CYP17A1 Maintains the Survival of Glioblastomas by Regulating SAR1-Mediated Endoplasmic Reticulum Health and Redox Homeostasis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
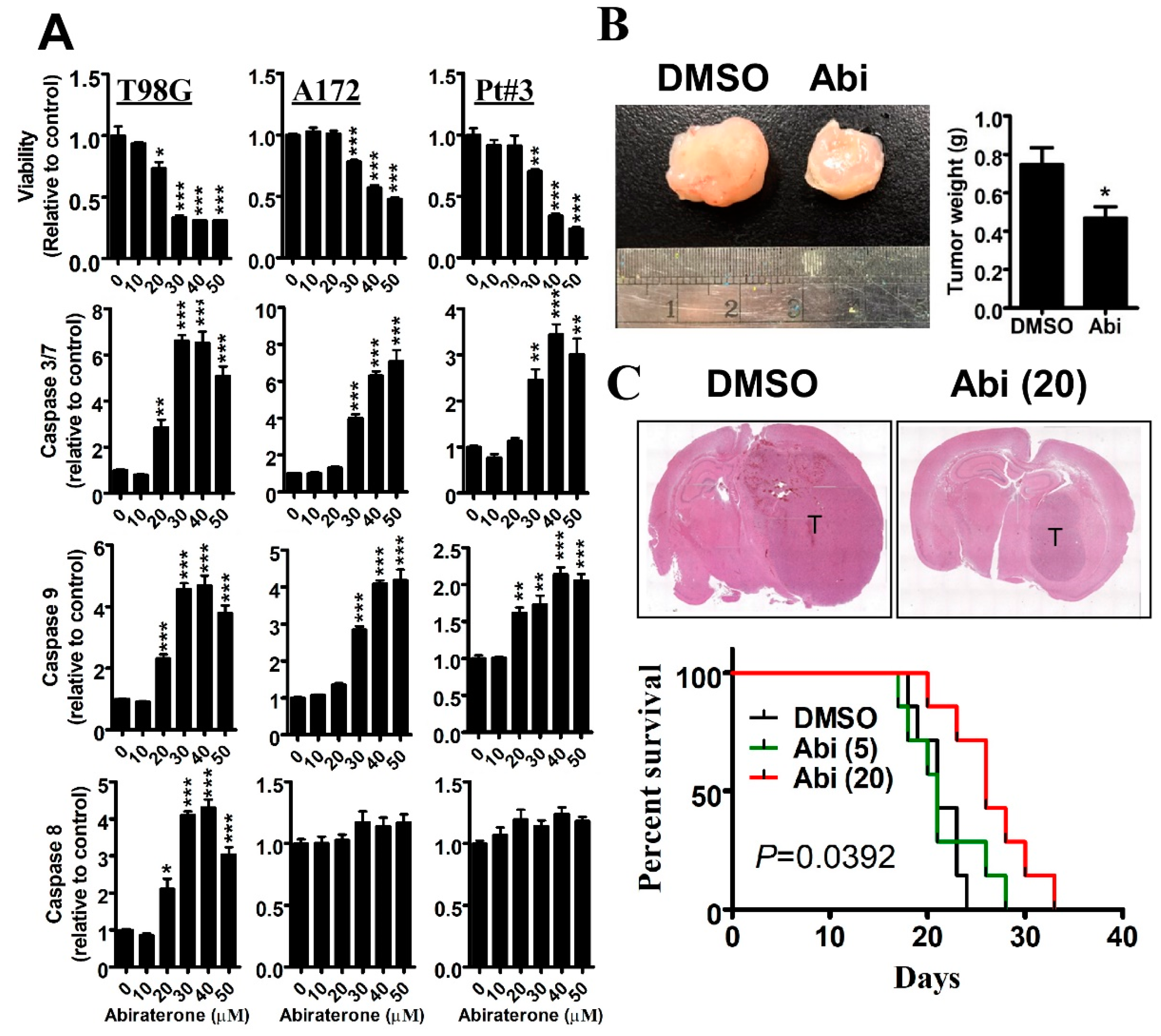
2.1. Inhibition of CYP17A1 by Abiraterone Exhibits a Tumor-Suppressive Effect on Glioblastomas in Vitro and in Vivo
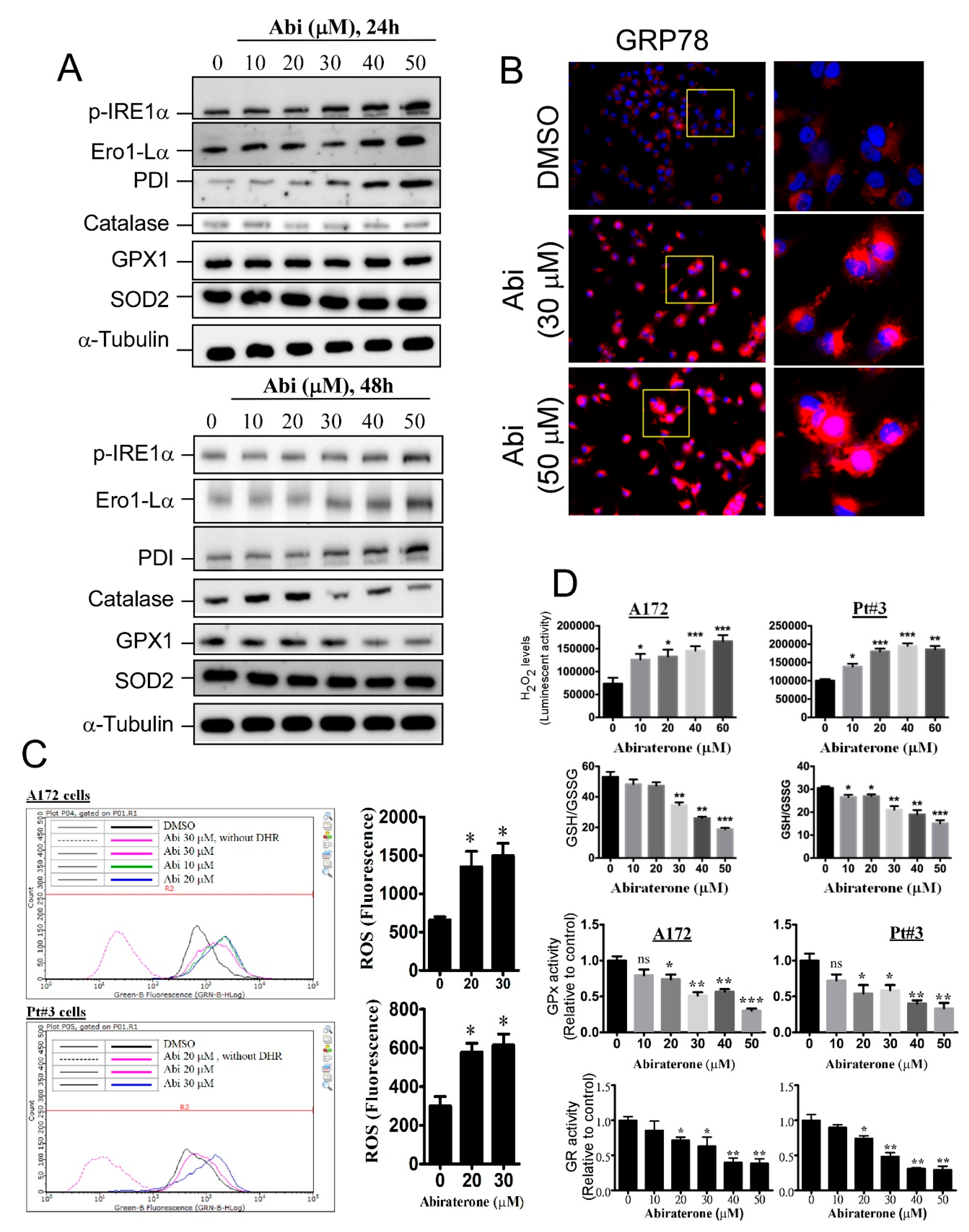
2.2. Abiraterone Induces Endoplasmic Reticulum Stress and Reactive Oxygen Species Accumulation by Impairing Redox Reactions
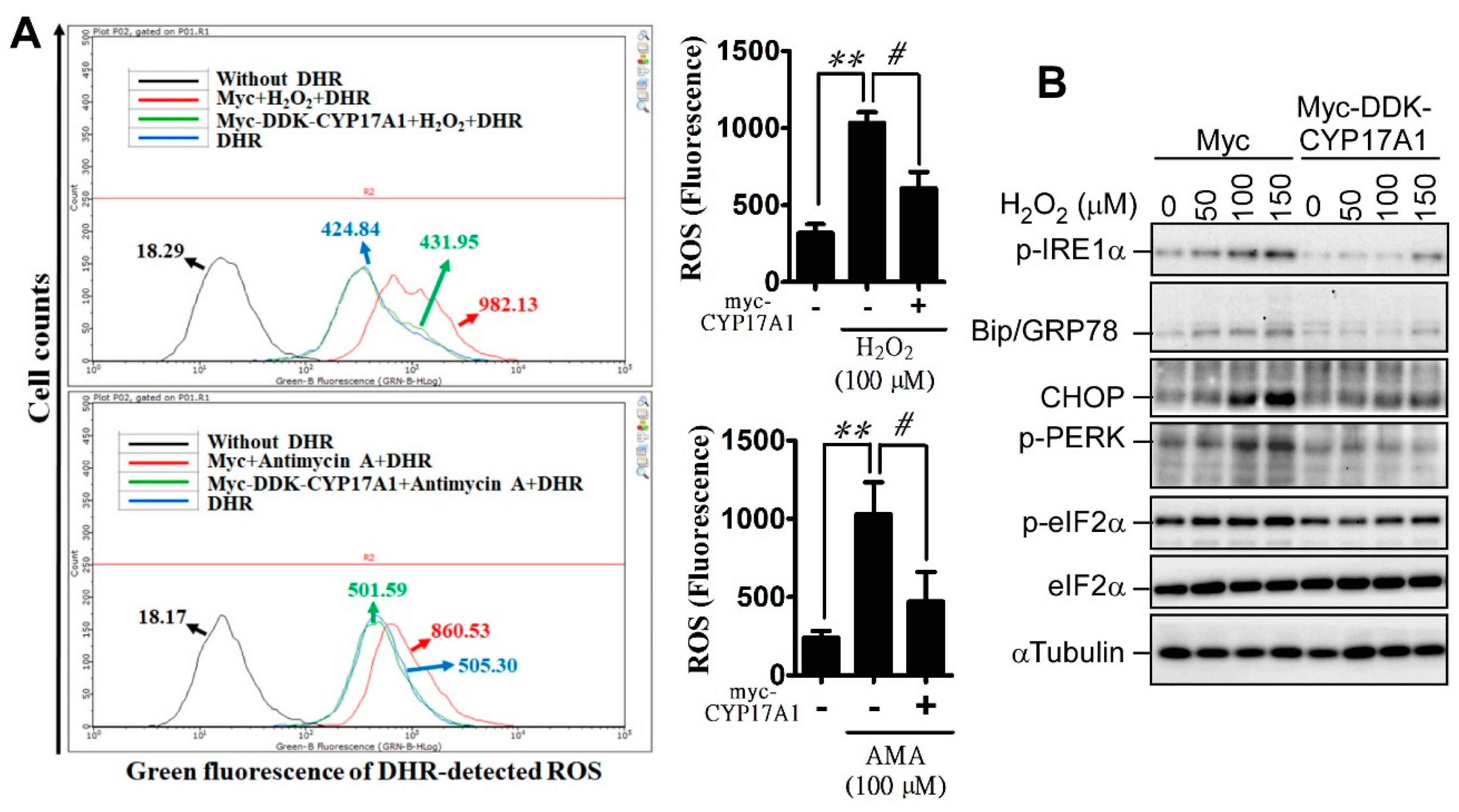
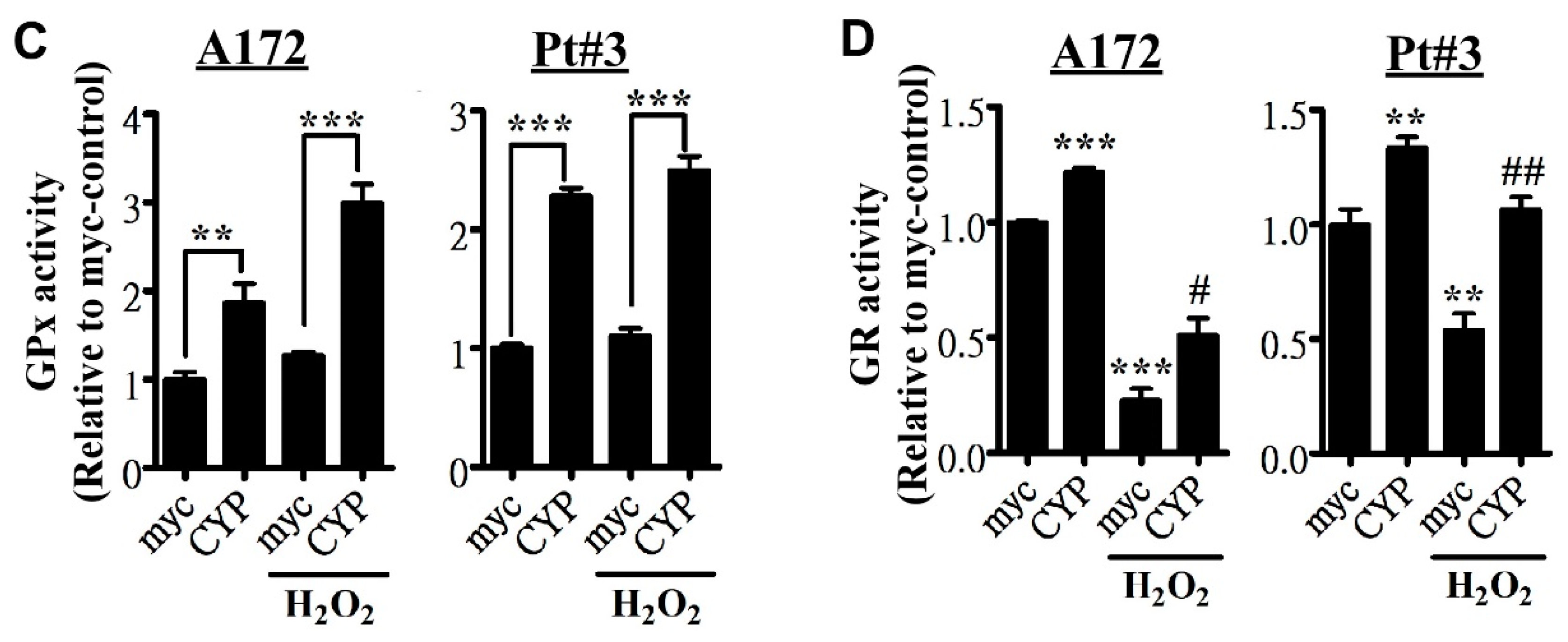
2.3. CYP17A1 Prevents Reactive Oxygen Species Accumulation and Attenuates Reactive Oxygen Species-Induced Endoplasmic Reticulum Stress
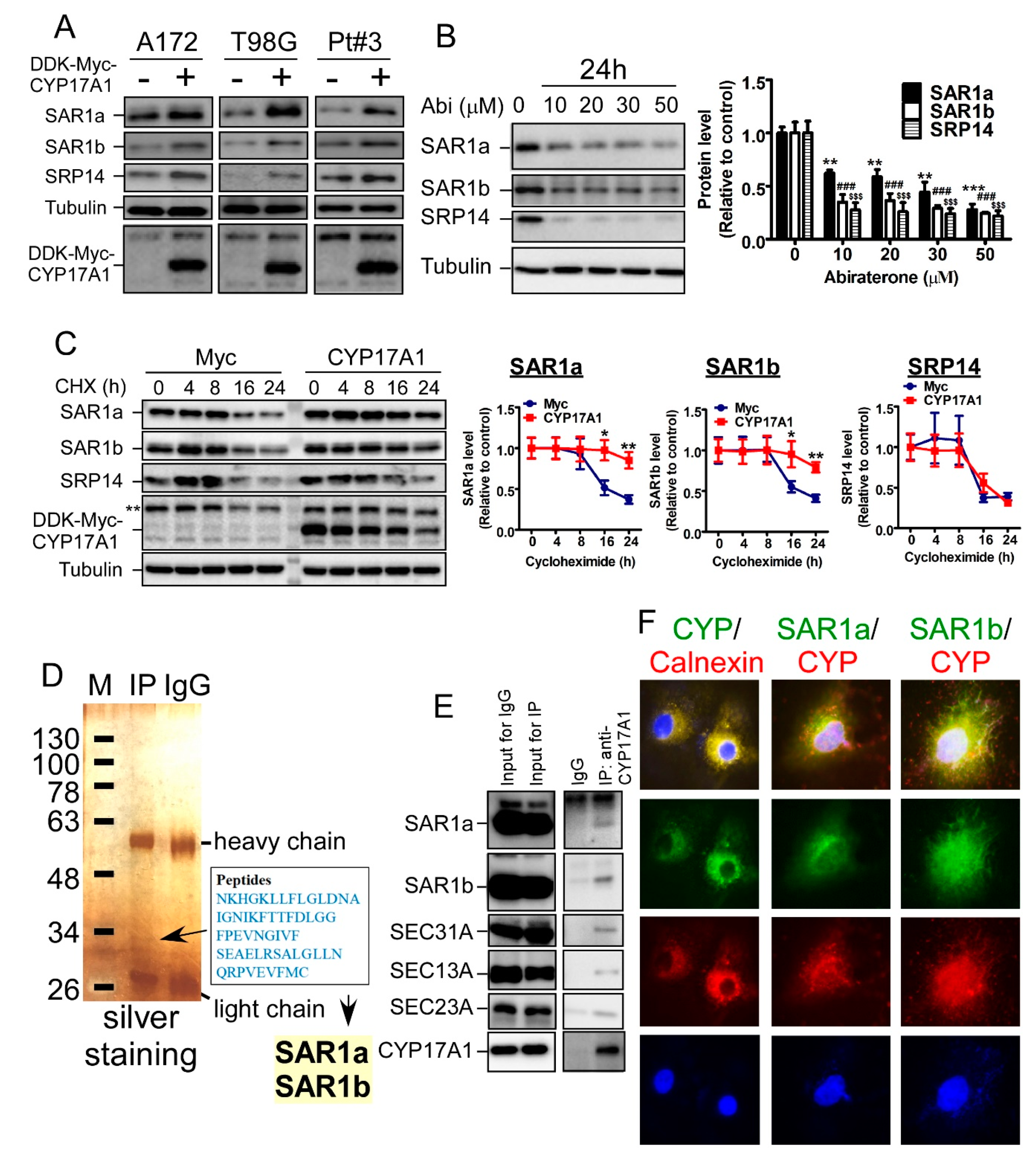
2.4. CYP17A1 Regulates the Protein Stability of SAR1
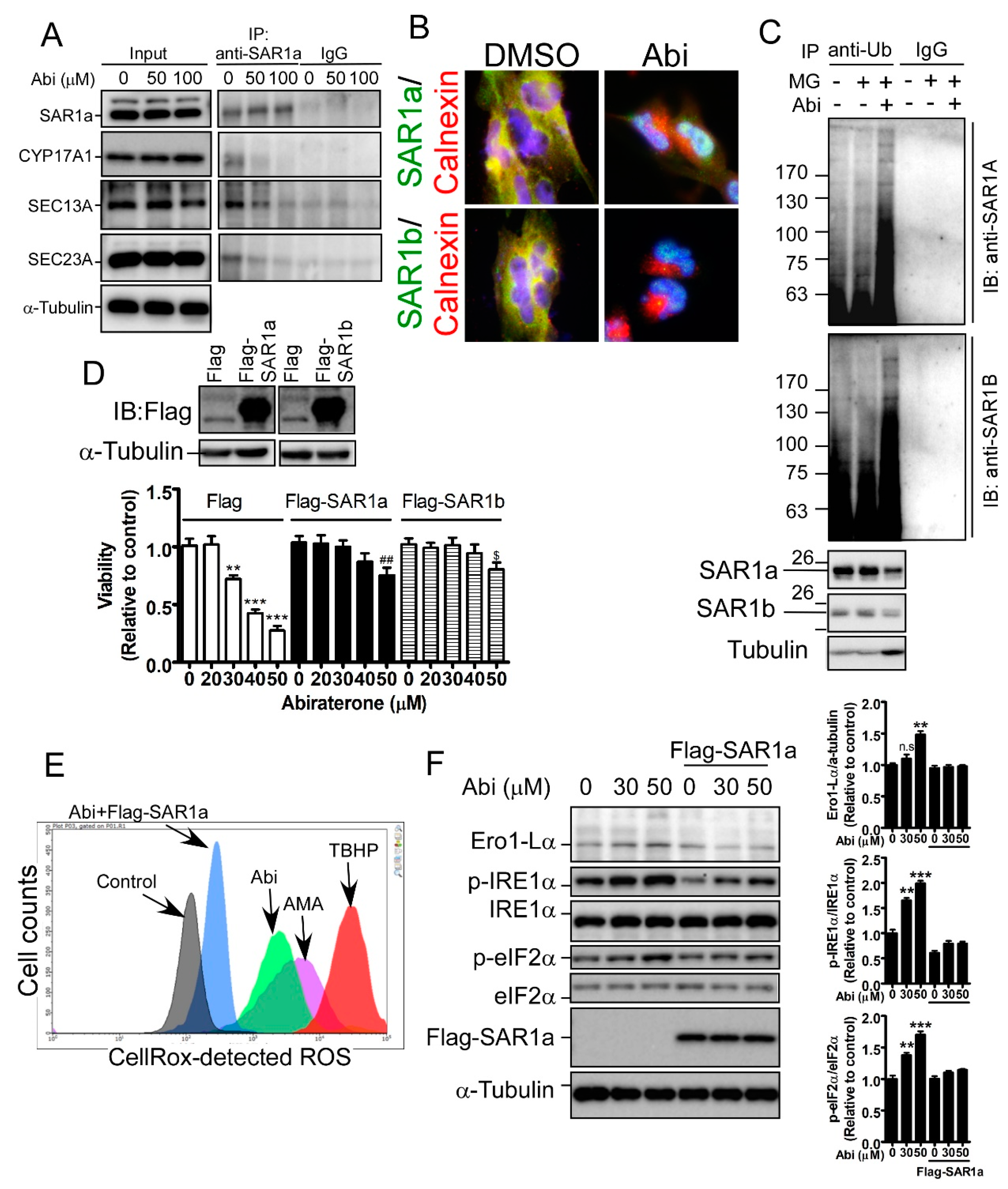
2.5. Inhibition of CYP17A1 Induces Oxidative Stress-Mediated Cell Death by Inducing SAR1 Ubiquitination
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Chemical Compounds, and cDNA Clone
4.2. Primary Glioblastoma Cells
4.3. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Assay
4.4. Immunoprecipitation and Western Blotting
