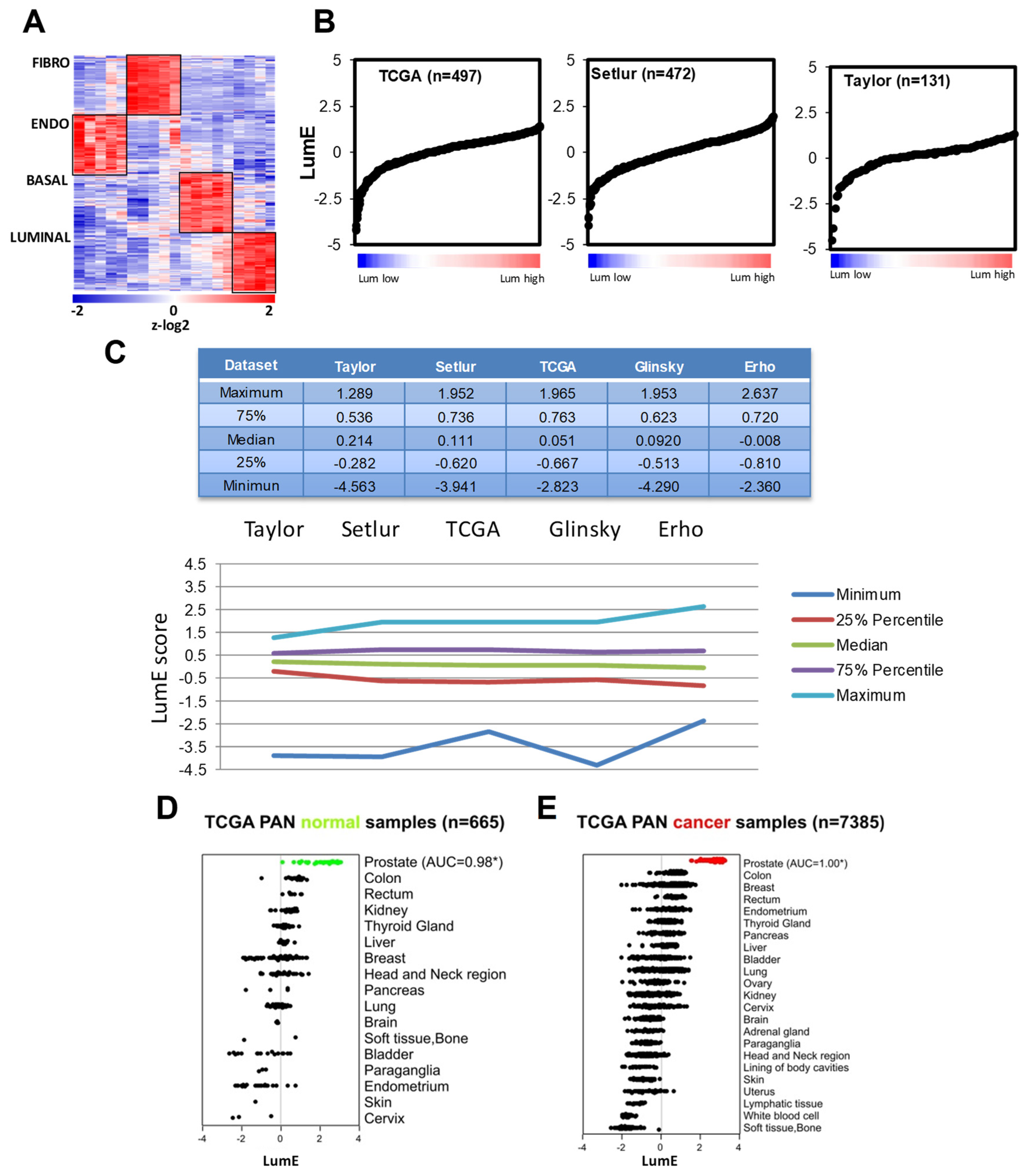
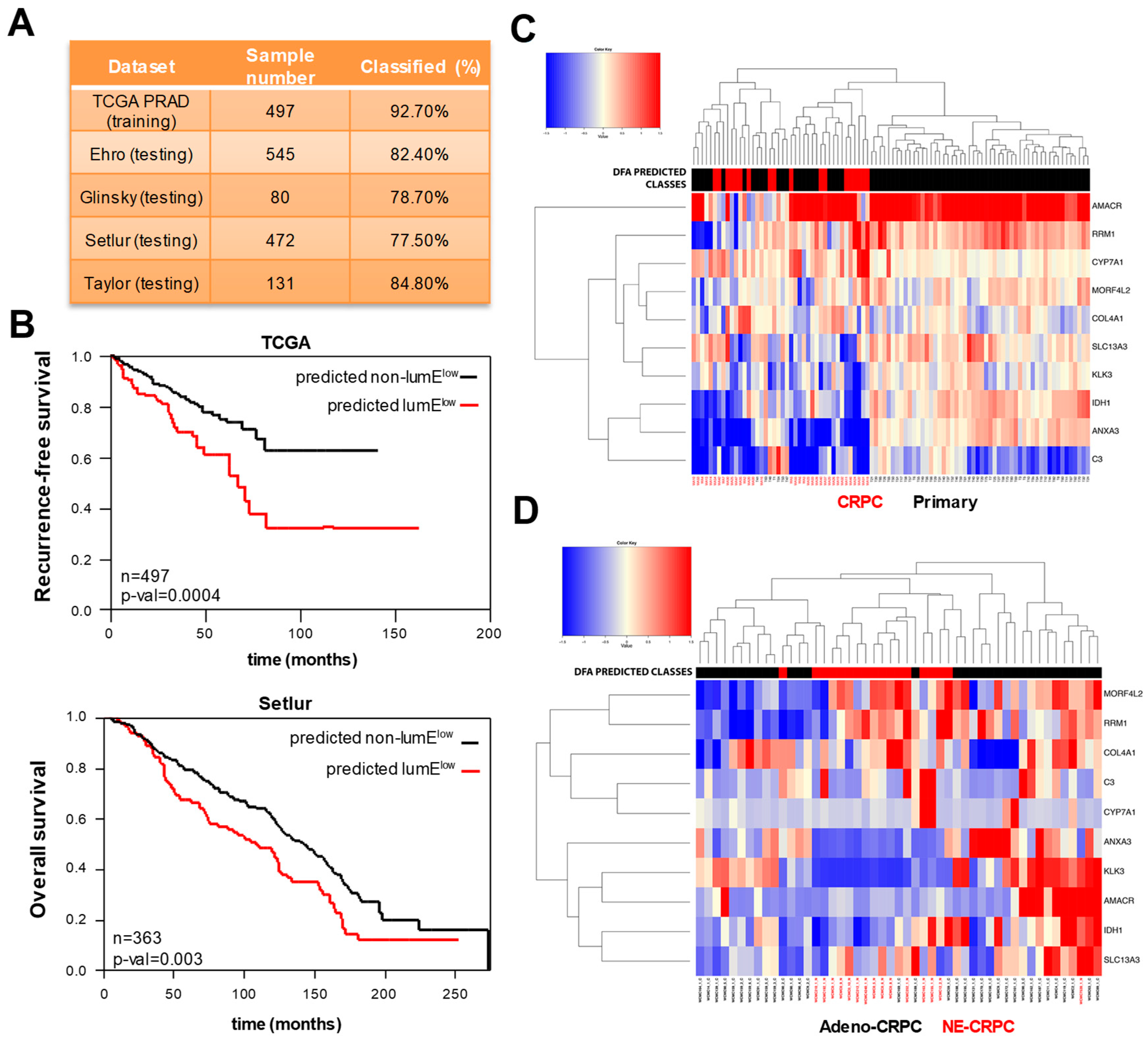
2.1. Prostate Cell-Specific Metagenes Dissect Tumor Phenotypic Variability
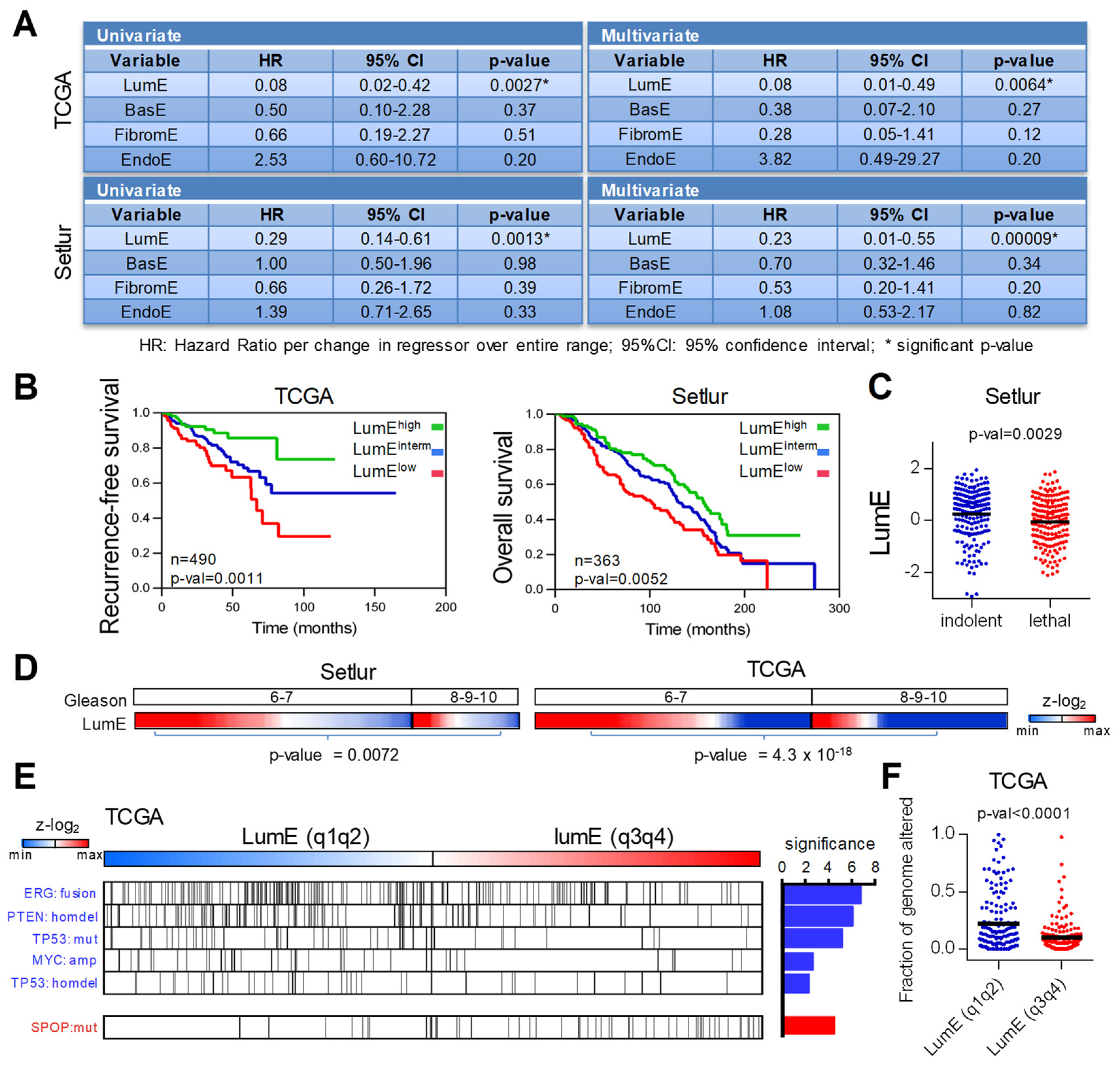
2.2. Low Luminal Tumors Exhibit Poor Clinical Outcome and Increased Mutational Burden
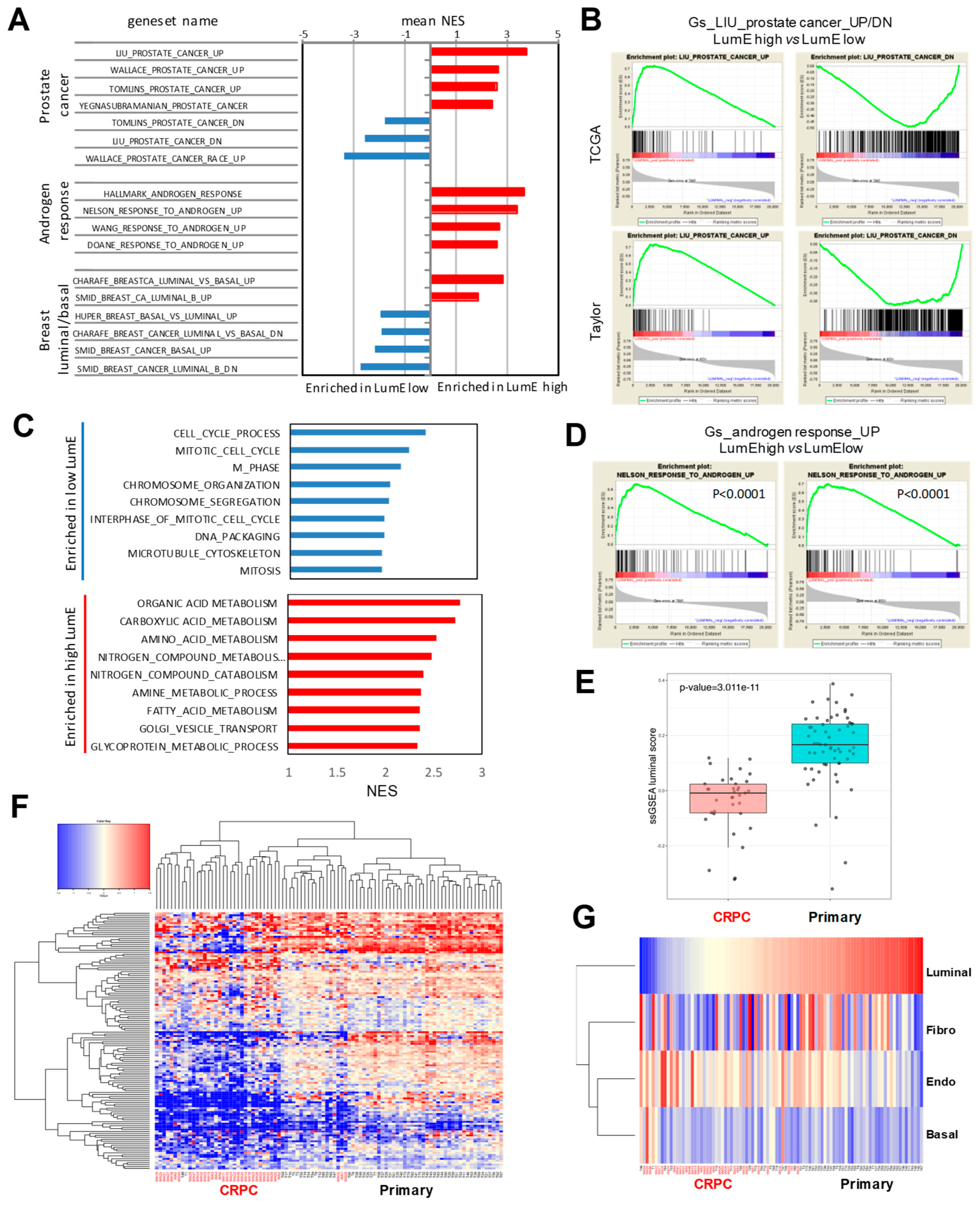
2.3. Association of Prostate-Specific Features in LumE-Defined Tumor Subtypes
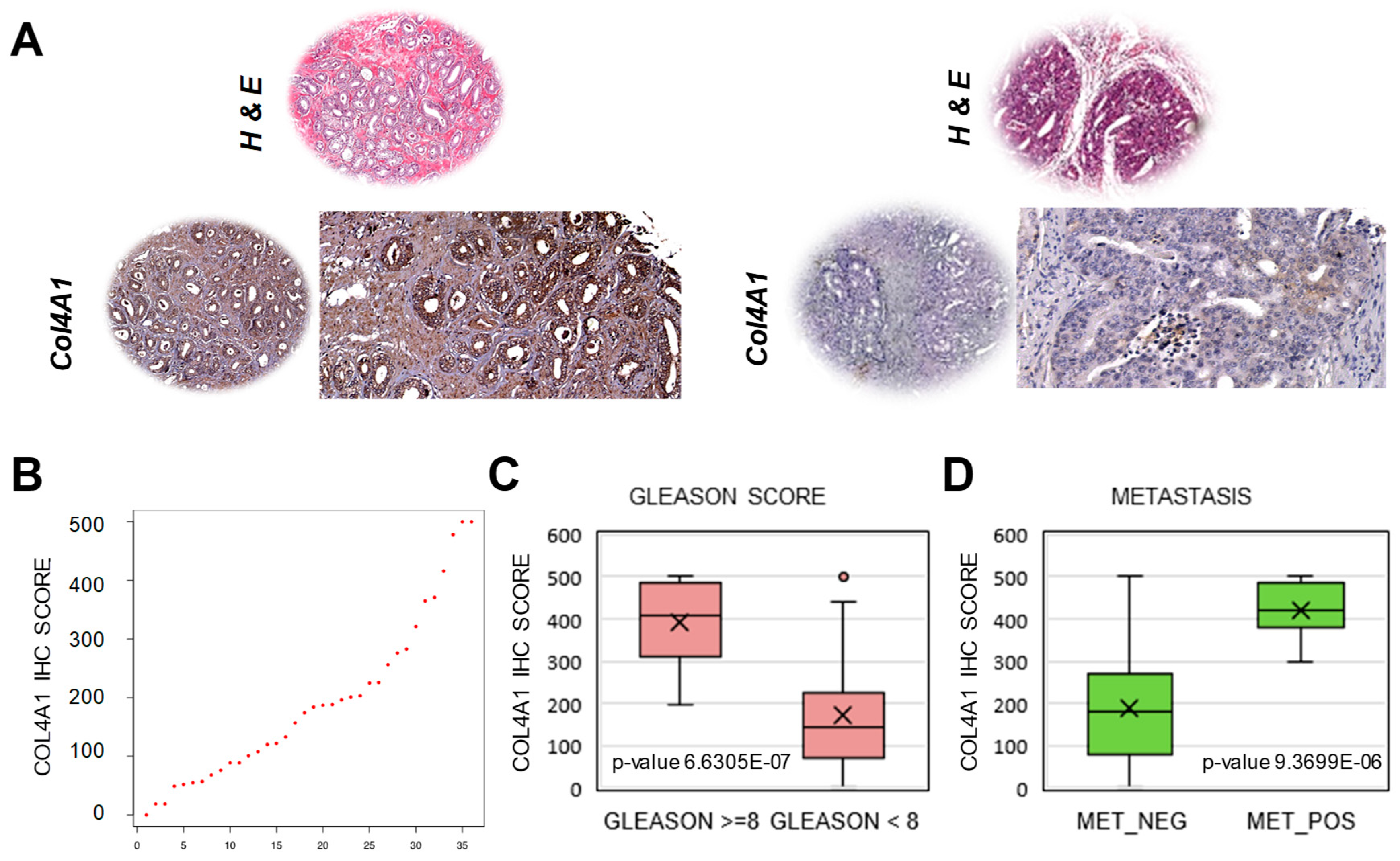
2.4. Extraction of a Mini-Classifier Gene Set and Orthogonal Validation by Immunohistochemistry
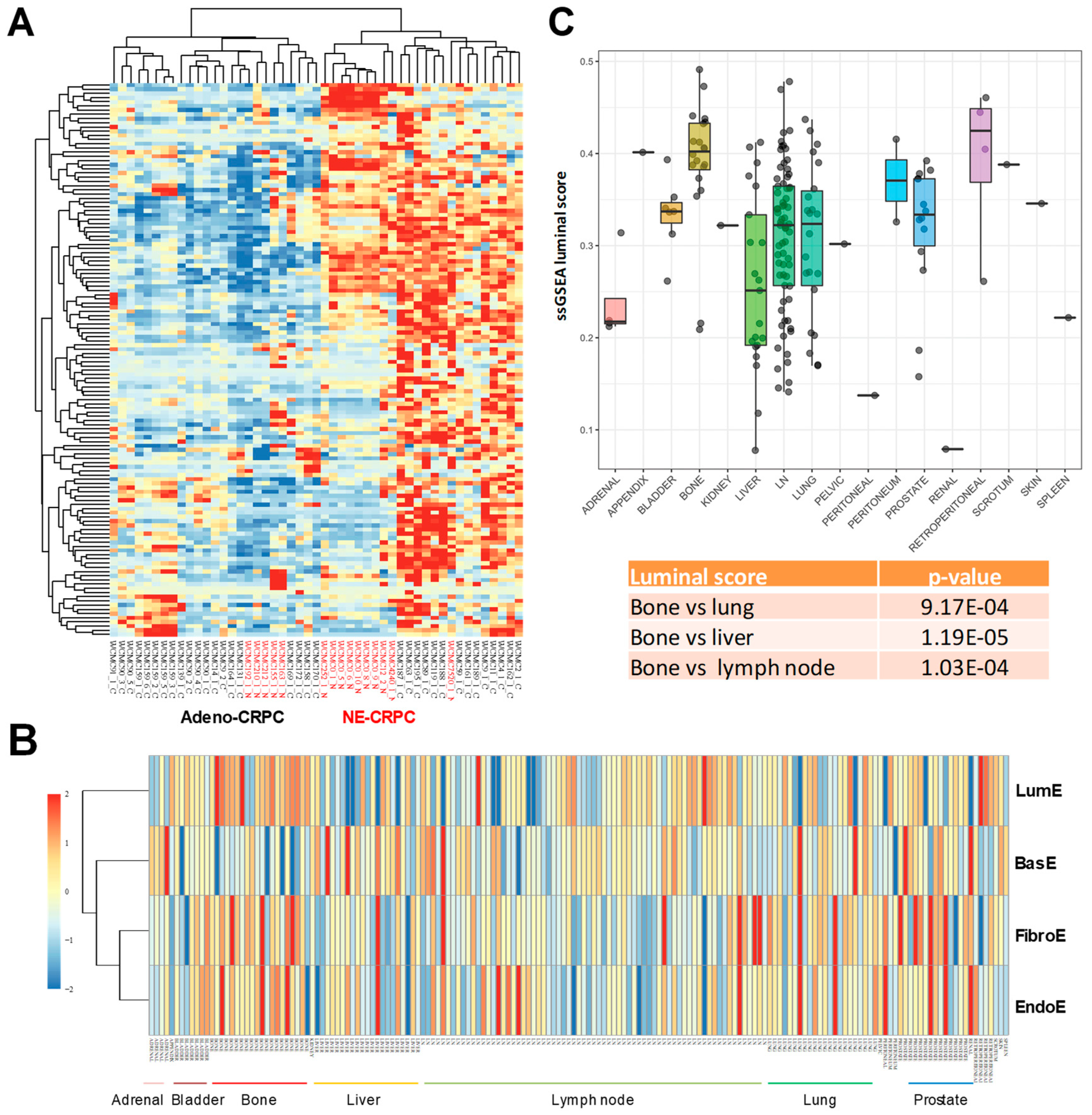
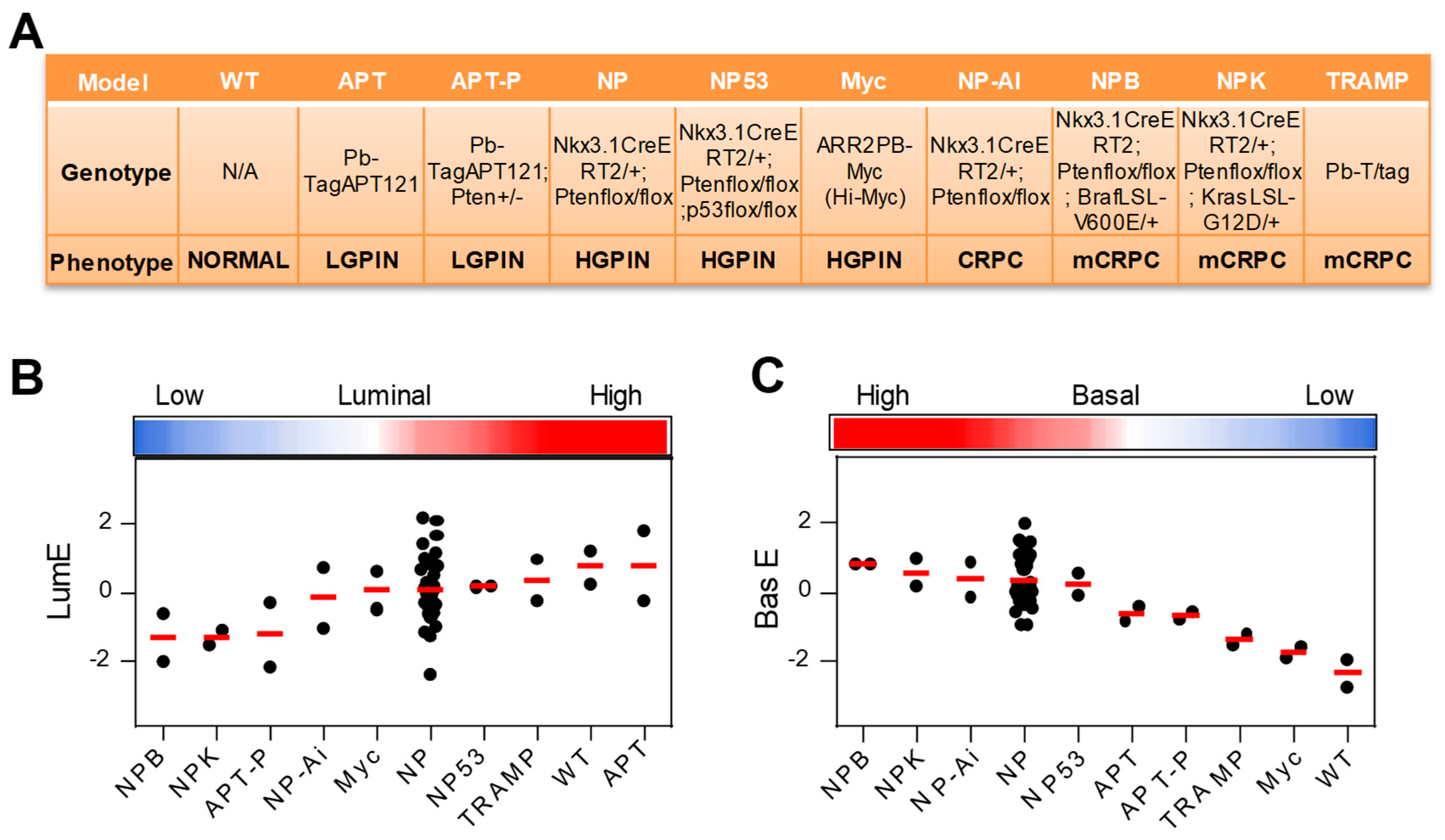
2.5. Application of Luminal Metagene to Mouse Experimental Models
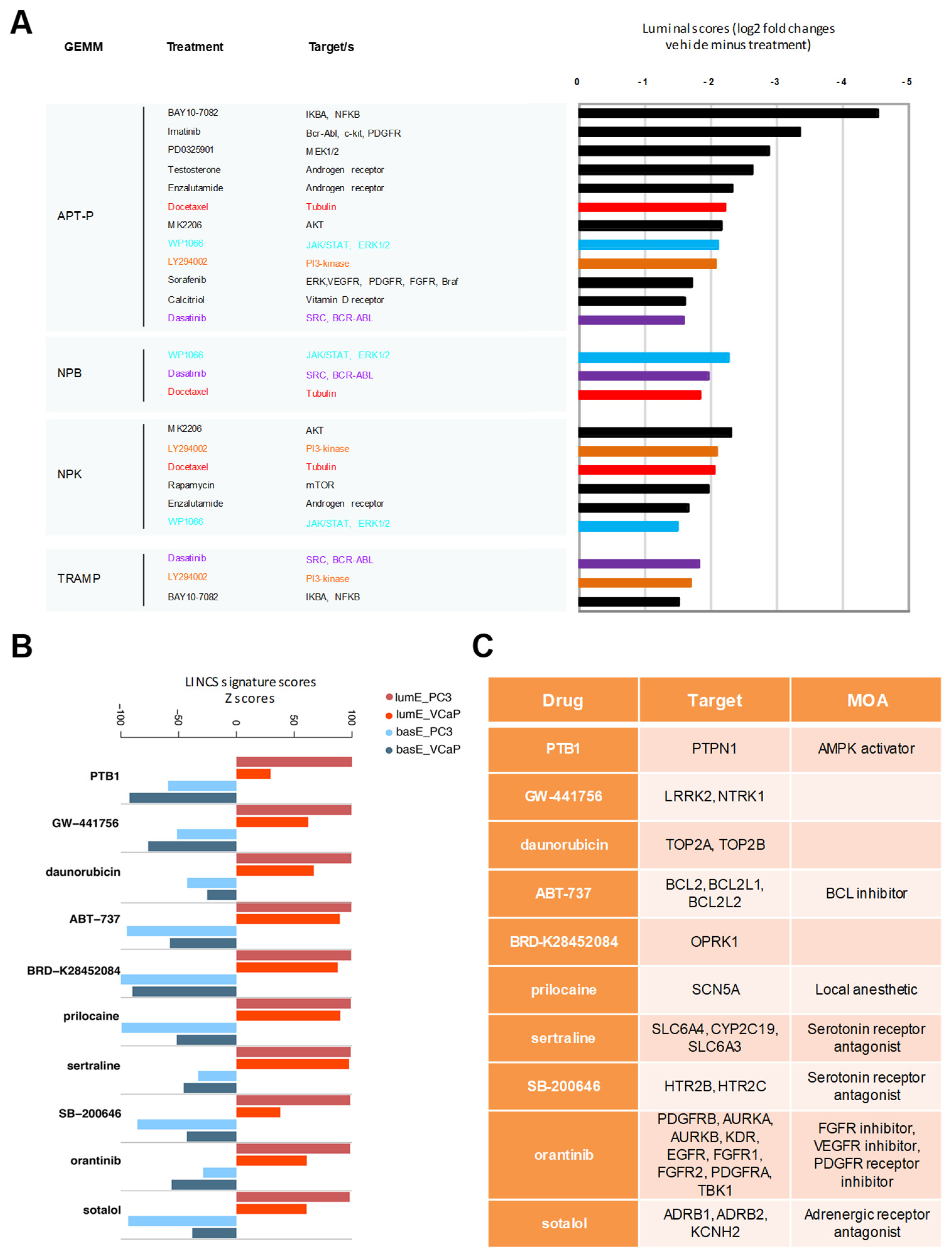
2.6. Luminal Metagene and Therapeutic Response in Human and Mouse Experimental Models
3. Discussion
4. Methods
4.1. Metagene Discovery
4.2. Patient Datasets
4.3. Metagene Scores Calculation
