Prediction of Drug Efficacy in Colon Cancer Preclinical Models Using a Novel Ranking Method of Gene Expression



Abstract
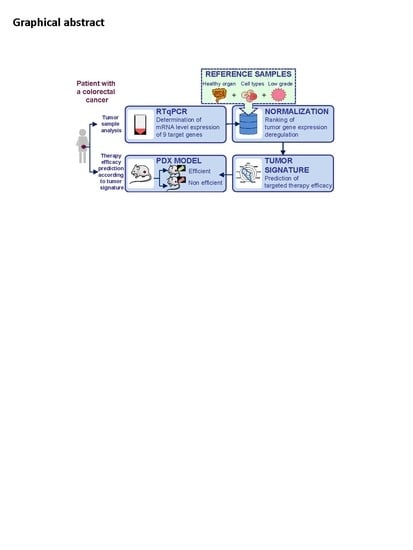
1. Introduction
2. Results
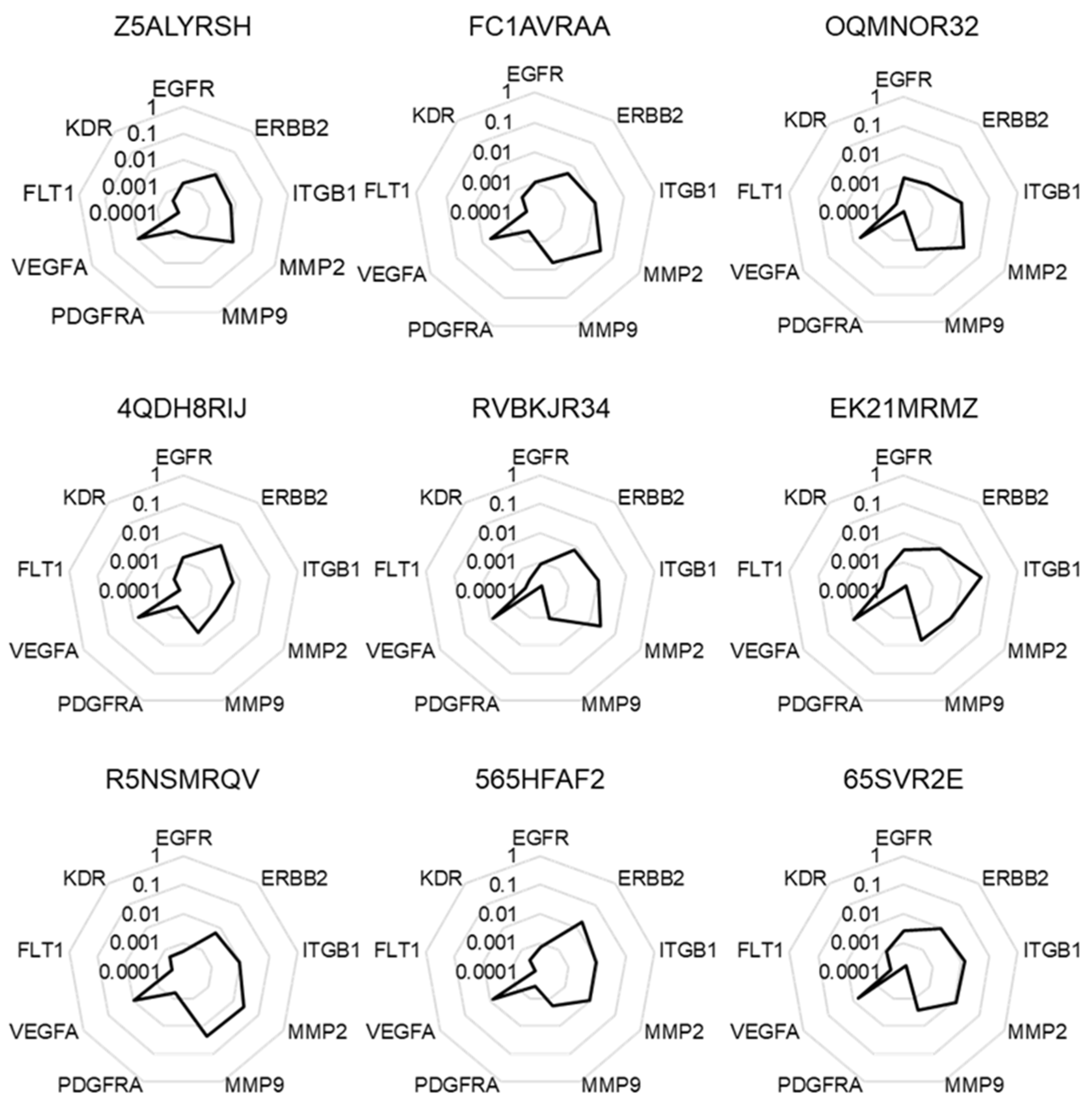
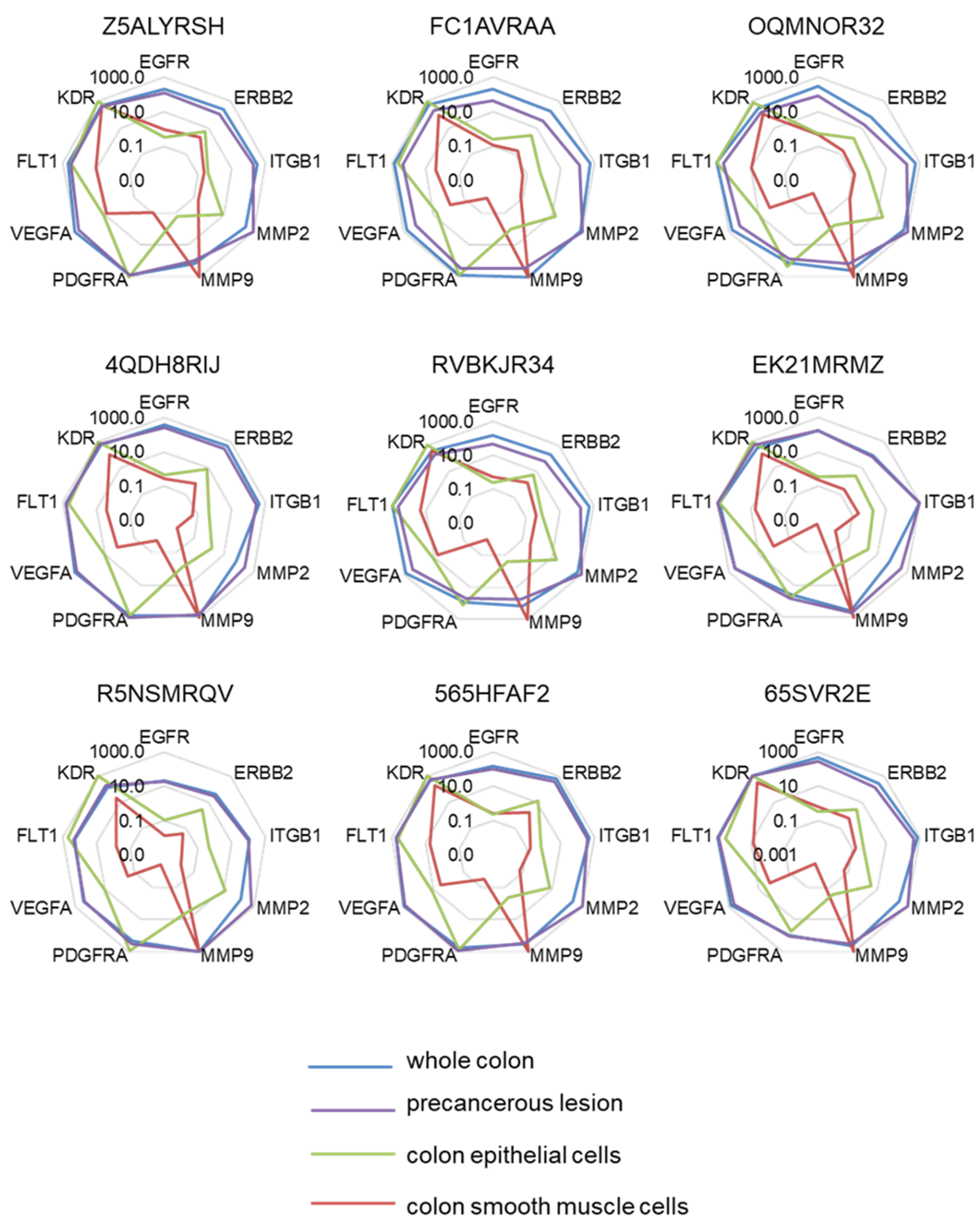
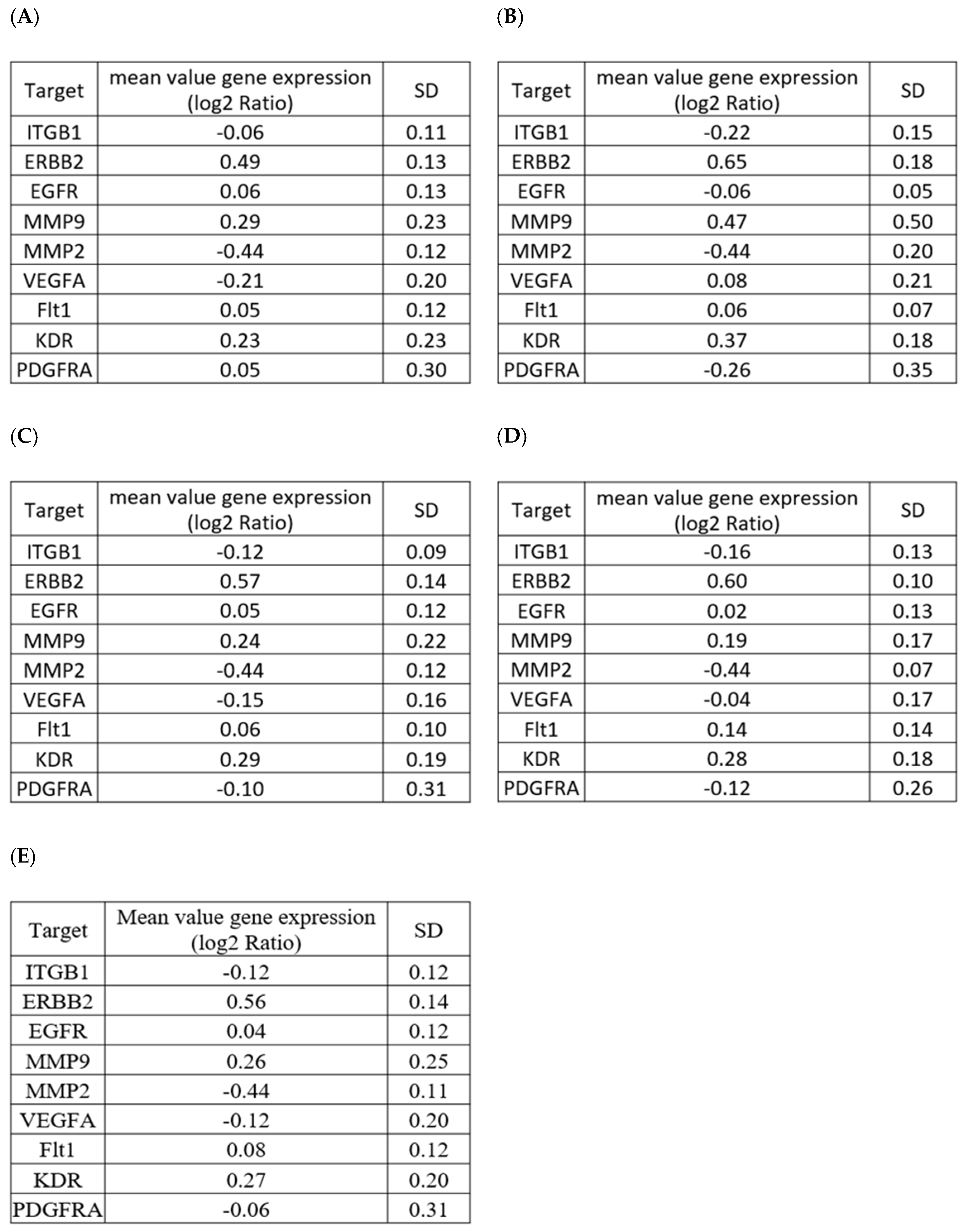
2.1. Selection of a Normalization Process Taking into Account Tumor Complexity
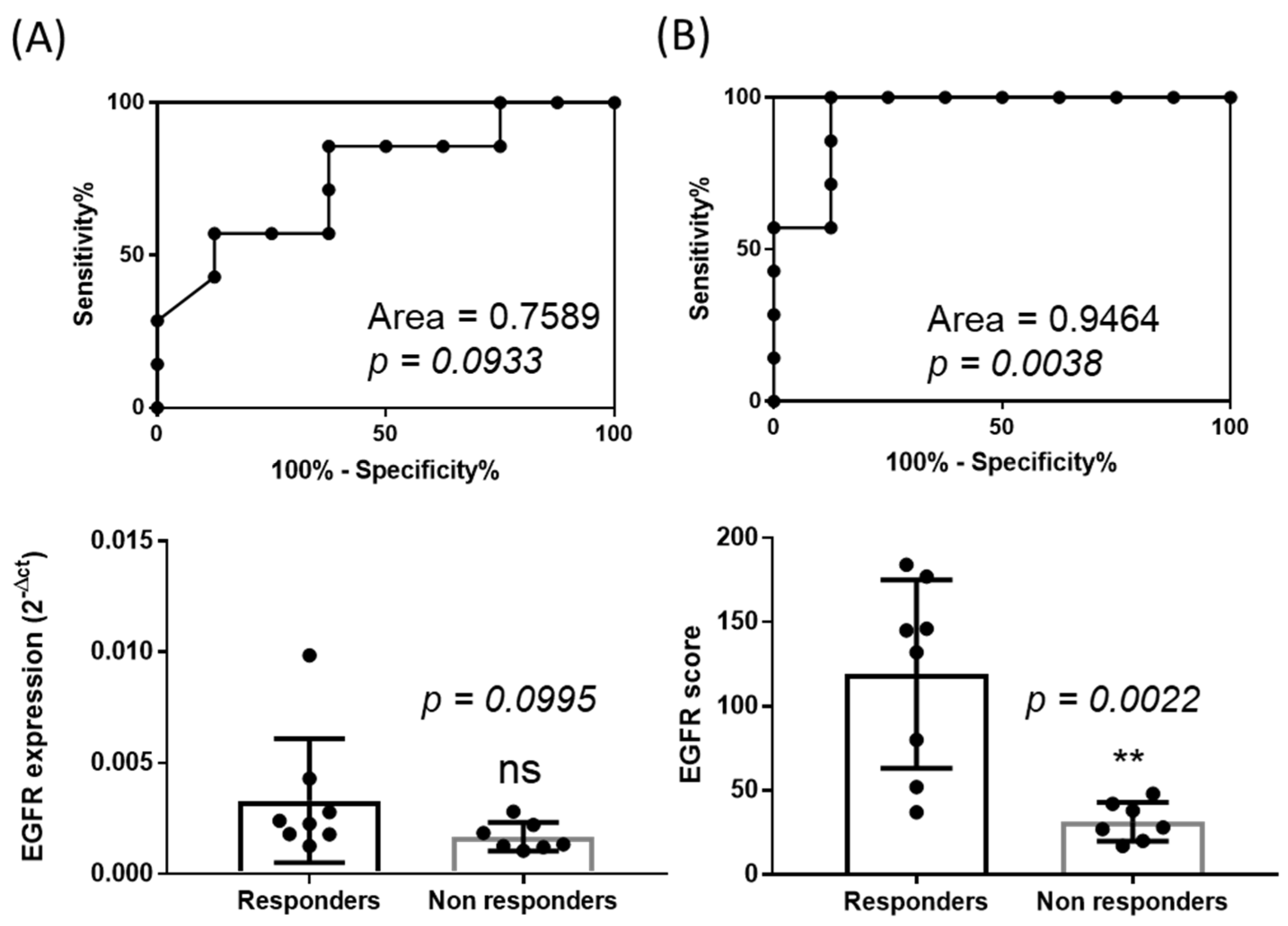
2.2. The Normalized Score of Expression of EGFR Correlates with Cetuximab Efficacy
2.3. The Score of Expression of EGFR Can Be Used to Discriminate Responders Versus Non Responders
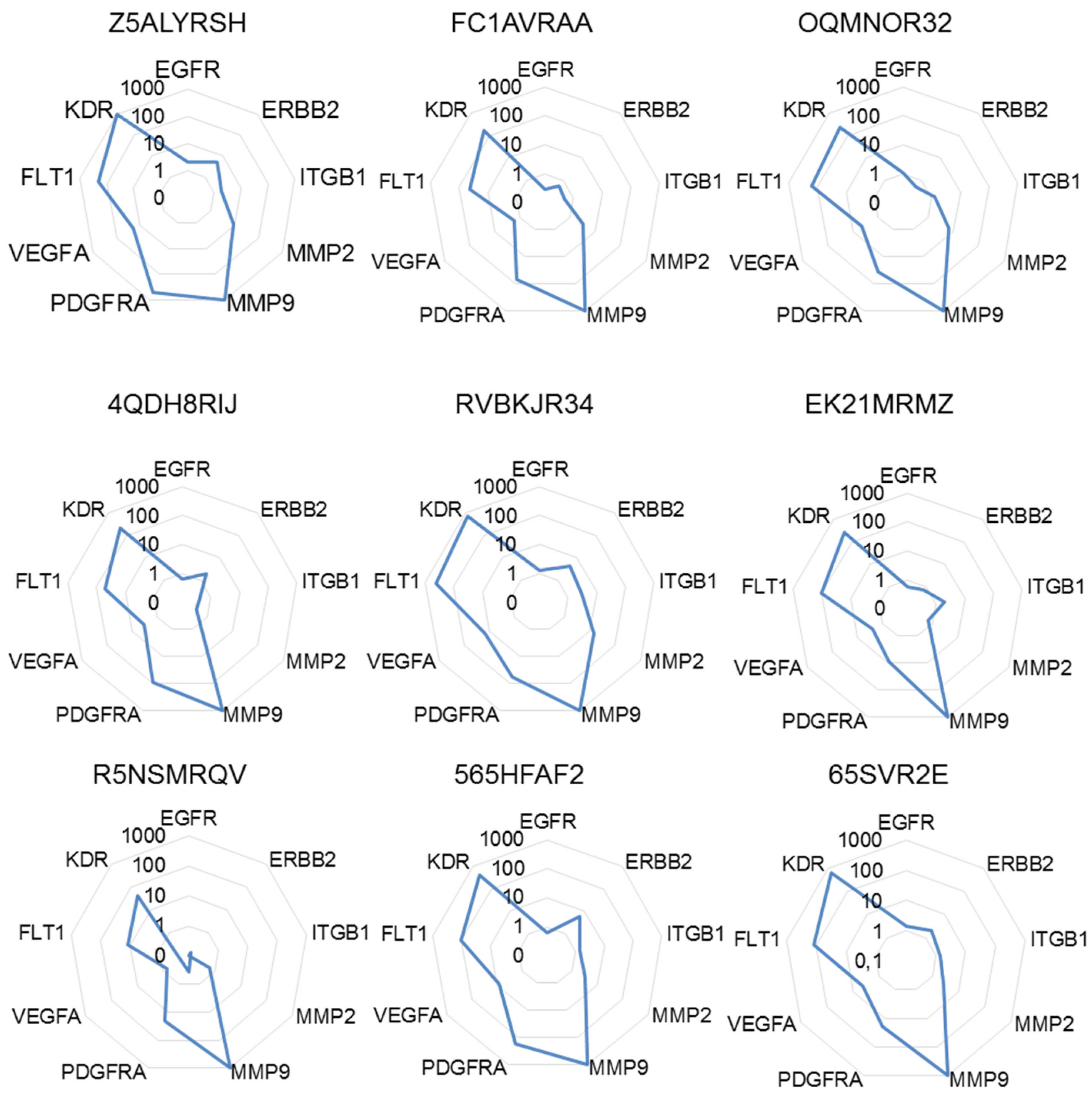
2.4. The Highest Score of the Normalized Signature is Predictive of Drug Efficacy
3. Discussion
4. Material and Methods
4.1. Tumor Samples Collection
4.2. RT-qPCR
4.3. Normalization Process
4.4. Heterotopic Grafting of Colorectal Cancer Patient Derived Xenograft
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hinkson, I.V.; Davidsen, T.M.; Klemm, J.D.; Chandramouliswaran, I.; Kerlavage, A.R.; Kibbe, W.A. A Comprehensive