Allosteric AKT Inhibitors Target Synthetic Lethal Vulnerabilities in E-Cadherin-Deficient Cells

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
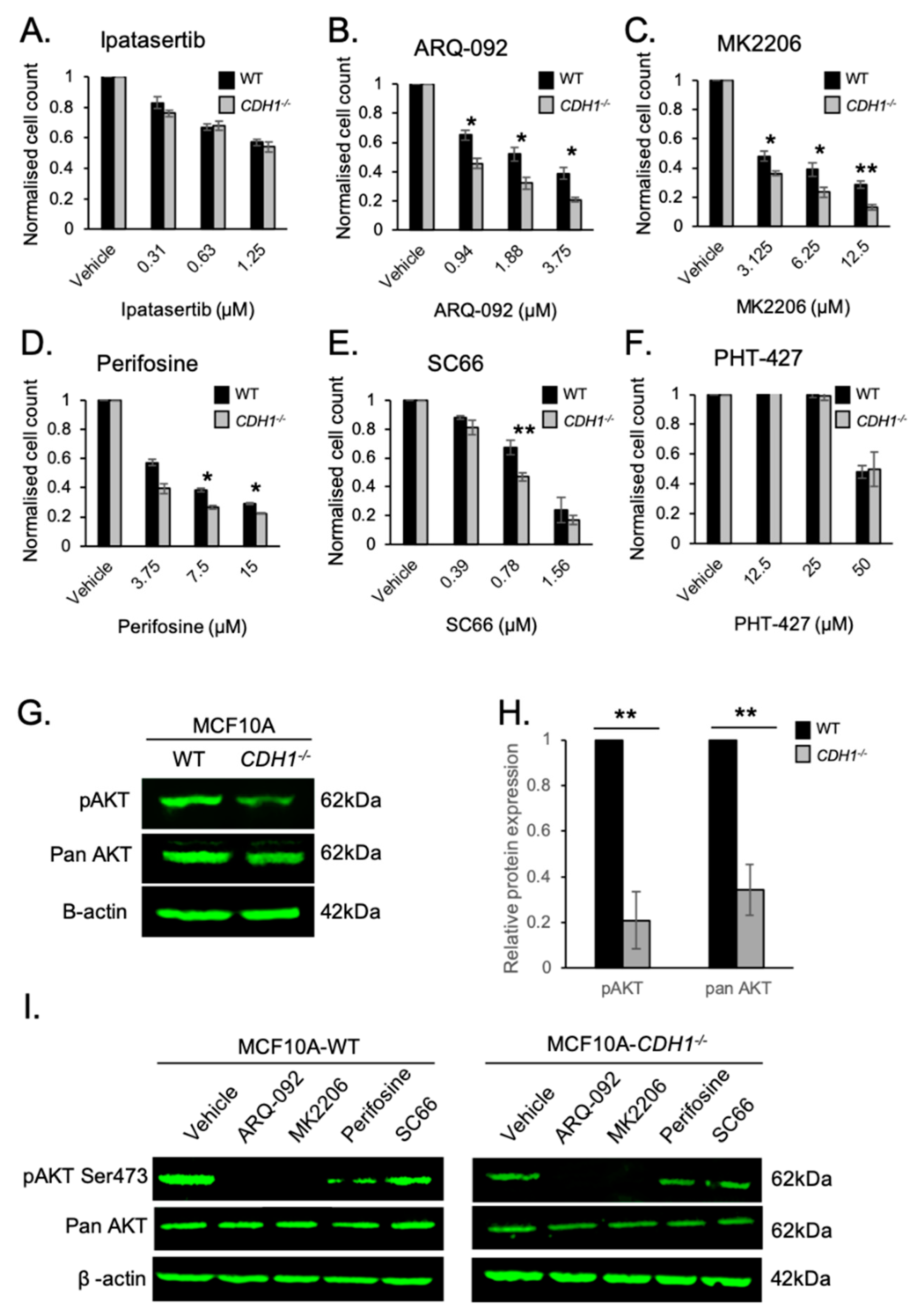
2.1. Allosteric AKT Inhibitors Preferentially Target Breast Cells Lacking CDH1
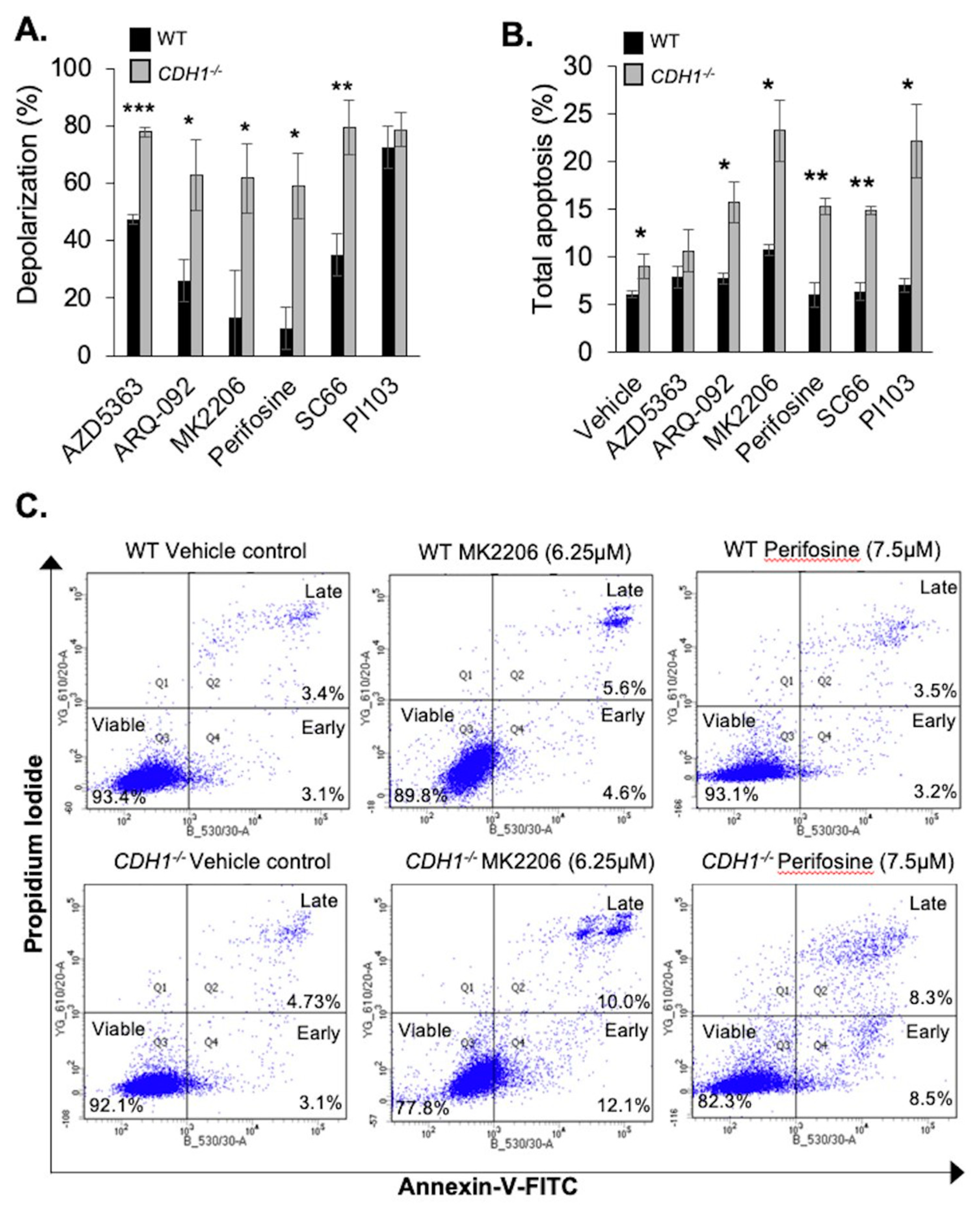
2.2. CDH1-Negative Breast Cells Have Enhanced Apoptotic Priming and Apoptosis Induction after Allosteric AKT Inhibitor Treatment
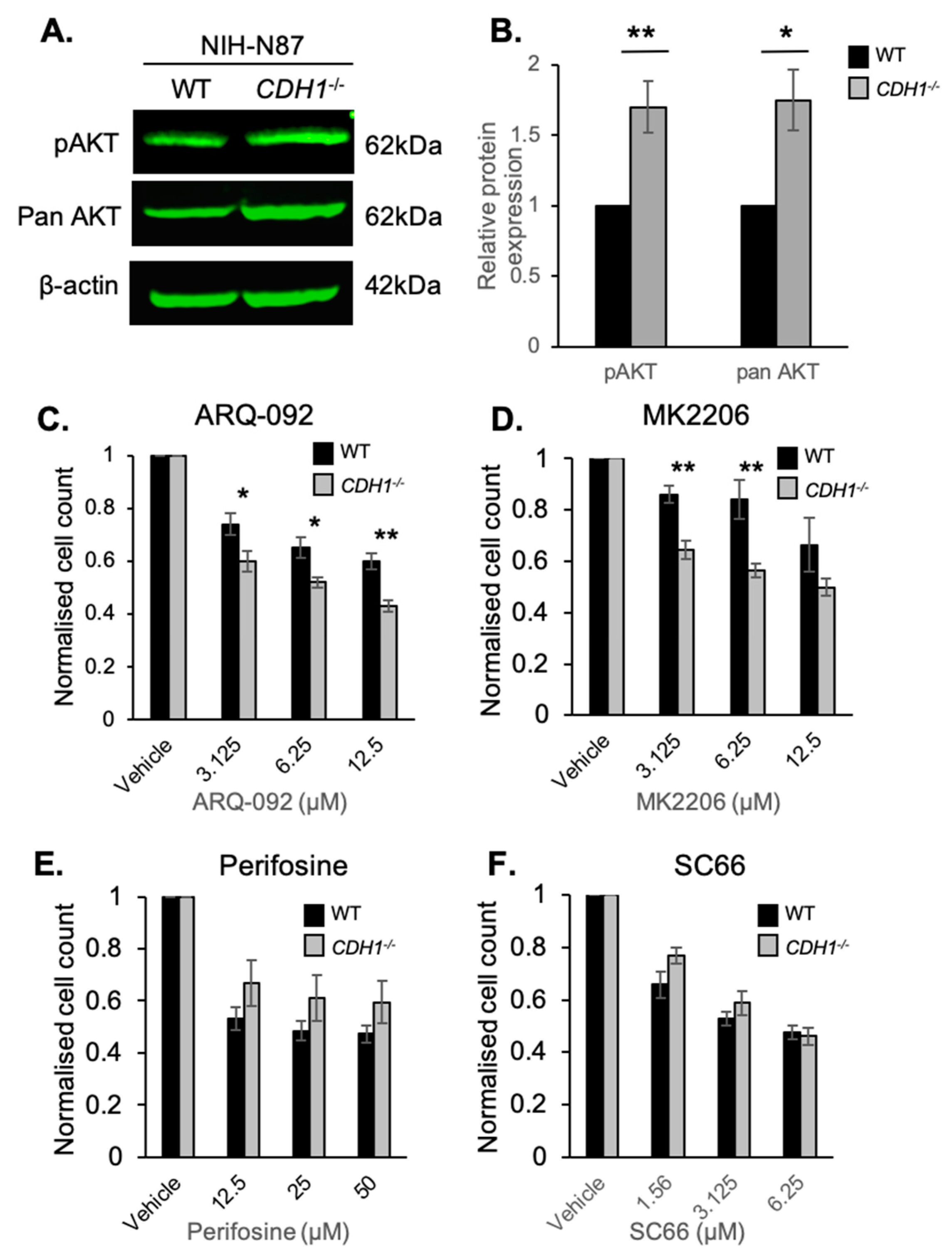
2.3. CDH1-Negative Gastric Cells Are More Sensitive to the Allosteric AKT Inhibitors ARQ-092 and MK2206
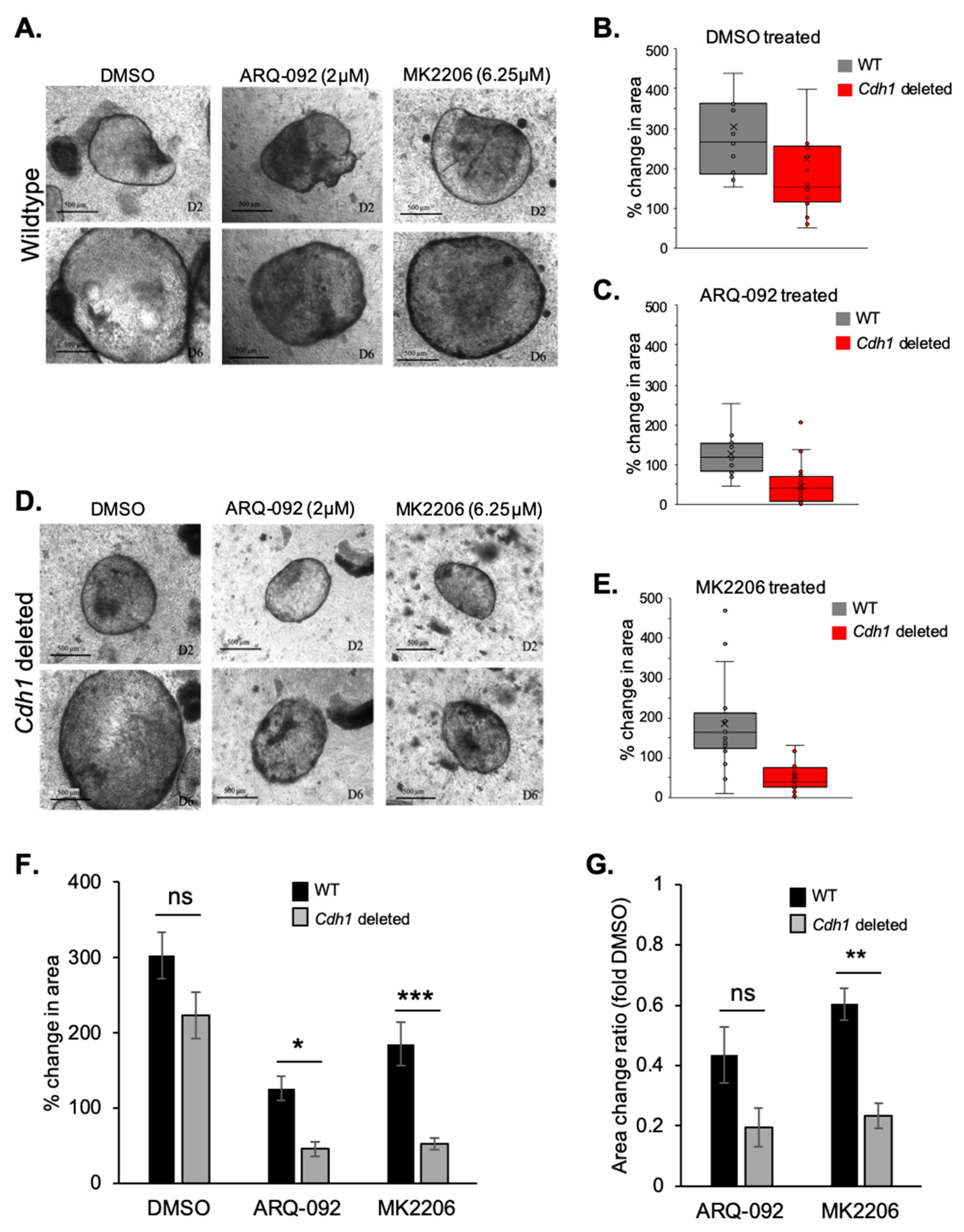
2.4. Allosteric AKT Inhibitors Preferentially Slow the Growth of Cdh1-Deleted Organoids
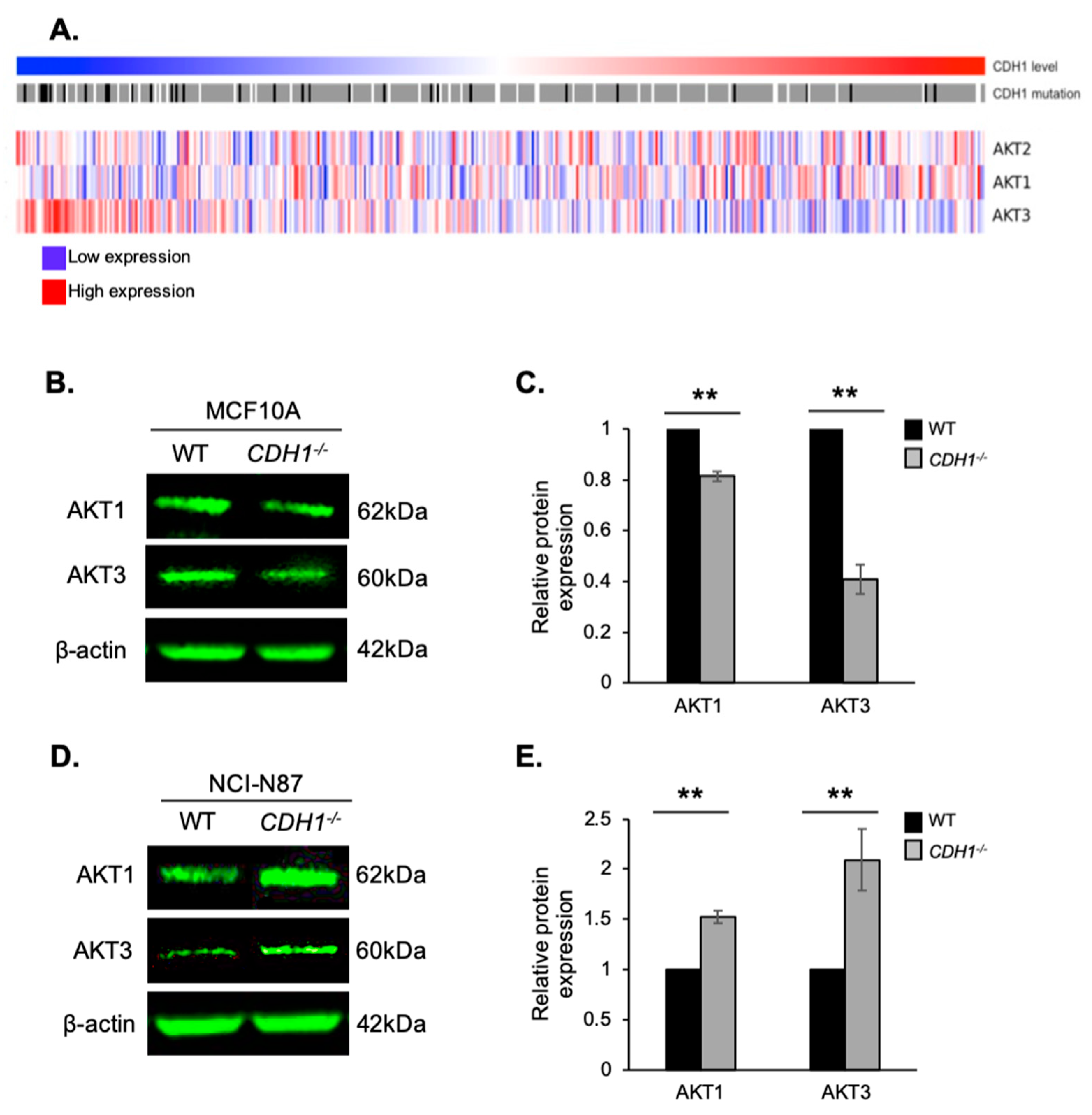
2.5. E-cadherin Expression Affects AKT Isoforms Differently in Breast and Gastric Cells (or in Normal and Cancer Cells)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Small Molecule Inhibitors and Chemicals
4.3. Nuclei Enumeration Assay
4.4. Western Blotting
4.5. FACS
4.6. Apoptosis Priming
4.7. Organoid Culture
4.8. Drug Treatment of Organoids
