Candidate Causal Variants at the 8p12 Breast Cancer Risk Locus Regulate DUSP4

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
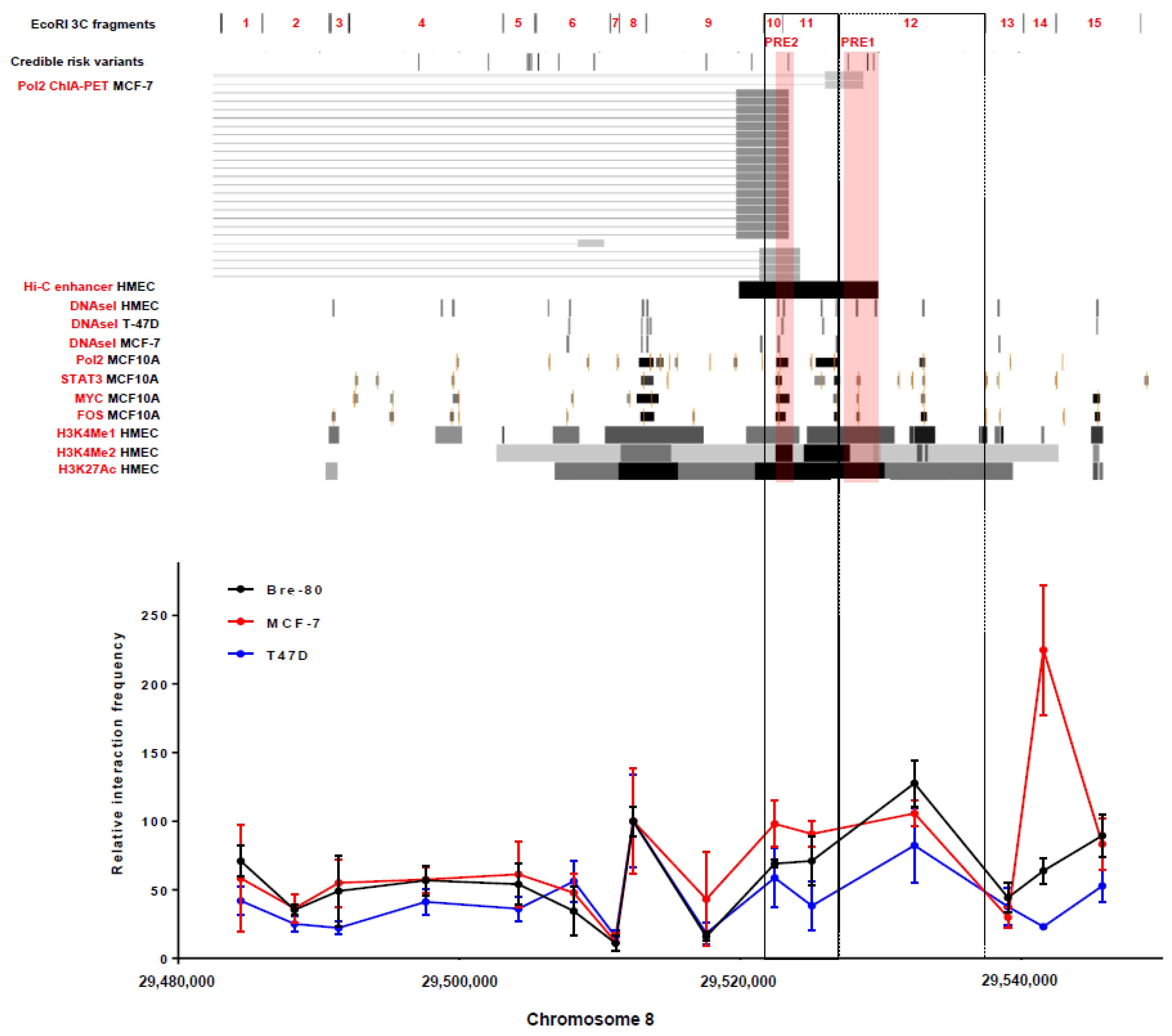
2.1. Chromatin Conformation Capture (3C) Analyses Confirmed That the 8p12 Risk Locus Physically Interacted with the Promoter Region of DUSP4
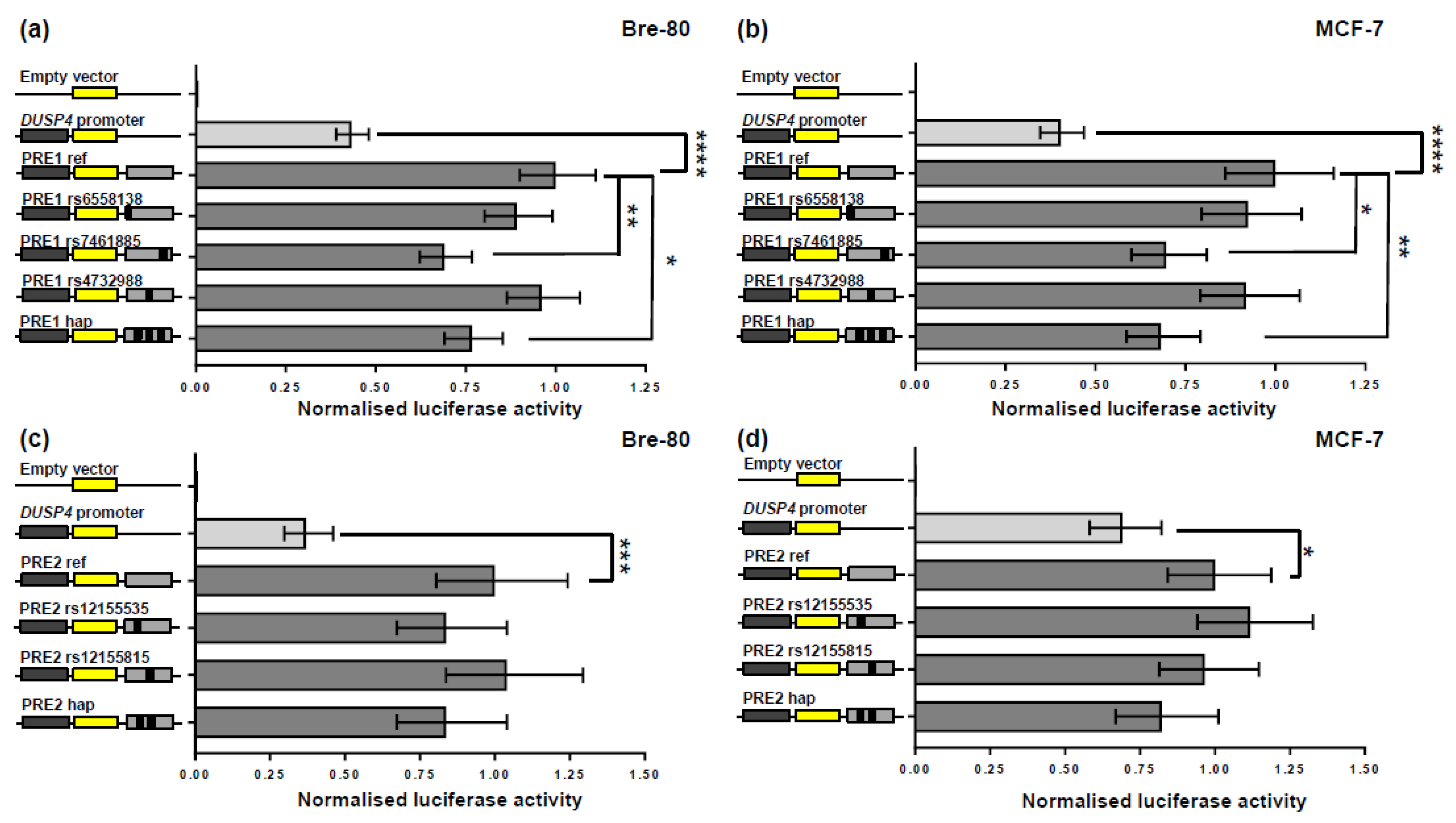
2.2. CCV rs7461885 Reduced the Enhancer Activity of PRE1 on the DUSP4 Promoter
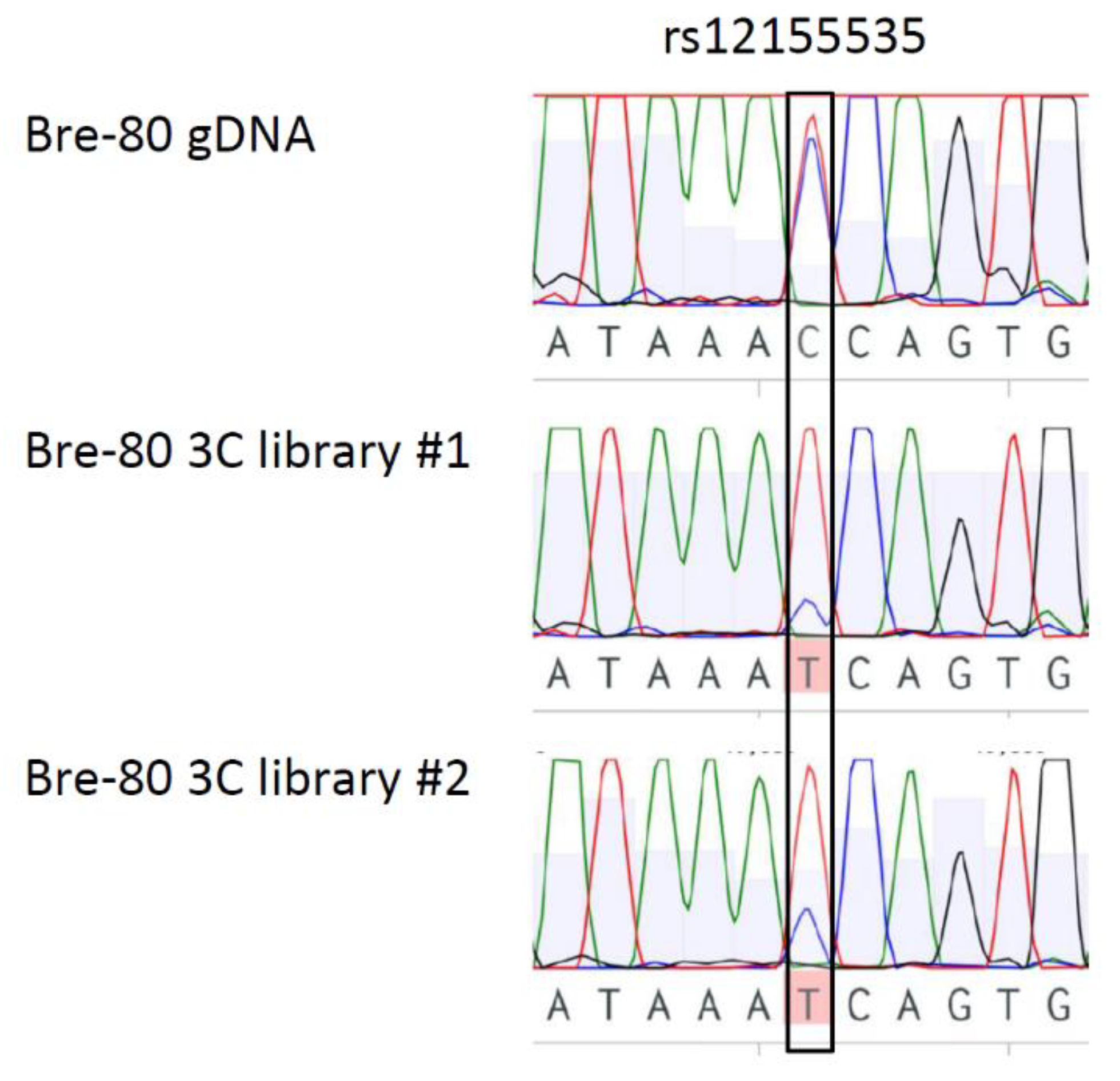
2.3. CCV rs12155535 (PRE2) Was Associated with Allele-Specific Looping to the DUSP4 Promoter Region
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. C Analysis
4.3. Reporter Gene Vector Construction
4.4. Reporter Gene Analysis
4.5. Allele-Specific 3C Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Mulligan, A.M.; Couch, F.J.; Barrowdale, D.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Robson, M.; Sherman, M.; Spurdle, A.B.; et al. Common breast cancer susceptibility alleles are associated with tumour subtypes in BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2. Breast Cancer Res. 2011, 13, R110. [Google Scholar] [CrossRef]