Infrastructure for Big Data in Cancer Research: Accelerating Cancer Research and Precision Medicine. Front. Cell Dev. Biol. 2017, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef]
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy—Leveraging a knowledge base for clinical decision-making. Mol. Case Stud. 2018, 4, a001578. [Google Scholar] [CrossRef]
- Ke, X.; Shen, L. Molecular targeted therapy of cancer: The progress and future prospect. Front. Lab. Med. 2017, 1, 69–75. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Köhne, C.-H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lenz, H.-J.; Köhne, C.-H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef]
- Soria, J.-C.; Rodon Ahnert, J.; Berger, R.; Miller, W.H.; Brana, I.; Loriot, Y.; Mughal, T.I.; Lazar, V.; Wunder, F.; Bresson, C.; et al. WINTHER: An international study to select rational therapeutics based on the analysis of matched tumor and normal biopsies in subjects with advanced malignancies. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Wagenaar-Miller, R.A.; Gorden, L.; Matrisian, L.M. Matrix metalloproteinases in colorectal cancer: Is it worth talking about? Cancer Metastasis Rev. 2004, 23, 119–135. [Google Scholar] [CrossRef]
- De Cuba, E.M.V.; Kwakman, R.; van Egmond, M.; Bosch, L.J.W.; Bonjer, H.J.; Meijer, G.A.; te Velde, E.A. Understanding molecular mechanisms in peritoneal dissemination of colorectal cancer: Future possibilities for personalised treatment by use of biomarkers. Virchows Arch. Int. J. Pathol. 2012, 461, 231–243. [Google Scholar] [CrossRef]
- La Salvia, A.; Lopez-Gomez, V.; Garcia-Carbonero, R. HER2-targeted therapy: An emerging strategy in advanced colorectal cancer. Expert Opin. Investig. Drugs 2019, 28, 29–38. [Google Scholar] [CrossRef]
- Cohen, R.B. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin. Colorectal Cancer 2003, 2, 246–251. [Google Scholar] [CrossRef]
- Manzat Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The Role of PDGFs and PDGFRs in Colorectal Cancer. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Winder, T.; Lenz, H.