4.9. RNA-seq Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.-H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Guilford, P.J.; Hopkins, J.B.; Grady, W.M.; Markowitz, S.D.; Willis, J.; Lynch, H.; Rajput, A.; Wiesner, G.L.; Lindor, N.M.; Burgart, L.J.; et al. E-cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum. Mutat. 1999, 14, 249–255. [Google Scholar] [CrossRef]
- van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.C.; Hardwick, R.H.; Ausems, M.G.E.M.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Van der Post, R.S.; Oliveira, C.; Guilford, P.; Carneiro, F. Hereditary gastric cancer: what’s new? Update 2013–2018. Fam. Cancer 2019, 15, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pattison, S.; Mitchell, C.; Lade, S.; Leong, T.; Busuttil, R.A.; Boussioutas, A. Early relapses after adjuvant chemotherapy suggests primary chemoresistance in diffuse gastric cancer. PLoS ONE 2017, 12, e0183891. [Google Scholar] [CrossRef] [PubMed]
- Telford, B.J.; Chen, A.; Beetham, H.; Frick, J.; Brew, T.P.; Gould, C.M.; Single, A.; Godwin, T.; Simpson, K.J.; Guilford, P. Synthetic Lethal Screens Identify Vulnerabilities in GPCR Signaling and Cytoskeletal Organization in E-Cadherin-Deficient Cells. Mol. Cancer Ther. 2015, 14, 1213–1223. [Google Scholar] [CrossRef]
