Acute Myeloid Leukemia: Aging and Epigenetics


Abstract
1. Introduction
2. AML Development
2.1. AML Initiating Events
2.2. Mutations in Epigenetic Modifiers
2.3. Pre-Leukemic State
2.4. Leukemic Stem Cells
2.5. Cell of Origin and “Cellular Context”
3. Contribution of Aging to AML
3.1. Accumulation of Somatic Mutations upon Aging
3.2. Clonal Hematopoiesis in Elderly
3.3. Age-Associated Epigenetic Changes
3.4. Epigenetic Polarity in Aged HSC
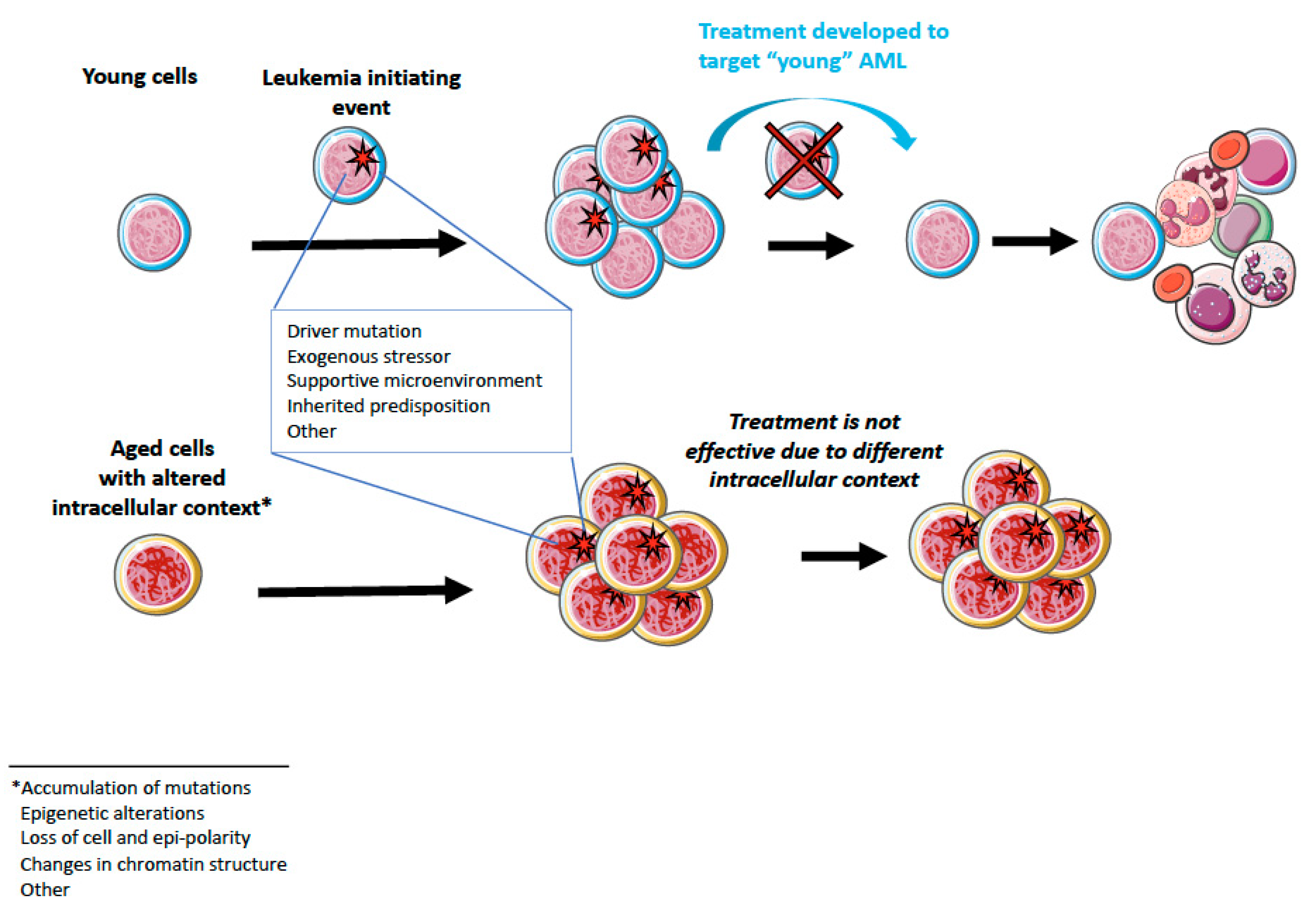
4. Aging and AML
5. Therapy
5.1. Currently Available Treatment Options
5.2. Possibilities for Aging-Targeted Therapies?
6. Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Bower, H.; Andersson, T.M.-L.; Björkholm, M.; Dickman, P.W.; Lambert, P.C.; Derolf, Å.R. Continued improvement in survival of acute myeloid leukemia patients: An application of the loss in expectation of life. Blood Cancer J. 2016, 6, e390. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Döhner, K.; Krauter, J.; Fröhling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Späth, D.; Morgan, M.; Benner, A.; et al. Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N. Engl. J. Med. 2008, 358, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Rozhok, A.I.; DeGregori, J. The Evolution of Lifespan and Age-Dependent Cancer Risk. Trends Cancer 2016, 2, 552–560. [Google Scholar] [CrossRef]
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.D.; Russell, S.M. Upsides and downsides to polarity and asymmetric cell division in leukemia. Oncogene 2008, 27, 7003–7017. [Google Scholar] [CrossRef]
- Ho, Y.-H.; Méndez-Ferrer, S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica 2019. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Saçma, M.; Pospiech, J.; Bogeska, R.; de Back, W.; Mallm, J.-P.; Sakk, V.; Soller, K.; Marka, G.; Vollmer, A.; Karns, R.; et al. Haematopoietic stem cells in perisinusoidal niches are protected from ageing. Nat. Cell Biol. 2019, 21, 1309–1320. [Google Scholar] [CrossRef]
- Wu, C.T.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Losman, J.-A.; Kaelin, W.G. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef]
- Nagel, G.; Weber, D.; Fromm, E.; Erhardt, S.; Lübbert, M.; Fiedler, W.; Kindler, T.; Krauter, J.; Brossart, P.; Kündgen, A.; et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann. Hematol. 2017, 96, 1993–2003. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D.; Harrington, D.H.; Theil, K.S.; Mohamed, A.; Paietta, E.; Willman, C.L.; Head, D.R.; Rowe, J.M.; et al. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef]
- PDQ Adult Treatment Editorial Board. Myelodysplastic Syndromes Treatment (PDQ®): Health Professional Version; National Cancer Institute (US): Bethesda, MD, USA, 2002.
- Cazzola, M.; Della Porta, M.G.; Malcovati, L.; Jhanwar, S.C.; Kernan, N.A.; Castro-Malaspina, H.; O’Reilly, R.J.; Bourhis, J.H. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef]
