Dendritic Cell-Based and Other Vaccination Strategies for Pediatric Cancer

 ,
,
Abstract
1. Introduction
2. Therapeutic Vaccination for Pediatric Cancer: The Past and the Present
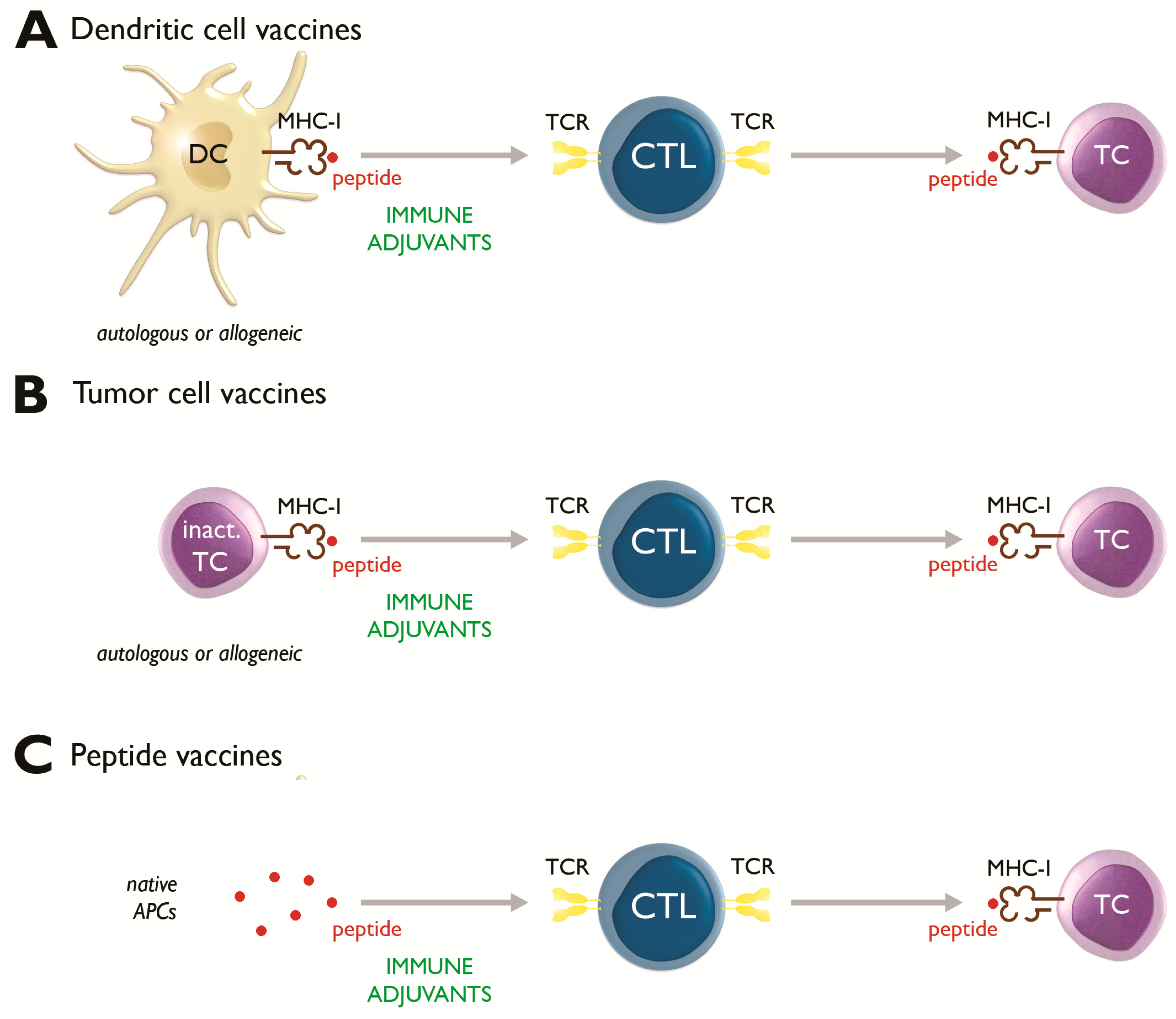
2.1. Dendritic Cell-Based Vaccines
2.2. Tumor Cell Vaccines
2.3. Peptide Vaccines
3. Therapeutic Vaccination for Pediatric Cancer: The Future
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- National Cancer Institute. A Snapshot of Adolescent and Young Adult Cancers. Available online: https://www.cancer.gov/types/aya (accessed on 1 September 2019).