-J. Vascular endothelial growth factor and epidermal growth factor signaling pathways as therapeutic targets for colorectal cancer. Gastroenterology 2010, 138, 2163–2176. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, L. The clinical application of fruquintinib on colorectal cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Tampellini, M.; Sonetto, C.; Scagliotti, G.V. Novel anti-angiogenic therapeutic strategies in colorectal cancer. Expert Opin. Investig. Drugs 2016, 25, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointest. Cancer Res. GCR 2012, 5, 19–27. [Google Scholar]
- Noble, C.L.; Abbas, A.R.; Cornelius, J.; Lees, C.W.; Ho, G.-T.; Toy, K.; Modrusan, Z.; Pal, N.; Zhong, F.; Chalasani, S.; et al. Regional variation in gene expression in the healthy colon is dysregulated in ulcerative colitis. Gut 2008, 57, 1398–1405. [Google Scholar] [CrossRef]
- Julien, S.; Merino-Trigo, A.; Lacroix, L.; Pocard, M.; Goéré, D.; Mariani, P.; Landron, S.; Bigot, L.; Nemati, F.; Dartigues, P.; et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5314–5328. [Google Scholar] [CrossRef]
- Easton, D.F.; Ford, D.; Bishop, D.T. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1995, 56, 265–271. [Google Scholar]
- Yesner, R.; Kelly, L.J.; Chan, Y.K. Minimum prostate-specific antigen (PSA) level diagnostic of prostate cancer. Connect. Med. 1996, 60, 399–404. [Google Scholar]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Izumchenko, E.; Aliper, A.M.; Makarev, E.; Paz, K.; Buzdin, A.A.; Zhavoronkov, A.A.; Sidransky, D. Pathway activation strength is a novel independent prognostic biomarker for cetuximab sensitivity in colorectal cancer patients. Hum. Genome Var. 2015, 2, 15009. [Google Scholar] [CrossRef]
- Snyder, M.; Bottiglieri, S.; Almhanna, K. Impact of Primary Tumor Location on First-line Bevacizumab or Cetuximab in Metastatic Colorectal Cancer. Rev. Recent Clin. Trials 2018, 13, 139–149. [Google Scholar] [CrossRef]
- Verlingue, L.; Baranger, M.; Gianesini, C.; Fritz, J.; Schaeffer, M.; Malka, D.; Jouffroy, C.; Loubaton, B.; Bagnard, D. PPREDMED®, a normalized expression signature of drug targets versus reference tissues aiming at generalizing treatment personalization. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Biological Function | Relevance in CRC | |