- Chen, J.; Kao, Y.R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Bejanyan, N.; Weisdorf, D.J.; Logan, B.R.; Wang, H.-L.; Devine, S.M.; de Lima, M.; Bunjes, D.W.; Zhang, M.-J. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: A center for international blood and marrow transplant research study. Biol. Blood Marrow Transplant. 2015, 21, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Weissman, I.L.; Akashi, K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8; 21 chromosomal translocation. Proc. Natl. Acad. Sci. USA 2000, 97, 7521–7526. [Google Scholar] [CrossRef] [PubMed]
- Jonas, D.; Lubbert, M.; Kawasaki, E.; Henke, M.; Bross, K.; Mertelsmann, R.; Herrmann, F. Clonal analysis of bcr-abl rearrangement in T lymphocytes from patients with chronic myelogenous leukemia. Blood 1992, 79, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Gunsilius, E.; Duba, H.-C.; Petzer, A.L.; Kahler, C.M.; Grunewald, K.; Stockhammer, G.; Gabl, C.; Dirnhofer, S.; Clausen, J.; Gastl, G. Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet 2000, 355, 1688–1691. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- George, J.; Uyar, A.; Young, K.; Kuffler, L.; Waldron-Francis, K.; Marquez, E.; Ucar, D.; Trowbridge, J.J. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat. Commun. 2016, 7, 12166. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.-J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef]
- Chen, W.; Kumar, A.R.; Hudson, W.A.; Li, Q.; Wu, B.; Staggs, R.A.; Lund, E.A.; Sam, T.N.; Kersey, J.H. Malignant Transformation Initiated by Mll-AF9: Gene Dosage and Critical Target Cells. Cancer Cell 2008, 13, 432–440. [Google Scholar] [CrossRef]
- Cozzio, A.; Passegué, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef]
- Huntly, B.J.P.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar] [CrossRef]
- Siriboonpiputtana, T.; Zeisig, B.B.; Zarowiecki, M.; Fung, T.K.; Mallardo, M.; Tsai, C.; Lau, P.N.I.; Hoang, Q.C.; Veiga, P.; Barnes, J.; et al. Transcriptional memory of cells of origin overrides β-catenin requirement of MLL cancer stem cells. EMBO J. 2017, 36, 3139–3155. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.; Neumann, M.; Schroeder, M.P.; Vosberg, S.; Schlee, C.; Isaakidis, K.; Ortiz-Tanchez, J.; Fransecky, L.R.; Hartung, T.; Türkmen, S.; et al. Acute myeloid leukemia in the elderly is characterized by a distinct genetic and epigenetic landscape. Leukemia 2017, 31, 1640–1644. [Google Scholar] [CrossRef] [PubMed]
- Verovskaya, E.V.; Dellorusso, P.V.; Passegué, E. Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol. Med. 2019, 25, 494–515. [Google Scholar] [CrossRef]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef]
- Moehrle, B.M.; Geiger, H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp. Hematol. 2016, 44, 895–901. [Google Scholar] [CrossRef]
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308–2316. [Google Scholar] [CrossRef]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Moehrle, B.M.; Nattamai, K.; Brown, A.; Florian, M.C.; Ryan, M.; Vogel, M.; Bliederhaeuser, C.; Soller, K.; Prows, D.R.; Abdollahi, A.; et al. Stem Cell-Specific Mechanisms Ensure Genomic Fidelity within HSCs and upon Aging of HSCs. Cell Rep. 2015, 13, 2412–2424. [Google Scholar] [CrossRef]