- Cancer Research UK. Young People’s Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/young-peoples-cancers (accessed on 1 September 2019).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Moll, A.J.; Byrd, T.T.; Louis, C.U.; Ahmed, N. Cellular immunotherapy for pediatric solid tumors. Cytotherapy 2015, 17, 3–17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, D.K.; Helenowski, I.; Hijiya, N. Secondary malignancies in pediatric cancer survivors: Perspectives and review of the literature. Int. J. Cancer 2014, 135, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Guinipero, T.; Finn, O.J. Cancer vaccines: Emphasis on pediatric cancers. Curr. Pharm. Des. 2010, 16, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Bryant, C.; Van Acker, H.H.; Goossens, H.; Lion, E.; Fromm, P.D.; Hart, D.N.; Van Tendeloo, V.F.; Berneman, Z.N. Dendritic cells as pharmacological tools for cancer immunotherapy. Pharmacol. Rev. 2015, 67, 731–753. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Mackall, C.L.; Merchant, M.S.; Fry, T.J. Immune-based therapies for childhood cancer. Nat. Rev. Clin. Oncol. 2014, 11, 693–703. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Ceppi, F.; Beck-Popovic, M.; Bourquin, J.P.; Renella, R. Opportunities and challenges in the immunological therapy of pediatric malignancy: A concise snapshot. Eur. J. Pediatr. 2017, 176, 1163–1172. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buque, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef]
- Banchereau, J.; Palucka, K. Immunotherapy: Cancer vaccines on the move. Nat. Rev. Clin. Oncol. 2018, 15, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.H.; Versteven, M.; Lichtenegger, F.S.; Roex, G.; Campillo-Davo, D.; Lion, E.; Subklewe, M.; Van Tendeloo, V.F.; Berneman, Z.N.; Anguille, S. Dendritic cell-based immunotherapy of acute myeloid leukemia. J. Clin. Med. 2019, 8, 579. [Google Scholar] [CrossRef] [PubMed]
- Capitini, C.M.; Mackall, C.L.; Wayne, A.S. Immune-based therapeutics for pediatric cancer. Expert Opin. Biol. Ther. 2010, 10, 163–178. [Google Scholar] [CrossRef] [PubMed]
- McDowell, K.A.; Hank, J.A.; DeSantes, K.B.; Capitini, C.M.; Otto, M.; Sondel, P.M. NK cell-based immunotherapies in pediatric oncology. J. Pediatr. Hematol. Oncol. 2015, 37, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Wayne, A.S.; Capitini, C.M.; Mackall, C.L. Immunotherapy of childhood cancer: From biologic understanding to clinical application. Curr. Opin. Pediatr. 2010, 22, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J. Pediatric high grade glioma: A review and update on tumor clinical characteristics and biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar]
- Liu, J.K. Anti-cancer vaccines—A one-hit wonder? Yale J. Biol. Med. 2014, 87, 481–489. [Google Scholar]
- Khatua, S.; Sadighi, Z.S.; Pearlman, M.L.; Bochare, S.; Vats, T.S. Brain tumors in children—Current therapies and newer directions. Indian J. Pediatr. 2012, 79, 922–927. [Google Scholar] [CrossRef]
- Wells, E.M.; Packer, R.J. Pediatric brain tumors. Continuum (Minneap Minn) 2015, 21, 373–396. [Google Scholar] [CrossRef]
- Mallhi, K.; Lum, L.G.; Schultz, K.R.; Yankelevich, M. Hematopoietic cell transplantation and cellular therapeutics in the treatment of childhood malignancies. Pediatr. Clin. N. Am. 2015, 62, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Lion, E.; Smits, E.L.; Berneman, Z.N.; Van Tendeloo, V.F. NK cells: Key to success of DC-based cancer vaccines? Oncologist 2012, 17, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.H.; Anguille, S.; Van Tendeloo, V.F.; Lion, E. Empowering gamma delta T cells with antitumor immunity by dendritic cell-based immunotherapy. Oncoimmunology 2015, 4, e1021538. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.A.; Orme, L.M.; Neale, A.M.; Radcliff, F.J.; Amor, G.M.; Maixner, W.; Downie, P.; Hassall, T.E.; Tang, M.L.; Ashley, D.M. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro Oncol. 2004, 6, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Himoudi, N.; Wallace, R.; Parsley, K.L.; Gilmour, K.; Barrie, A.U.; Howe, K.; Dong, R.; Sebire, N.J.; Michalski, A.; Thrasher, A.J.; et al. Lack of T-cell responses following autologous tumour lysate pulsed dendritic cell vaccination, in patients with relapsed osteosarcoma. Clin. Transl. Oncol. 2012, 14, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.L., 3rd; Panosyan, E.H.; Plant, A.; Davidson, T.; Yong, W.H.; Prins, R.M.; Liau, L.M.; Moore, T.B. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res. 2013, 33, 2047–2056. [Google Scholar] [PubMed]
- Felzmann, T.; Witt, V.; Wimmer, D.; Ressmann, G.; Wagner, D.; Paul, P.; Huttner, K.; Fritsch, G. Monocyte enrichment from leukapharesis products for the generation of DCs by plastic adherence, or by positive or negative selection. Cytotherapy 2003, 5, 391–398. [Google Scholar] [CrossRef]
- Barth, R.J., Jr.; Mule, J.J.; Spiess, P.J.; Rosenberg, S.A. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J. Exp. Med. 1991, 173, 647–658. [Google Scholar] [CrossRef]
- Mohme, M.; Neidert, M.C.; Regli, L.; Weller, M.; Martin, R. Immunological challenges for peptide-based immunotherapy in glioblastoma. Cancer Treat. Rev. 2014, 40, 248–258. [Google Scholar] [CrossRef]
- de Vries, I.J.; Lesterhuis, W.J.; Barentsz, J.O.; Verdijk, P.; van Krieken, J.H.; Boerman, O.C.; Oyen, W.J.; Bonenkamp, J.J.; Boezeman, J.B.; Adema, G.J.; et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat. Biotechnol. 2005, 23, 1407–1413. [Google Scholar] [CrossRef]
- Hegde, M.; Bielamowicz, K.J.; Ahmed, N. Novel approaches and mechanisms of immunotherapy for glioblastoma. Discov. Med. 2014, 17, 145–154. [Google Scholar] [PubMed]
- Tuyaerts, S.; Aerts, J.L.; Corthals, J.; Neyns, B.; Heirman, C.; Breckpot, K.; Thielemans, K.; Bonehill, A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol. Immunother. 2007, 56, 1513–1537. [Google Scholar] [CrossRef] [PubMed]
- Benitez-Ribas, D.; Cabezon, R.; Florez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V.; Cruz, O.; et al. Immune response generated with the administration of autologous dendritic cells pulsed with an allogenic tumoral cell-lines lysate in patients with newly diagnosed diffuse intrinsic pontine glioma. Front. Oncol. 2018, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Long, L.M.; Read, E.J.; Leitman, S.F.; Carter, C.S.; Tsokos, M.; Goletz, T.J.; Avila, N.; Berzofsky, J.A.; Helman, L.J.; et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: An inter-institute NIH study. Med. Pediatr. Oncol. 2002, 38, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Mackall, C.L.; Rhee, E.H.; Read, E.J.; Khuu, H.M.; Leitman, S.F.; Bernstein, D.; Tesso, M.; Long, L.M.; Grindler, D.; Merino, M.; et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin. Cancer Res. 2008, 14, 4850–4858. [Google Scholar] [CrossRef] [PubMed]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. 2015, 64, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Yanagisawa, R.; Yoshikawa, K.; Higuchi, Y.; Koya, T.; Yoshizawa, K.; Tanaka, M.; Sakashita, K.; Kobayashi, T.; Kurata, T.; et al. Safety and tolerability of allogeneic dendritic cell vaccination with induction of Wilms tumor 1-specific T cells in a pediatric donor and pediatric patient with relapsed leukemia: A case report and review of the literature. Cytotherapy 2015, 17, 330–335. [Google Scholar] [PubMed]
- Shah, N.N.; Loeb, D.M.; Khuu, H.; Stroncek, D.; Ariyo, T.; Raffeld, M.; Delbrook, C.; Mackall, C.L.; Wayne, A.S.; Fry, T.J. Induction of immune response after allogeneic Wilms’ tumor 1 dendritic cell vaccination and donor lymphocyte infusion in patients with hematologic malignancies and post-transplantation relapse. Biol. Blood Marrow Transplant. 2016, 22, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Hashii, Y.; Sato, E.; Ohta, H.; Oka, Y.; Sugiyama, H.; Ozono, K. WT1 peptide immunotherapy for cancer in children and young adults. Pediatr. Blood Cancer 2010, 55, 352–355. [Google Scholar] [CrossRef]