4.5. Immunofluorescence
4.6. Caspase 3/7, 8, and 9 Detection
4.7. Reactive Oxygen Species (ROS) Analysis
4.8. H2O2-Glo and Glutathione/Oxidized Glutathione-Glo Analysis, and Glutathione Peroxidase (GPx) and Glutathione Reductase (GR) Activity Analysis
4.9. Proteomics
4.10. Protein Sample Preparation and iTRAQ Labeling
4.11. In-Gel Digestion
4.12. LC-MS/MS Analysis
4.13. Protein Identification
4.14. Tumor Transplantation
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Carver, C.M.; Reddy, D.S. Neurosteroid interactions with synaptic and extrasynaptic GABA(A) receptors: Regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacology 2013, 230, 151–188. [Google Scholar] [CrossRef]
- Faroni, A.; Magnaghi, V. The neurosteroid allopregnanolone modulates specific functions in central and peripheral glial cells. Front. Endocrinol. (Lausanne) 2011, 2, 103. [Google Scholar] [CrossRef]
- Mellon, S.H. Neurosteroid regulation of central nervous system development. Pharmacol. Ther. 2007, 116, 107–124. [Google Scholar] [CrossRef]
- Mtchedlishvili, Z.; Kapur, J. A presynaptic action of the neurosteroid pregnenolone sulfate on GABAergic synaptic transmission. Mol. Pharmacol. 2003, 64, 857–864. [Google Scholar] [CrossRef]