- Godwin, T.D.; Kelly, S.T.; Brew, T.P.; Bougen-Zhukov, N.M.; Single, A.B.; Chen, A.; Stylianou, C.E.; Harris, L.D.; Currie, S.K.; Telford, B.J.; et al. E-cadherin-deficient cells have synthetic lethal vulnerabilities in plasma membrane organisation, dynamics and function. Gastric Cancer 2018, 22, 273–286. [Google Scholar] [CrossRef]
- Canaani, D. Methodological approaches in application of synthetic lethality screening towards anticancer therapy. Br. J. Cancer 2009, 100, 1213–1218. [Google Scholar] [CrossRef]
- Pece, S.; Chiariello, M.; Murga, C.; Gutkind, J.S. Activation of the Protein Kinase Akt/PKB by the Formation of E-cadherin-mediated cell-cell junctions evidence for the association of phosphatidylinositol 3-kinase with the e-cadherin adhesion complex. J. Biol. Chem. 1999, 274, 19347–19351. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-Cadherin-Mediated Cell-Cell Adhesion Activates Akt and Mitogen Activated Protein Kinase via Phosphatidylinositol 3 Kinase and Ligand-Independent Activation of Epidermal Growth Factor Receptor in Ovarian Cancer Cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Xia, X.; Ding, C.; Zhen, B.; Zhou, Q.; Feng, J.; Yuan, J.; Chen, R.; Li, Y.; Ge, Z.; et al. A proteomic landscape of diffuse-type gastric cancer. Nat. Commun. 