|---|---|---|---|
| MMP9 | Extracellular matrix remodeling | Proliferation | [16] |
| MMP2 | [16] | ||
| ITGB1 | [17] | ||
| ERBB2 | [18] | ||
| EGFR | [19] | ||
| PDGFRA | [20] | ||
| VEGFA | Angiogenesis | [21] | |
| Flt1 | [22] | ||
| KDR | [23] | ||
| Sample. | Z5ALYRSH | FC1AVRAA | OQMNOR32 | ||||||
| Reference | SMC | EPIC | POL | SMC | EPIC | POL | SMC | EPIC | POL |
| NORM | ** | * | ns | *** | * | ns | **** | ns | ns |
| SMC | ns | * | * | ** | * | ** | |||
| EPIC | ns | ns | ns | ||||||
| Sample | 4QDH8RIJ | RVBKJR34 | EK21MRMZ | ||||||
| Reference | SMC | EPIC | POL | SMC | EPIC | POL | SMC | EPIC | POL |
| NORM | *** | ns | ns | *** | ns | ns | ** | ns | ns |
| SMC | * | *** | ns | * | * | **** | |||
| EPIC | ns | ns | * | ||||||
| Sample | R5NSMRQV | 565HFAF2 | 65SVR2E | ||||||
| Reference | SMC | EPIC | POL | SMC | EPIC | POL | SMC | EPIC | POL |
| NORM | *** | ns | ns | *** | ns | ns | **** | ** | ns |
| SMC | ** | *** | ns | *** | ns | *** | |||