- Hashii, Y.; Sato-Miyashita, E.; Matsumura, R.; Kusuki, S.; Yoshida, H.; Ohta, H.; Hosen, N.; Tsuboi, A.; Oji, Y.; Oka, Y.; et al. WT1 peptide vaccination following allogeneic stem cell transplantation in pediatric leukemic patients with high risk for relapse: Successful maintenance of durable remission. Leukemia 2012, 26, 530–532. [Google Scholar] [CrossRef]
- Anguille, S.; Fujiki, F.; Smits, E.L.; Oji, Y.; Lion, E.; Oka, Y.; Berneman, Z.N.; Sugiyama, H. Identification of a Wilms’ tumor 1-derived immunogenic CD4(+) T-cell epitope that is recognized in the context of common Caucasian HLA-DR haplotypes. Leukemia 2013, 27, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van Tendeloo, V.F.; Berneman, Z.N. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia 2012, 26, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Fieuws, S.; Rutkowski, S.; Van Calenbergh, F.; Van Loon, J.; Goffin, J.; Sciot, R.; Wilms, G.; Demaerel, P.; Warmuth-Metz, M.; et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.S.; Bernstein, D.; Amoako, M.; Baird, K.; Fleisher, T.A.; Morre, M.; Steinberg, S.M.; Sabatino, M.; Stroncek, D.F.; Venkatasan, A.M.; et al. Adjuvant immunotherapy to improve outcome in high-risk pediatric sarcomas. Clin. Cancer Res. 2016, 22, 3182–3191. [Google Scholar] [CrossRef]
- Dohnal, A.M.; Witt, V.; Hugel, H.; Holter, W.; Gadner, H.; Felzmann, T. Phase I study of tumor Ag-loaded IL-12 secreting semi-mature DC for the treatment of pediatric cancer. Cytotherapy 2007, 9, 755–770. [Google Scholar] [CrossRef]
- Ridolfi, R.; Riccobon, A.; Galassi, R.; Giorgetti, G.; Petrini, M.; Fiammenghi, L.; Stefanelli, M.; Ridolfi, L.; Moretti, A.; Migliori, G.; et al. Evaluation of in vivo labelled dendritic cell migration in cancer patients. J. Transl. Med. 2004, 2, 27. [Google Scholar] [CrossRef]
- Geiger, J.D.; Hutchinson, R.J.; Hohenkirk, L.F.; McKenna, E.A.; Yanik, G.A.; Levine, J.E.; Chang, A.E.; Braun, T.M.; Mule, J.J. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001, 61, 8513–8519. [Google Scholar]
- Rutkowski, S.; De Vleeschouwer, S.; Kaempgen, E.; Wolff, J.E.; Kuhl, J.; Demaerel, P.; Warmuth-Metz, M.; Flamen, P.; Van Calenbergh, F.; Plets, C.; et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br. J. Cancer 2004, 91, 1656–1662. [Google Scholar] [CrossRef]
- Suminoe, A.; Matsuzaki, A.; Hattori, H.; Koga, Y.; Hara, T. Immunotherapy with autologous dendritic cells and tumor antigens for children with refractory malignant solid tumors. Pediatr. Transplant. 2009, 13, 746–753. [Google Scholar] [CrossRef]
- Bai, Y.; Zheng, J.E.; Wang, N.; Cai, H.H.; Zhai, L.N.; Wu, Y.H.; Wang, F.; Jin, R.M.; Zhou, D.F. Effects of dendritic cell-activated and cytokine-induced killer cell therapy on 22 children with acute myeloid leukemia after chemotherapy. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van Acker, H.H.; Van den Bergh, J.; Willemen, Y.; Goossens, H.; Van Tendeloo, V.F.; Smits, E.L.; Berneman, Z.N.; Lion, E. Interleukin-15 dendritic cells harness NK cell cytotoxic effector function in a contact- and IL-15-dependent manner. PLoS ONE 2015, 10, e0123340. [Google Scholar] [CrossRef] [PubMed]
- Van Elssen, C.H.; Oth, T.; Germeraad, W.T.; Bos, G.M.; Vanderlocht, J. Natural killer cells: The secret weapon in dendritic cell vaccination strategies. Clin. Cancer Res. 2014, 20, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Ardon, H.; De Vleeschouwer, S.; Van Calenbergh, F.; Claes, L.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.; Van Gool, S.W. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 2010, 54, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J.; Hodge, J.W.; Palena, C.; Tsang, K.Y.; Jochems, C.; Greiner, J.W.; Farsaci, B.; Madan, R.A.; Heery, C.R.; Gulley, J.L. Therapeutic cancer vaccines. Adv. Cancer Res. 2014, 121, 67–124. [Google Scholar] [PubMed]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Dumba, M.; Jawad, N.; McHugh, K. Neuroblastoma and nephroblastoma: A radiological review. Cancer Imaging 2015, 15, 5. [Google Scholar] [CrossRef]