2018, 9, 1012. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.R.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.F.; Xu, R.; Bencsik, J.R.; Xiao, D.; Kallan, N.C.; Schlachter, S.; Mitchell, I.S.; Spencer, K.L.; Banka, A.L.; Wallace, E.M.; et al. Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J. Med. Chem. 2012, 55, 8110–8127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, D.; Chen, B.; Tang, H.; Xie, X.; Wei, W. Efficacy of PI3K/AKT/mTOR pathway inhibitors for the treatment of advanced solid cancers: A literature-based meta-analysis of 46 randomised control trials. PLOS ONE 2018, 13, e0192464. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2015, 48, 869–885. [Google Scholar] [CrossRef]
- Chan, T.C.K.; Lapierre, J.-M.; Ashwell, M.A.; France, D.S.; Chen, C.-R.; Cornell-Kennon, S.; Bull, C.; Eathiraj, S.; Palma, R.; Liu, Y.; et al. Abstract A230: Discovery and characterization of ARQ 092, an ATP-independent, potent and selective inhibitor of AKT kinases. Mol. Cancer Ther. 2011, 10, A230. [Google Scholar]
- Wu, W.-I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal Structure of Human AKT1 with an Allosteric Inhibitor Reveals a New Mode of Kinase Inhibition. PLOS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Poradosu, E.; Lemmon, M.; Keleti, D. Perifosine selectively inhibits binding of Akt PH domain to PtdIns(3,4)P2. Cancer Res. 2007, 67, 1645. [Google Scholar]
- Jo, H.; Lo, P.-K.; Li, Y.; Loison, F.; Green, S.; Wang, J.; Silberstein, L.E.; Ye, K.; Chen, H.; Luo, H.R. Deactivation of Akt by a small molecule inhibitor targeting pleckstrin homology domain and facilitating Akt ubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 6486–6491. [Google Scholar] [CrossRef] [PubMed]
- Meuillet, E.J. Novel Inhibitors of AKT: Assessment of a Different Approach Targeting the Pleckstrin Homology Domain. Curr. Med. Chem. 2011, 18, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Okuzumi, T.; Fiedler, D.; Zhang, C.; Gray, D.C.; Aizenstein, B.; Hoffman, R.; Shokat, K.M. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 2009, 5, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Meuillet, E.J.; Zuohe, S.; Lemos, R.; Ihle, N.; Kingston, J.; Watkins, R.; Moses, S.A.; Zhang, S.; Du-Cuny, L.; Herbst, R.; et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol. Cancer Ther. 2010, 9, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-W.; Lin, Y.-M.; Lai, Y.-L.; Chen, C.-Y.; Hu, C.