- Djeghloul, D.; Kuranda, K.; Kuzniak, I.; Barbieri, D.; Naguibneva, I.; Choisy, C.; Bories, J.-C.; Dosquet, C.; Pla, M.; Vanneaux, V.; et al. Age-Associated Decrease of the Histone Methyltransferase SUV39H1 in HSC Perturbs Heterochromatin and B Lymphoid Differentiation. Stem Cell Rep. 2016, 6, 970–984. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Maegawa, S.; Gough, S.M.; Watanabe-Okochi, N.; Lu, Y.; Zhang, N.; Castoro, R.J.; Estecio, M.R.H.; Jelinek, J.; Liang, S.; Kitamura, T.; et al. Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res. 2014, 24, 580–591. [Google Scholar] [CrossRef]
- Adelman, E.R.; Huang, H.-T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef]
- Grigoryan, A.; Guidi, N.; Senger, K.; Liehr, T.; Soller, K.; Marka, G.; Vollmer, A.; Markaki, Y.; Leonhardt, H.; Buske, C.; et al. LaminA/C regulates epigenetic and chromatin architecture changes upon aging of hematopoietic stem cells. Genome Biol. 2018, 19, 189. [Google Scholar] [CrossRef]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.-D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- Failla, G. The aging process and cancerogenesis. Ann. N. Y. Acad. Sci. 1958, 71, 1124–1140. [Google Scholar] [CrossRef] [PubMed]
- Szilard, L. On the nature of the aging process. Proc. Natl. Acad. Sci. USA 1959, 45, 30. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Jamieson, C.H.M.; Weissman, I.L. Stems Cells and the Pathways to Aging and Cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef]
- McKerrell, T.; Vassiliou, G.S. Aging as a driver of leukemogenesis. Sci. Transl. Med. 2015, 7, 306fs38. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-H.; del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; García-García, A.; Macías, D.; González-Gómez, C.; del Monte, A.; Wittner, M.; et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 2019, 25, 407–418.e6. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Mäe, M.A.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Akunuru, S.; Geiger, H. Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends Mol. Med. 2016, 22, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015, 314, 811. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg-Thurley, M.; Amler, S.; Goerlich, D.; Köhnke, T.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Herold, T.; Hubmann, M.; Ksienzyk, B.; et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia 2018, 32, 1598–1608. [Google Scholar] [CrossRef]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Søraas, A.; Matsuyama, M.; de Lima, M.; Wald, D.; Buechner, J.; Gedde-Dahl, T.; Søraas, C.L.; Chen, B.; Ferrucci, L.; Dahl, J.A.; et al. Epigenetic age is a cell-intrinsic property in transplanted human hematopoietic cells. Aging Cell 2019, 18, e12897. [Google Scholar] [CrossRef]