- Chang, Y.; Hsieh, H.L.; Huang, S.K.; Wang, S.J. Neurosteroid allopregnanolone inhibits glutamate release from rat cerebrocortical nerve terminals. Synapse 2019, 73, e22076. [Google Scholar] [CrossRef]
- Borowicz, K.K.; Piskorska, B.; Banach, M.; Czuczwar, S.J. Neuroprotective actions of neurosteroids. Front. Endocrinol. (Lausanne) 2011, 2, 50. [Google Scholar] [CrossRef]
- Chuang, J.Y.; Lo, W.L.; Ko, C.Y.; Chou, S.Y.; Chen, R.M.; Chang, K.Y.; Hung, J.J.; Su, W.C.; Chang, W.C.; Hsu, T.I. Upregulation of CYP17A1 by Sp1-mediated DNA demethylation confers temozolomide resistance through DHEA-mediated protection in glioma. Oncogenesis 2017, 6, e339. [Google Scholar] [CrossRef]
- Yang, W.B.; Chuang, J.Y.; Ko, C.Y.; Chang, W.C.; Hsu, T.I. Dehydroepiandrosterone Induces Temozolomide Resistance Through Modulating Phosphorylation and Acetylation of Sp1 in Glioblastoma. Mol. Neurobiol. 2019, 56, 2301–2313. [Google Scholar] [CrossRef]
- Sato, K.; Nakano, A. Mechanisms of COPII vesicle formation and protein sorting. FEBS Lett. 2007, 581, 2076–2082. [Google Scholar] [CrossRef]