- Rousseau, R.F.; Brenner, M.K. Vaccine therapies for pediatric malignancies. Cancer J. 2005, 11, 331–339. [Google Scholar] [CrossRef]
- Bowman, L.C.; Grossmann, M.; Rill, D.; Brown, M.; Zhong, W.Y.; Alexander, B.; Leimig, T.; Coustan-Smith, E.; Campana, D.; Jenkins, J.; et al. Interleukin-2 gene-modified allogeneic tumor cells for treatment of relapsed neuroblastoma. Hum. Gene Ther. 1998, 9, 1303–1311. [Google Scholar] [CrossRef]
- Bowman, L.; Grossmann, M.; Rill, D.; Brown, M.; Zhong, W.Y.; Alexander, B.; Leimig, T.; Coustan-Smith, E.; Campana, D.; Jenkins, J.; et al. IL-2 adenovector-transduced autologous tumor cells induce antitumor immune responses in patients with neuroblastoma. Blood 1998, 92, 1941–1949. [Google Scholar] [PubMed]
- Russell, H.V.; Strother, D.; Mei, Z.; Rill, D.; Popek, E.; Biagi, E.; Yvon, E.; Brenner, M.; Rousseau, R. A phase 1/2 study of autologous neuroblastoma tumor cells genetically modified to secrete IL-2 in patients with high-risk neuroblastoma. J. Immunother. 2008, 31, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Russell, H.V.; Strother, D.; Mei, Z.; Rill, D.; Popek, E.; Biagi, E.; Yvon, E.; Brenner, M.; Rousseau, R. Phase I trial of vaccination with autologous neuroblastoma tumor cells genetically modified to secrete IL-2 and lymphotactin. J. Immunother. 2007, 30, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, R.F.; Haight, A.E.; Hirschmann-Jax, C.; Yvon, E.S.; Rill, D.R.; Mei, Z.; Smith, S.C.; Inman, S.; Cooper, K.; Alcoser, P.; et al. Local and systemic effects of an allogeneic tumor cell vaccine combining transgenic human lymphotactin with interleukin-2 in patients with advanced or refractory neuroblastoma. Blood 2003, 101, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Dilloo, D.; Bacon, K.; Holden, W.; Zhong, W.; Burdach, S.; Zlotnik, A.; Brenner, M. Combined chemokine and cytokine gene transfer enhances antitumor immunity. Nat. Med. 1996, 2, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Haining, W.N.; Cardoso, A.A.; Keczkemethy, H.L.; Fleming, M.; Neuberg, D.; DeAngelo, D.J.; Stone, R.M.; Galinsky, I.; Silverman, L.B.; Sallan, S.E.; et al. Failure to define window of time for autologous tumor vaccination in patients with newly diagnosed or relapsed acute lymphoblastic leukemia. Exp. Hematol. 2005, 33, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, R.F.; Biagi, E.; Dutour, A.; Yvon, E.S.; Brown, M.P.; Lin, T.; Mei, Z.; Grilley, B.; Popek, E.; Heslop, H.E.; et al. Immunotherapy of high-risk acute leukemia with a recipient (autologous) vaccine expressing transgenic human CD40L and IL-2 after chemotherapy and allogeneic stem cell transplantation. Blood 2006, 107, 1332–1341. [Google Scholar] [CrossRef]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Potter, D.M.; Connelly, A.K.; Dibridge, S.A.; Whiteside, T.L.; Okada, H. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J. Clin. Oncol. 2014, 32, 2050–2058. [Google Scholar]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Normolle, D.P.; Connelly, A.K.; Dibridge, S.; Mason, G.; Whiteside, T.L.; et al. Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro Oncol. 2016, 18, 1157–1168. [Google Scholar] [CrossRef]
- Sawada, A.; Inoue, M.; Kondo, O.; Yamada-Nakata, K.; Ishihara, T.; Kuwae, Y.; Nishikawa, M.; Ammori, Y.; Tsuboi, A.; Oji, Y.; et al. Feasibility of cancer immunotherapy with WT1 peptide vaccination for solid and hematological malignancies in children. Pediatr. Blood Cancer 2016, 63, 234–241. [Google Scholar] [CrossRef]