4.4. Patient Stratification for Kaplan-Meier Curves
4.5. Time Series Analysis
4.6. Differential Expression between LumElow and Non-LumElow Human Primary Tumors
4.7. Development of LumElow Gene Mini-Classifier
4.8. Immunohistochemistry
4.9. Statistical Analysis
4.10. Impact of Drug Treatments in GEMMs and Human Cancer Cell Lines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.J.; Autio, K.A.; Roach, M., 3rd; Scher, H.I. High-risk prostate cancer-classification and therapy. Nat. Rev. Clin. Oncol. 2014, 11, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperbeg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Al-Deen Ashab, H.; Davicioni, E.; Karnrs, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Walker, S.M.; Knight, L.A.; McCavigan, A.M.; Logan, G.E.; Berge, V.; Sherif, A.; Pandha, H.; Warren, A.Y.; Davidson, C.; Uprichard, A.; et al. Molecular Subgroup of Primary Prostate Cancer Presenting with Metastatic Biology. Eur. Urol. 2017, 72, 509–518. [Google Scholar] [CrossRef]
- Oudes, A.J.; Campbell, D.S.; Sorensen, C.M.; Walashek, L.S.; True, L.D.; Liu, A.Y. Transcriptomes of human prostate cells. BMC Genom. 2006, 7, 92. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a cell of origin for human prostate cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abata-Shen, C.; Shen, M.M.; et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 2012, 21, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Mizushima, T.; Miyamoto, H. Role of the androgen receptor in urothelial cancer. Mol. Cell Endocrinol. 2018, 465, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Wen, Y.; Wei, Y.; Zhang, S.; Li, S.; Liu, H.; Wang, F.; Zhao, Y.; Zhang, D.; Zhang, Y. Cell subpopulation deconvolution reveals breast cancer heterogeneity based on DNA methylation signature. Brief. Bioinform. 