- Fang, J.; Liu, M.; Zhang, X.; Sakamoto, T.; Taatjes, D.J.; Jena, B.P.; Sun, F.; Woods, J.; Bryson, T.; Kowluru, A.; et al. COPII-Dependent ER Export: A Critical Component of Insulin Biogenesis and beta-Cell ER Homeostasis. Mol. Endocrinol. 2015, 29, 1156–1169. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hazama, K.; Ishida, K.; Komori, M.; Nishimura, K.; Matsuo, S. Inhibition of PLD1 activity causes ER stress via regulation of COPII vesicle formation. Biochem. Biophys. Res. Commun. 2017, 490, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Bird, I.M.; Abbott, D.H. The hunt for a selective 17,20 lyase inhibitor; learning lessons from nature. J. Steroid. Biochem. Mol. Biol. 2016, 163, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Karnovsky, A.; Sans, M.D.; Andrews, P.C.; Williams, J.A. Molecular characterization of the endoplasmic reticulum: Insights from proteomic studies. Proteomics 2010, 10, 4040–4052. [Google Scholar] [CrossRef] [PubMed]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome. Biol. 2000, 1, REVIEWS3003. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Ramassamy, C.; Poirier, J.; Quirion, R. Dehydroepiandrosterone (DHEA) protects hippocampal cells from oxidative stress-induced damage. Brain. Res. Mol. Brain. Res. 1999, 66, 35–41. [Google Scholar] [CrossRef]