- Hirabayashi, K.; Yanagisawa, R.; Saito, S.; Higuchi, Y.; Koya, T.; Sano, K.; Koido, S.; Okamoto, M.; Sugiyama, H.; Nakazawa, Y.; et al. Feasibility and immune response of WT1 peptide vaccination in combination with OK-432 for paediatric solid tumors. Anticancer Res. 2018, 38, 2227–2234. [Google Scholar]
- Ceschin, R.; Kurland, B.F.; Abberbock, S.R.; Ellingson, B.M.; Okada, H.; Jakacki, R.I.; Pollack, I.F.; Panigrahy, A. Parametric response mapping of apparent diffusion coefficient as an imaging biomarker to distinguish pseudoprogression from true tumor progression in peptide-based vaccine therapy for pediatric diffuse intrinsic pontine glioma. AJNR Am. J. Neuroradiol. 2015, 36, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Hwu, W.J.; Kefford, R.; Weber, J.S.; Daud, A.; Hamid, O.; Patnaik, A.; Ribas, A.; Robert, C.; Gangadhar, T.C.; et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J. Clin. Oncol. 2016, 34, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Garnett-Benson, C.; Hodge, J.W.; Gameiro, S.R. Combination regimens of radiation therapy and therapeutic cancer vaccines: Mechanisms and opportunities. Semin. Radiat. Oncol. 2015, 25, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Versteven, M.; Van den Bergh, J.M.J.; Marcq, E.; Smits, E.L.J.; Van Tendeloo, V.F.I.; Hobo, W.; Lion, E. Dendritic cells and programmed death-1 blockade: A joint venture to combat cancer. Front. Immunol. 2018, 9, 394. [Google Scholar] [CrossRef]


{kind=link}
| Malignancy | Ph. | N (age) | Vaccine Type | Admin. Route | Adjuvant Treatment | Toxicity | Immune Response | Clinical Response | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Brain | |||||||||
| Relapsed brain tumors | I | 7/9 (9–22 y) | autologous immature DCs whole tumor RNA-pulsed | IV/ID | / | No significant toxicity | Humoral immune response (2/7); No T-cell reactivity to DCs |
| Caruso et al., 2004 [26] |
| Relapsed malignant glioma | I | 12 (11–78 y) | autologous mature DCs tumor lysate-pulsed | ID | / | Gr. 4 neurotoxicity (1); Gr. 2 hematotoxicity (2); other minor toxicities (2) | Positive DTH (6/8) |
| Rutkowski et al., 2004 [51] |
| Recurrent GBM | I/II | 56 (7–77 y) | autologous mature DCs tumor lysate-pulsed | ID | / | Gr. 4 neurotoxicity (1); Gr. 2 hematotoxicity (2) | Positive DTH |
| De Vleeschouwer et al., 2008 [46] |
| Brain tumors # | I | 45 (children, age n.s.) | autologous mature DCs tumor lysate-pulsed | ID | IMQ | Only minor toxicities (including fatigue, headache, fever, vomitus) | ND |
| Ardon et al., 2010 [56] |
| Newly diagnosed or recurrent HGG | I | 3/7 (1–18 y) | autologous immature DCs tumor lysate-pulsed | ID | / | Gr. 4 ↑ alkaline phosphatase (1/3) | ND |
| Lasky et al., 2013 [28] |
| Newly diagnosed DIPG | I | 5/9 (4–10 y) | autologous mature DCs tumor lysate-pulsed (allogeneic) | ID | / | No significant toxicity | ↑ T-cell reactivity to TL (8/9) and to TL in CSF (2/9) | ND | Benitez-Ribas et al., 2018 [35] |
| Solid | |||||||||
| Relapsed solid tumors | I | 10/15 (3–17 y) | autologous immature DCs tumor lysate-pulsed | ID | KLH | No significant toxicity | Positive DTH (3/6); ↑ T-cell reactivity (3/7) |
| Geiger et al., 2001 [50] |
| Recurrent AR and ES | I | 15/16 (8–30 y) | autologous mature DCs peptide-pulsed (breakpoint) | IV | IL-2 | Gr. 3 toxicity attributed to IL-2 including fever (2/15), nausea/vomiting (1/15), ↑ bilirubin (2/15), hematotoxicity (4/15) | No T-cell reactivity to peptides |
| Dagher et al., 2002 [36] |
| Advanced solid extra-cranial tumors | I | 20 (7–22 y) | autologous semimature DCs tumor lysate-pulsed | SC/IN | KLH | Only minor toxicities (including fever and local injection site pain) | Positive DTH (2/9 SC, 3/6 IN); ↑ T-cell reactivity (0/3 SC, 8/8 IN) | ● SC treated patients (14): - CR patients (5): CCR (4), SD (1) - PR patient (1): PD (1) - PD patients (8): MR (1), SD (1) ● IN treated patients (8): - CR patients (4): CCR (3), PD (1) - PD patients (4): PD (4) | Dohnal et al., 2007 [48] |