- Fachal, L.; Aschard, H.; Beesley, J.; Barnes, D.R.; Allen, J.; Kar, S.; Pooley, K.A.; Dennis, J.; Michailidou, K.; Turman, C.; et al. Fine-mapping of 150 breast cancer risk regions identifies 178 high confidence target genes. bioRxiv 2019, 521054. [Google Scholar] [CrossRef]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Beesley, J.; Sivakumaran, H.; Marjaneh, M.M.; Lima, L.G.; Hillman, K.M.; Kaufmann, S.; Tuano, N.; Hussein, N.; Ham, S.; Mukhopadhyay, P.; et al. Chromatin interactome mapping at 139 independent breast cancer risk signals. bioRxiv 2019, 520916. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Glubb, D.M.; Maranian, M.J.; Michailidou, K.; Pooley, K.A.; Meyer, K.B.; Kar, S.; Carlebur, S.; O’Reilly, M.; Betts, J.A.; Hillman, K.M.; et al. Fine-Scale Mapping of the 5q11.2 Breast Cancer Locus Reveals at Least Three Independent Risk Variants Regulating MAP3K1. Am. J. Hum. Genet. 2015, 96, 5–20. [Google Scholar] [CrossRef]
- Ghoussaini, M.; Edwards, S.L.; Michailidou, K.; Nord, S.; Cowper-Sal Lari, R.; Desai, K.; Kar, S.; Hillman, K.M.; Kaufmann, S.; Glubb, D.M.; et al. Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat. Commun. 2014, 4, 4999. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F.; et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- O’Mara, T.A.; Glubb, D.M.; Kho, P.F.; Thompson, D.J.; Spurdle, A. Genome-wide association studies of endometrial cancer: Latest developments and future directions. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1095–1102. [Google Scholar] [CrossRef]