- Yoshimoto, F.K.; Auchus, R.J. The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J. Steroid. Biochem. Mol. Biol. 2015, 151, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, R.; Uzilday, B.; Iwata, Y.; Koizumi, N.; Turkan, I. Interplay between the unfolded protein response and reactive oxygen species: A dynamic duo. J. Exp. Bot. 2018, 69, 3333–3345. [Google Scholar] [CrossRef] [PubMed]
- Malikova, J.; Brixius-Anderko, S.; Udhane, S.S.; Parween, S.; Dick, B.; Bernhardt, R.; Pandey, A.V. CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2. J. Steroid. Biochem. Mol. Biol. 2017, 174, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Udhane, S.S.; Dick, B.; Hu, Q.; Hartmann, R.W.; Pandey, A.V. Specificity of anti-prostate cancer CYP17A1 inhibitors on androgen biosynthesis. Biochem. Biophys. Res. Commun. 2016, 477, 1005–1010. [Google Scholar] [CrossRef]
- Fischer, R.; Konkel, A.; Mehling, H.; Blossey, K.; Gapelyuk, A.; Wessel, N.; von Schacky, C.; Dechend, R.; Muller, D.N.; Rothe, M.; et al. Dietary omega-3 fatty acids modulate the eicosanoid profile in man primarily via the CYP-epoxygenase pathway. J. Lipid. Res. 2014, 55, 1150–1164. [Google Scholar] [CrossRef]