| Metastatic or recurrent AR and ES | II | 30/52 (1–39 y) | autologous mature DCs peptide-pulsed (breakpoint) | IV/ID | ± IL-2 | Gr. IV thrombocytopenia (1); grade 3 neutropenia (6); diarrhea (2); ↑ bilirubin (1), abdominal pain (1), skin rash (3) | ↑ T-cell reactivity to peptide (9/23) | ● OS at 60m: - Vaccinated patients: 43% - Non-vaccinated patients: 15% | Mackall et al., 2008 [37] |
| Relapsed/refractory solid tumors (ES, SS, NB) | ND | 5 (3–11y) | autologous mature DCs tumor lysate-pulsed | SC | KLH | No significant toxicity | Positive DTH to TL (1/5); ↑ activated CD8+ T cells (2/5); ↑ NK cell cytotoxic activity (3/5) |
| Suminoe et al., 2009 [52] |
| Relapsed solid tumors (OSa, NB, ES, MB) | I | 15/16 (14–30.5 y) | autologous mature DCs tumor lysate-pulsed | ID | KLH and IL-2 | No DC vaccine-related toxicities | ↑ T-cell reactivity to TL (4/15) |
| Himoudi et al., 2012 [27] |
| Relapsed/refractory solid tumors (NB and sarcoma) | I/II | 10/15 (2.5–15 y) | autologous matured DCs peptide-pulsed (MAGE-A1, MAGE-A3 and NY-ESO-1) | IV | IMQ | Hematotoxicity attributed to DAC (5/10); urticaria multiforme attributed to DC vaccine (1/10) | ↑ T-cell reactivity (6/9) |
| Krishnadas et al., 2015 [38] |
| Metastatic and recurrent high-risk sarcomas | II | 29/43 (6–33 y) | autologous mature DCs tumor lysate-pulsed | SC/ID | KLH and IL-7 | Gr. 2 injection site reactions attributed to DC vaccine (5/29); Transaminitis (9/29), gr. 4 fever (1) and gr. 4 anaphylaxis (1) attributed to IL-7 | ↑ T-cell reactivity (16/26); | ● OS and PFS at 60m, respectively: - All patients: 51% and 32% - ES/RMS: 63% and 40% - NDMD: 77% | Merchant et al., 2016 [47] |
| Hematological | |||||||||
| AML | ND | 22 (3–14 y) | autologous mature DCs + CIKs | SC | IL-2 | Only minor toxicities (including fever and hives) (7) | ↑ CD8+ T cells |
| Bai et al., 2015 [53] |
| Relapsed ALL | I | 1 (15 y) | allogeneic mature DCs peptide-pulsed (WT1) | ID | OK-432 | No significant toxicity | ↑ T-cell reactivity |
| Saito et al., 2015 [39] |
| Post-HSCT relapse of hematological malignancies (ALL, AML, HL) | I/II | 5 (9–19 y) | allogeneic mature DCs peptide-pulsed (WT1) + DLI | SC/ID | KLH | No significant toxicity | Positive DTH to WT1 (2/5) ↑ T-cell reactivity to WT1 (3/5) |
| Shah et al., 2016 [40] |
| Malignancy | Ph. | N (age) | Vaccine Type | Admin. Route | Adjuvant Treatment | Toxicity | Immune Response | Clinical Response | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Solid | |||||||||
| Relapsed stage IV NB | I | 12/13 (2.9–11.9 y) | IL-2 gene-modified allogeneic tumor cells | SC | IL-2 | Only minor toxicities (including gr. 1 inflam-matory response and panniculitis) | No ↑ CD4+ or CD8+ T cells No eosinophilia ↑ CTL activity (3/5) |
| Bowman et al., 1998 [62] |
| Advanced NB with measurable disease | I | 10 (11 m–17 y) | AAV IL-2 gene-modified autologous tumor cells | SC | AAV IL-2 | Only minor toxicities (including gr. 1 inflam-matory response and panniculitis) | ↑ CD3+CD4+, CD16+, eosinophilia; ↑ IgG antibodies (4/9); ↑ CTL activity (4/9) |
| Bowman et al., 1998 [63] |
| Advanced/refractory NB | I | 21 (2–17 y) | IL-2/Lptn gene-modified allogeneic tumor cells | SC | IL-2 Lptn | Injection site reactions (20); Flu-like symptoms (myalgia and fever) (10) | ↑ CD4+ T cells, NK cells, eosinophilia; ↑ IgG antibodies (15/17); ↑ IL-5 (9/13) |
| Rousseau et al., 2003 [66] |
| Recurrent stage IV NB | I | 1/7 (6–13 y) | IL-2/Lptn gene-modified autologous tumor cells | SC | IL-2 Lptn | No significant toxicity | IFN-γ (2/6) and IL-5 (3/6) tumor-specific immune response |
| Russell et al., 2007 [65] |
| Stage IV NB in remission | I/II | 13 (2–9 y) | AAV IL-2 gene-modified autologous tumor cells | SC | AAV IL-2 | Gr. 1-2 injection site reactions attributed to DC vaccine | ↑ CD3+CD4+, eosinophilia; IFN-γ (4/10) and IL-5 (11/12) tumor-specific immune response |