- Liu, W.; Du, W.; Shang, X.; Wang, L.; Evelyn, C.; Florian, M.C.; Ryan, M.A.; Rayes, A.; Zhao, X.; Setchell, K.; et al. Rational identification of a Cdc42 inhibitor presents a new regimen for long-term hematopoietic stem cell mobilization. Leukemia 2019, 33, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Klose, M.; Sacma, M.; Jablanovic, J.; Knudson, L.; Nattamai, K.J.; Marka, G.; Vollmer, A.; Soller, K.; Sakk, V.; et al. Aging alters the epigenetic asymmetry of HSC division. PLoS Biol. 2018, 16, e2003389. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yu, R.; Dang, W. Chromatin Architectural Changes during Cellular Senescence and Aging. Genes 2018, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, A.; Li, W. Global heterochromatin loss. Epigenetics 2012, 7, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Kosak, S.T.; Scalzo, D.; Alworth, S.V.; Li, F.; Palmer, S.; Enver, T.; Lee, J.S.J.; Groudine, M. Coordinate Gene Regulation during Hematopoiesis Is Related to Genomic Organization. PLoS Biol. 2007, 5, e309. [Google Scholar] [CrossRef]
- Galm, O.; Herman, J.G.; Baylin, S.B. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev. 2006, 20, 1–13. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY) 2013, 5, 867–883. [Google Scholar] [CrossRef]
- Geiger, H.; de Haan, G.; Florian, M.C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar] [CrossRef]
- de Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef]
- Basheer, F.; Giotopoulos, G.; Meduri, E.; Yun, H.; Mazan, M.; Sasca, D.; Gallipoli, P.; Marando, L.; Gozdecka, M.; Asby, R.; et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med. 2019, 216, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Prassek, V.V.; Rothenberg-Thurley, M.; Sauerland, M.C.; Herold, T.; Janke, H.; Ksienzyk, B.; Konstandin, N.P.; Goerlich, D.; Krug, U.; Faldum, A.; et al. Genetics of acute myeloid leukemia in the elderly: Mutation spectrum and clinical impact in intensively treated patients aged 75 years or older. Haematologica 2018, 103, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Mizukawa, B.; O’Brien, E.; Moreira, D.C.; Wunderlich, M.; Hochstetler, C.L.; Duan, X.; Liu, W.; Orr, E.; Grimes, H.L.; Mulloy, J.C.; et al. The cell polarity determinant CDC42 controls division symmetry to block leukemia cell differentiation. Blood 2017, 130, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Klenk, J.; Marka, G.; Soller, K.; Kiryakos, H.; Peter, R.; Herbolsheimer, F.; Rothenbacher, D.; Denkinger, M.; Geiger, H. Expression and Activity of the Small RhoGTPase Cdc42 in Blood Cells of Older Adults Are Associated with Age and Cardiovascular Disease. J. Gerontol. Ser. A 2017, 72, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Clinical Implications of Clonal Hematopoiesis. Mayo Clin. Proc. 2018, 93, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.; Bill, M.; Jentzsch, M.; Beinicke, S.; Häntschel, J.; Goldmann, K.; Schulz, J.; Cross, M.; Franke, G.; Behre, G.; et al. Clinical impact of clonal hematopoiesis in acute myeloid leukemia patients receiving allogeneic transplantation. Bone Marrow Transplant. 2019, 54, 1189–1197. [Google Scholar] [CrossRef]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X.; et al. Clearance of Somatic Mutations at Remission and the Risk of Relapse in Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Colla, S.; Ong, D.S.T.; Ogoti, Y.; Marchesini, M.; Mistry, N.A.; Clise-Dwyer, K.; Ang, S.A.; Storti, P.; Viale, A.; Giuliani, N.; et al. Telomere Dysfunction Drives Aberrant Hematopoietic Differentiation and Myelodysplastic Syndrome. Cancer Cell 2015, 27, 644–657. [Google Scholar] [CrossRef]