| EPIC | ns | ns | * | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritz, J.; Lefebvre, O.; Fernandez, A.; Schmidt, J.; Bagnard, D. Prediction of Drug Efficacy in Colon Cancer Preclinical Models Using a Novel Ranking Method of Gene Expression. Cancers 2020, 12, 149. https://doi.org/10.3390/cancers12010149
Fritz J, Lefebvre O, Fernandez A, Schmidt J, Bagnard D. Prediction of Drug Efficacy in Colon Cancer Preclinical Models Using a Novel Ranking Method of Gene Expression. Cancers. 2020; 12(1):149. https://doi.org/10.3390/cancers12010149
Chicago/Turabian StyleFritz, Justine, Olivier Lefebvre, Aurore Fernandez, Jordane Schmidt, and Dominique Bagnard. 2020. "Prediction of Drug Efficacy in Colon Cancer Preclinical Models Using a Novel Ranking Method of Gene Expression" Cancers 12, no. 1: 149. https://doi.org/10.3390/cancers12010149
APA StyleFritz, J., Lefebvre, O., Fernandez, A., Schmidt, J., & Bagnard, D. (2020). Prediction of Drug Efficacy in Colon Cancer Preclinical Models Using a Novel Ranking Method of Gene Expression. Cancers, 12(1), 149. https://doi.org/10.3390/cancers12010149