| Russell et al., 2008 [64] |
| Hematological | |||||||||
| Newly diagnosed or relapsed/refractory ALL | I | 2/9 (5–60 y) | Autologous CD40L cells | SC | / | No significant toxicity | ↑ allogeneic and peptide-specific T cell reactivity in vitro |
| Haining et al., 2005 [68] |
| High risk AML or ALL in cytologic remission | ND | 10/44 (4–56 y) | Autologous IL-2/CD40L cells | SC | AAV IL-2 | Abscess locally at the injection site (1/10) | ↑ IgG antibodies (2/10) ↑ cytotoxic (5/8), Th1- (4/8) and Th2-cell (3/8) reactivity; ↑ IFN-γ and IL-5 secretion |
| Rousseau et al., 2006 [69] |
| Malignancy | Ph. | N (age) | Vaccine Type | Admin. Route | Adjuvant Treatment | Toxicity | Immune Response | Clinical Response | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Brain | |||||||||
| High risk glioma | I | 26 (1–21 y) | GAA peptides | SC | Montanide poly-ICLC | Gr. 1–2 injection site reactions (26); flu-like symptoms (24); gr. 1 GI toxicity (8); gr. 1 leukopenia (4) | ↑ T-cell reactivity to epitopes (10 to 13Rα2, 11 to EphA2 and 3 to survivin) |
| Pollack et al., 2014 [70] |
| Recurrent LGG | I | 14 (1.9–19 y) | GAA peptides | SC | Montanide poly-ICLC | Gr. 1–2 injection site reactions (14), flu-like symptoms (13); gr. 1–2 GI toxicity (6); gr. 2 urticaria (1) | ↑ T-cell reactivity to epitopes (3 to 13Rα2, 11 to EphA2 and 3 to survivin) |
| Pollack et al., 2016 [71] |
| Solid | |||||||||
| Solid tumors (RS, OSa, LS, SS) | I/II | 4/23 (7–19 y) | WT1 peptide | ID | Montanide | Injection site reactions (4) | ↑ WT1-specific CTLs (3/4) |
| Hashii et al., 2010 [41] |
| Relapsed/refractory solid tumors | I/II | 9/26 (0–17 y) | WT1 peptide | ID | Montanide | Injection site reactions (9) | ↑ WT1-specific CTL reactivity (4/4) | ● Overt disease (4): - MR (1) - SD (1) ● High risk in CR (5): - CCR (4) | Sawada et al., 2016 [72] |
| Solid tumors | ND | 18/24 (2–19 y) | WT1 peptide | ID | OK-432 | Only minor toxicities (including gr. 1–2 injection site reactions and fever) except gr. 3 anaphylaxis (1/18) | WT1 EliSPOT (4/18) | ND | Hirabayashi et al., 2018 [73] |
| Hematological | |||||||||
| ALL | I/II | 1/23 (9 y) | WT1 peptide | ID | Montanide | Injection site reactions (1) | No ↑ WT1-specific CTLs |
| Hashii et al., 2010 [41] |
| High risk hematological malignancies | II | 3 (1–13 y) | WT1 peptide | ID | Montanide | Injection site reactions (3) | ↑ WT1-specific CTLs (3) |
| Hashii et al., 2012 [42] |
| Relapsed/refractory hematological malignancies | I/II | 4/26 (0–17 y) | WT1 peptide | ID | Montanide | Injection site reactions (4) | ↑ WT1-specific CTL reactivity (4/4) | ● MRD positive (3): - CR (3) ● High risk in CR (1): - CCR (1) | Sawada et al., 2016 [72] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bruijn, S.; Anguille, S.; Verlooy, J.; Smits, E.L.; van Tendeloo, V.F.; de Laere, M.; Norga, K.; Berneman, Z.N.; Lion, E. Dendritic Cell-Based and Other Vaccination Strategies for Pediatric Cancer. Cancers 2019, 11, 1396. https://doi.org/10.3390/cancers11091396
de Bruijn S, Anguille S, Verlooy J, Smits EL, van Tendeloo VF, de Laere M, Norga K, Berneman ZN, Lion E. Dendritic Cell-Based and Other Vaccination Strategies for Pediatric Cancer. Cancers. 2019; 11(9):1396. https://doi.org/10.3390/cancers11091396
Chicago/Turabian Stylede Bruijn, Sévérine, Sébastien Anguille, Joris Verlooy, Evelien L. Smits, Viggo F. van Tendeloo, Maxime de Laere, Koenraad Norga, Zwi N. Berneman, and Eva Lion. 2019. "Dendritic Cell-Based and Other Vaccination Strategies for Pediatric Cancer" Cancers 11, no. 9: 1396. https://doi.org/10.3390/cancers11091396
APA Stylede Bruijn, S., Anguille, S., Verlooy, J., Smits, E. L., van Tendeloo, V. F., de Laere, M., Norga, K., Berneman, Z. N., & Lion, E. (2019). Dendritic Cell-Based and Other Vaccination Strategies for Pediatric Cancer. Cancers, 11(9), 1396. https://doi.org/10.3390/cancers11091396