- Mangaonkar, A.A.; Ferrer, A.; Pinto e Vairo, F.; Cousin, M.A.; Kuisle, R.J.; Klee, E.W.; Kennedy, C.C.; Peters, S.G.; Scott, J.P.; Utz, J.P.; et al. Clinical Correlates and Treatment Outcomes for Patients with Short Telomere Syndromes. Mayo Clin. Proc. 2018, 93, 834–839. [Google Scholar] [CrossRef]
- Mangaonkar, A.A.; Patnaik, M.M. Short Telomere Syndromes in Clinical Practice: Bridging Bench and Bedside. Mayo Clin. Proc. 2018, 93, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.E.; Fielding, A.K.; Harrison, C.N.; Linch, D.C. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. Br. J. Haematol. 1997, 98, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA Is a Potent Suppressor of Hematologic Cancer in Mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.R.; Holland, J.F.; Glidewell, O.J.; Weinberg, V.; Brunner, K.; Obrecht, J.P.; Preisler, H.D.; Nawabi, I.W.; Prager, D.; Carey, R.W.; et al. Treatment of acute myelocytic leukemia: A study by cancer and leukemia group B. Blood 1981, 58, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Midostaurin FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/midostaurin (accessed on 20 September 2019).
- FDA Approves Gilteritinib for Relapsed or Refractory Acute Myeloid Leukemia (AML) with a FLT3 Mutation. Available online: https://www.fda.gov/drugs/fda-approves-gilteritinib-relapsed-or-refractory-acute-myeloid-leukemia-aml-flt3-mutatation (accessed on 20 September 2019).
- Knapper, S.; Mills, K.I.; Gilkes, A.F.; Austin, S.J.; Walsh, V.; Burnett, A.K.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: The induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood 2006, 108, 3494–3503. [Google Scholar] [CrossRef]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef]
- FDA Approves Ivosidenib as First-Line Treatment for AML with IDH1 Mutation. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-first-line-treatment-aml-idh1-mutation (accessed on 20 September 2019).
- FDA Granted Regular Approval to Enasidenib for the Treatment of Relapsed or Refractory AML. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-granted-regular-approval-enasidenib-treatment-relapsed-or-refractory-aml (accessed on 20 September 2019).
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- FDA Approves Gemtuzumab Ozogamicin for CD33-positive AML. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-gemtuzumab-ozogamicin-cd33-positive-aml (accessed on 20 September 2019).
- FDA Approves Venetoclax in Combination for AML in Adults. Available online: https://www.fda.gov/drugs/fda-approves-venetoclax-combination-aml-adults (accessed on 20 September 2019).
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes On-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Glasdegib for AML in Adults Age 75 or Older or Who Have Comorbidities. Available online: https://www.fda.gov/drugs/fda-approves-glasdegib-aml-adults-age-75-or-older-or-who-have-comorbidities (accessed on 20 September 2019).