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [PubMed]
- Ivanova, E.; Berger, A.; Scherrer, A.; Alkalaeva, E.; Strub, K. Alu RNA regulates the cellular pool of active ribosomes by targeted delivery of SRP9/14 to 40S subunits. Nucleic. Acids. Res. 2015, 43, 2874–2887. [Google Scholar] [CrossRef] [PubMed]
- Markolovic, S.; Wilkins, S.E.; Schofield, C.J. Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 2015, 290, 20712–20722. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, G.; Guo, J.; Takada, M.; Wei, W.; Zhang, Q. New Insights into Protein Hydroxylation and Its Important Role in Human Diseases. Biochim. Biophys. Acta 2016, 1866, 208–220. [Google Scholar] [CrossRef]
- Feng, T.; Yamamoto, A.; Wilkins, S.E.; Sokolova, E.; Yates, L.A.; Munzel, M.; Singh, P.; Hopkinson, R.J.; Fischer, R.; Cockman, M.E.; et al. Optimal translational termination requires C4 lysyl hydroxylation of eRF1. Mol. Cell 2014, 53, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, Z.N.; Blagosklonny, M.V. The purpose of the HIF-1/PHD feedback loop: To limit mTOR-induced HIF-1alpha. Cell Cycle 2011, 10, 1557–1562. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci. Transl. Med. 2019, 11, eaao5253. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Rubin, J.B.; Lathia, J.D.; Berens, M.E.; Barnholtz-Sloan, J.S. Females have the survival advantage in glioblastoma. Neuro. Oncol. 2018, 20, 576–577. [Google Scholar] [CrossRef]
- Gittleman, H.; Lim, D.; Kattan, M.W.; Chakravarti, A.; Gilbert, M.R.; Lassman, A.B.; Lo, S.S.; Machtay, M.; Sloan, A.E.; Sulman, E.P.; et al. An independently validated nomogram for individualized estimation of survival among patients with newly diagnosed glioblastoma: NRG Oncology RTOG 0525 and 0825. Neuro. Oncol. 2017, 19, 669–677. [Google Scholar] [CrossRef]
- Auchus, R.J. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid. Biochem. Mol. Biol. 2017, 165, 71–78. [Google Scholar] [CrossRef]
- Chang, K.Y.; Hsu, T.I.; Hsu, C.C.; Tsai, S.Y.; Liu, J.J.; Chou, S.W.; Liu, M.S.; Liou, J.P.; Ko, C.Y.; Chen, K.Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O(6)-methylguanine-DNA methyltransferase. Redox. Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-Y.; Ko, C.-Y.; Kao, T.-J.; Yang, W.-B.; Tsai, Y.-T.; Chuang, J.-Y.; Hu, S.-L.; Yang, P.-Y.; Lo, W.