- Lawrenson, K.; Li, Q.; Kar, S.; Seo, J.H.; Tyrer, J.; Spindler, T.J.; Lee, J.; Chen, Y.; Karst, A.; Drapkin, R.; et al. Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat. Commun. 2015, 6, 8234. [Google Scholar] [CrossRef]
- Guo, X.; Lin, W.; Bao, J.; Cai, Q.; Pan, X.; Bai, M.; Yuan, Y.; Shi, J.; Sun, Y.; Han, M.-R.; et al. A Comprehensive cis-eQTL Analysis Revealed Target Genes in Breast Cancer Susceptibility Loci Identified in Genome-wide Association Studies. Am. J. Hum. Genet. 2018, 102, 890–903. [Google Scholar] [CrossRef]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Boulding, T.; Wu, F.; McCuaig, R.; Dunn, J.; Sutton, C.R.; Hardy, K.; Tu, W.; Bullman, A.; Yip, D.; Dahlstrom, J.E.; et al. Differential Roles for DUSP Family Members in Epithelial-to-Mesenchymal Transition and Cancer Stem Cell Regulation in Breast Cancer. PLoS ONE 2016, 11, e0148065. [Google Scholar] [CrossRef]
- Armes, J.E.; Hammet, F.; de Silva, M.; Ciciulla, J.; Ramus, S.J.; Soo, W.K.; Mahoney, A.; Yarovaya, N.; Henderson, M.A.; Gish, K.; et al. Candidate tumor-suppressor genes on chromosome arm 8p in early-onset and high-grade breast cancers. Oncogene 2004, 23, 5697–5702. [Google Scholar] [CrossRef]