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Xu, L.; Brunet, A. Turning back time with emerging rejuvenation strategies. Nat. Cell Biol. 2019, 21, 32–43. [Google Scholar] [CrossRef]
- Guidi, N.; Geiger, H. Rejuvenation of aged hematopoietic stem cells. Semin. Hematol. 2017, 54, 51–55. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal 2009, 2, ra75. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old (hematopoietic) stem cells. Nature 2017, 543, 205. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Büchner, T.; Berdel, W.E.; Haferlach, C.; Haferlach, T.; Schnittger, S.; Müller-Tidow, C.; Braess, J.; Spiekermann, K.; Kienast, J.; Staib, P.; et al. Age-related risk profile and chemotherapy dose response in acute myeloid leukemia: A study by the german acute myeloid leukemia cooperative group. J. Clin. Oncol. 2009, 27, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Velasco, G.; López-Otín, C. Mouse Models to Disentangle the Hallmarks of Human Aging. Circ. Res. 2018, 123, 905–924. [Google Scholar] [CrossRef] [PubMed]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating Mitochondrial DNA Mutations Drive Premature Hematopoietic Aging Phenotypes Distinct from Physiological Stem Cell Aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Category | Type of Alteration | References |
|---|---|---|
| Mutations | 2–3 fold increase in mutation load | [58,59,60,61] |
| Protein expression | Altered expression of epigenetic regulators | [62,63,64] |
| Epigenetic drift | DNA methylation alteration (redistribution, level change) | [65,66,67,68,69] |
| Changes in histone modifications | [63,67] | |
| Nuclear/chromatin structure alterations | Reduced lamin A/C level, changed nuclear size and shape, global changes in heterochromatin mark deposition | [62,70] |
| Epipolarity | CDC42 activity, H4K16ac polarity | [71] |
| Others (not discussed in the review) | Increased expression of repetitive elements | [63] |
| Alternative protein isoforms | [63] | |
| Altered expression of non-epigenetic cellular regulators | [63,72] |
| Gene | Type pf Mutation and Mechanism | Frequency in Adult AML (<70 y) | Frequency in Elderly AML (>75 y) | Clinical Features | Reference |
|---|---|---|---|---|---|
| TET2 | Missense, nonsense, and frame shift mutations which cause a loss-of-function phenotype and the impairment of the catalytic activity, resulting in low levels of 5-hmc in genomic DNA | 8–12% | 42% | Debated prognostic value (associated with poor prognosis in AML patients with intermediate-risk cytogenetic but also reported with no clinical impact in other studies); mutually exclusive with IDH1/2 mutation | [6,7,11,103] |
| DNMT3A | Missense, nonsense, and frame shift heterozygote mutations which cause a dominant negative loss of function | 19–26% | 35% | Frequently occurring in AML with a normal karyotype; unfavorable prognosis; the loss of methylase activity results in hypomethylation and uncontrolled expression of multiple HOX genes | [11,103] |
| ASXL1 | Missense, nonsense, and frame shift loss-of-function mutations; ASXL1 interacts with PCR2 and mutations decrease recruitment of PRC2 to its targets | 11% | 21% | Early mutation which tends to be associated with an aggressive disease and a poor overall survival | [103] |
| IDH2/IDH1 | Heterozygous missense mutations which result in reduced production of α-KG and in a neomorphic gain-of-function effect, catalyzing the conversion of α-KG to 2-HG resulting in inhibition of TET2. | 12–20% | 15% | Hypermethylation signature, altered gene expression, and impaired hematopoietic differentiation | [11,28,103] |
| STAG2, RAD21, SMC3, SMC1A | Frameshift and missense mutations which disrupt cohesin complex assembly; these mutations act at least partially as dominant negatives | 9–15% | n.d. | Mutually exclusive with unfavorable-risk cytogenetics as well as complex chromosomal changes; independent favorable risk factors in AML but associated with a shorter survival in MDS | [11,103] |
| EZH2 | Missense, nonsense, and frame shift, loss-of-function mutations: EZH2 is a histone H3K27 methyl-transferase and part of PRC2. Loss-of-function mutations occur in the catalytic SET domain | 1–2% | n.d. | EZH2 inactivating mutations are associated to induction of HOXA9 expression | [11,103] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zjablovskaja, P.; Florian, M.C. Acute Myeloid Leukemia: Aging and Epigenetics. Cancers 2020, 12, 103. https://doi.org/10.3390/cancers12010103
Zjablovskaja P, Florian MC. Acute Myeloid Leukemia: Aging and Epigenetics. Cancers. 2020; 12(1):103. https://doi.org/10.3390/cancers12010103
Chicago/Turabian StyleZjablovskaja, Polina, and Maria Carolina Florian. 2020. "Acute Myeloid Leukemia: Aging and Epigenetics" Cancers 12, no. 1: 103. https://doi.org/10.3390/cancers12010103
APA StyleZjablovskaja, P., & Florian, M. C. (2020). Acute Myeloid Leukemia: Aging and Epigenetics. Cancers, 12(1), 103. https://doi.org/10.3390/cancers12010103