-L.; Hsu, T.-I. CYP17A1 Maintains the Survival of Glioblastomas by Regulating SAR1-Mediated Endoplasmic Reticulum Health and Redox Homeostasis. Cancers 2019, 11, 1378. https://doi.org/10.3390/cancers11091378
Lin H-Y, Ko C-Y, Kao T-J, Yang W-B, Tsai Y-T, Chuang J-Y, Hu S-L, Yang P-Y, Lo W-L, Hsu T-I. CYP17A1 Maintains the Survival of Glioblastomas by Regulating SAR1-Mediated Endoplasmic Reticulum Health and Redox Homeostasis. Cancers. 2019; 11(9):1378. https://doi.org/10.3390/cancers11091378
Chicago/Turabian StyleLin, Hong-Yi, Chiung-Yuan Ko, Tzu-Jen Kao, Wen-Bin Yang, Yu-Ting Tsai, Jian-Ying Chuang, Siou-Lian Hu, Pei-Yu Yang, Wei-Lun Lo, and Tsung-I Hsu. 2019. "CYP17A1 Maintains the Survival of Glioblastomas by Regulating SAR1-Mediated Endoplasmic Reticulum Health and Redox Homeostasis" Cancers 11, no. 9: 1378. https://doi.org/10.3390/cancers11091378
APA StyleLin, H.-Y., Ko, C.-Y., Kao, T.-J., Yang, W.-B., Tsai, Y.-T., Chuang, J.-Y., Hu, S.-L., Yang, P.-Y., Lo, W.-L., & Hsu, T.-I. (2019). CYP17A1 Maintains the Survival of Glioblastomas by Regulating SAR1-Mediated Endoplasmic Reticulum Health and Redox Homeostasis. Cancers, 11(9), 1378. https://doi.org/10.3390/cancers11091378