- Mazumdar, A.; Poage, G.M.; Shepherd, J.; Tsimelzon, A.; Hartman, Z.C.; Den Hollander, P.; Hill, J.; Zhang, Y.; Chang, J.; Hilsenbeck, S.G.; et al. Analysis of phosphatases in ER-negative breast cancers identifies DUSP4 as a critical regulator of growth and invasion. Breast Cancer Res. Treat. 2016, 158, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Schwarz, L.J.; Bhola, N.E.; Kurupi, R.; Owens, P.; Miller, T.W.; Gomez, H.; Cook, R.S.; Arteaga, C.L. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013, 73, 6346–6358. [Google Scholar] [CrossRef] [PubMed]
- Gaggianesi, M.; Turdo, A.; Chinnici, A.; Lipari, E.; Apuzzo, T.; Benfante, A.; Sperduti, I.; Di Franco, S.; Meraviglia, S.; Lo Presti, E.; et al. IL4 Primes the Dynamics of Breast Cancer Progression via DUSP4 Inhibition. Cancer Res. 2017, 77, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.B.; O’Reilly, M.; Michailidou, K.; Carlebur, S.; Edwards, S.L.; French, J.D.; Prathalingham, R.; Dennis, J.; Bolla, M.K.; Wang, Q.; et al. Fine-Scale Mapping of the FGFR2 Breast Cancer Risk Locus: Putative Functional Variants Differentially Bind FOXA1 and E2F1. Am. J. Hum. Genet. 2013, 93, 1046–1060. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glubb, D.M.; Shi, W.; Beesley, J.; Fachal, L.; Pritchard, J.-L.; McCue, K.; Barnes, D.R.; Antoniou, A.C.; Dunning, A.M.; Easton, D.F.; et al. Candidate Causal Variants at the 8p12 Breast Cancer Risk Locus Regulate DUSP4. Cancers 2020, 12, 170. https://doi.org/10.3390/cancers12010170
Glubb DM, Shi W, Beesley J, Fachal L, Pritchard J-L, McCue K, Barnes DR, Antoniou AC, Dunning AM, Easton DF, et al. Candidate Causal Variants at the 8p12 Breast Cancer Risk Locus Regulate DUSP4. Cancers. 2020; 12(1):170. https://doi.org/10.3390/cancers12010170
Chicago/Turabian StyleGlubb, Dylan M., Wei Shi, Jonathan Beesley, Laura Fachal, Jayne-Louise Pritchard, Karen McCue, Daniel R. Barnes, Antonis C. Antoniou, Alison M. Dunning, Douglas F. Easton, and et al. 2020. "Candidate Causal Variants at the 8p12 Breast Cancer Risk Locus Regulate DUSP4" Cancers 12, no. 1: 170. https://doi.org/10.3390/cancers12010170
APA StyleGlubb, D. M., Shi, W., Beesley, J., Fachal, L., Pritchard, J.-L., McCue, K., Barnes, D. R., Antoniou, A. C., Dunning, A. M., Easton, D. F., & Chenevix-Trench, G. (2020). Candidate Causal Variants at the 8p12 Breast Cancer Risk Locus Regulate DUSP4. Cancers, 12(1), 170. https://doi.org/10.3390/cancers12010170