2017, 18, 426–440. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Liang, C.; Wang, S.; Qin, C.; Bao, M.; Cheng, G.; Liu, B.; Shao, P.; Lv, Q.; Song, N.; Hua, L.; et al. TRIM36, a novel androgen-responsive gene, enhances anti-androgen efficacy against prostate cancer by inhibiting MAPK/ERK signaling pathways. Cell Death Dis. 2018, 9, 155. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad Sci.USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Setlur, S.R.; Mertz, K.D.; Hoshida, Y.; Demichelis, F.; Lupien, M.; Perner, S.; Sboner, A.; Pawitan, Y.; Andren, O.; Johnson, L.A.; et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J. Natl. Cancer Inst. 2008, 100, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataral, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. eLife 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Best, C.J.; Gillespie, J.W.; Yi, Y.; Chandramouli, G.V.; Perlmutter, M.A.; Gathright, Y.; Erickson, H.S.; Georgevich, L.; Tangrea, M.A.; Duray, P.H.; et al. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clin. Cancer Res. 2005, 11, 6823–6834. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Civenni, G.; Carbone, G.M.; Catapano, C.V. Overview of Genetically Engineered Mouse Models of Prostate Cancer and Their Applications in Drug Discovery. Curr. Protoc. Pharmacol. 2018, 81, e39. [Google Scholar] [CrossRef]
- Aytes, A.; Mitrofanova, A.; Lefebvre, C.; Alvarez, M.J.; Castillo-Martin, M.; Zheng, T.; Eastham, J.A.; Gopalan, A.; Pienta, K.J.; Shen, M.M.; et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 2014, 25, 638–651. [Google Scholar] [CrossRef]
- Li, J.J.; Shen, M.M. Prostate Stem Cells and Cancer Stem Cells. Cold Spring Harb. Perspect. Med. 2018, 9, a030395. [Google Scholar] [CrossRef]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrai, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Dizeyi, N.; Bjartell, A.; Hedlund, P.; Tasken, K.A.; Gadaleanu, V.; Abrahamsson, P.A. Expression of serotonin receptors 2B and 4 in human prostate cancer tissue and effects of their antagonists on prostate cancer cell lines. Eur. Urol. 2005, 47, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. beta-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2014, 4, 375. [Google Scholar] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The beta2-adrenergic receptor is a molecular switch for neuroendocrine transdifferentiation of prostate cancer cells. Mol. Cancer Res. 2019, 17, 2154–2168. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Arnal-Estape, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Wareen, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Scimeca, M.; Bonfiglio, R.; Urbano, N.; Cerroni, C.; Anemona, L.; Montanaro, M.; Fazi, S.; Schillaci, O.; Mauriello, A.; Bonanno, E. Programmed death ligand 1 expression in prostate cancer cells is associated with deep changes of the tumor inflammatory infiltrate composition. Urol. Oncol. 2019, 37, 297.e19–e31. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mapelli, S.N.; Albino, D.; Mello-Grand, M.; Shinde, D.; Scimeca, M.; Bonfiglio, R.; Bonanno, E.; Chiorino, G.; Garcia-Escudero, R.; Catapano, C.V.; et al. A Novel Prostate Cell Type-Specific Gene Signature to Interrogate Prostate Tumor Differentiation Status and Monitor Therapeutic Response. Cancers 2020, 12, 176. https://doi.org/10.3390/cancers12010176
Mapelli SN, Albino D, Mello-Grand M, Shinde D, Scimeca M, Bonfiglio R, Bonanno E, Chiorino G, Garcia-Escudero R, Catapano CV, et al. A Novel Prostate Cell Type-Specific Gene Signature to Interrogate Prostate Tumor Differentiation Status and Monitor Therapeutic Response. Cancers. 2020; 12(1):176. https://doi.org/10.3390/cancers12010176
Chicago/Turabian StyleMapelli, Sarah N., Domenico Albino, Maurizia Mello-Grand, Dheeraj Shinde, Manuel Scimeca, Rita Bonfiglio, Elena Bonanno, Giovanna Chiorino, Ramon Garcia-Escudero, Carlo V. Catapano, and et al. 2020. "A Novel Prostate Cell Type-Specific Gene Signature to Interrogate Prostate Tumor Differentiation Status and Monitor Therapeutic Response" Cancers 12, no. 1: 176. https://doi.org/10.3390/cancers12010176
APA StyleMapelli, S. N., Albino, D., Mello-Grand, M., Shinde, D., Scimeca, M., Bonfiglio, R., Bonanno, E., Chiorino, G., Garcia-Escudero, R., Catapano, C. V., & Carbone, G. M. (2020). A Novel Prostate Cell Type-Specific Gene Signature to Interrogate Prostate Tumor Differentiation Status and Monitor Therapeutic Response. Cancers, 12(1), 176. https://doi.org/10.3390/cancers12010176