-Y.; Tien, H.-F.; Ou, D.-L.; Lin, L.-I. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med. Oncol. Northwood Lond. Engl. 2015, 32, 206. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, Y.; Zhang, L.; Ren, X.; Huber-Keener, K.J.; Liu, X.; Zhou, L.; Liao, J.; Keihack, H.; Yan, L.; et al. MK-2206, a Novel Allosteric Inhibitor of Akt, Synergizes with Gefitinib against Malignant Glioma via Modulating Both Autophagy and Apoptosis. Mol. Cancer Ther. 2012, 11, 154–164. [Google Scholar] [CrossRef]
- Ryan, J.A.; Brunelle, J.K.; Letai, A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 12895–12900. [Google Scholar] [CrossRef]
- Ryan, J.; Letai, A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013, 61, 156–164. [Google Scholar] [CrossRef]
- Nagoshi, T.; Matsui, T.; Aoyama, T.; Leri, A.; Anversa, P.; Li, L.; Ogawa, W.; del Monte, F.; Gwathmey, J.K.; Grazette, L.; et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J. Clin. Investig. 2005, 115, 2128–2138. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein Kinase B Inhibits Cell Death by Preventing the Release of Cytochrome c from Mitochondria. Mol. Cell. Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Marsters, S.; Ye, X.; Luis, E.; Gonzalez, L.; Ashkenazi, A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell 2014, 54, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Chunhacha, P.; Sriuranpong, V.; Chanvorachote, P. Epithelial-mesenchymal transition mediates anoikis resistance and enhances invasion in pleural effusion-derived human lung cancer cells. Oncol. Lett. 2013, 5, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Anderson, K.; Stokoe, D.; Erdjument-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Belham, C.; Wu, S.; Avruch, J. Intracellular signalling: PDK1—A kinase at the hub of things. Curr. Biol. 1999, 9, R93–R96. [Google Scholar] [CrossRef]
- Cinti, C.; Vindigni, C.; Zamparelli, A.; La Sala, D.; Epistolato, M.C.; Marrelli, D.; Cevenini, G.; Tosi, P. Activated Akt as an indicator of prognosis in gastric cancer. Virchows Arch. Int. J. Pathol. 2008, 453, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Kennecke, H.F.; Iqbal, S.; Baranda, J.C.; Seery, T.E.; Lim, H.J.; Hezel, A.F.; Vaccaro, G.M.; Blanke, C.D. Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: A SWOG cooperative group trial (S1005). Cancer 2015, 121, 2193–2197. [Google Scholar] [CrossRef]
- Dasari, A.; Overman, M.J.; Fogelman, D.R.; Kee, B.K.; Menter, D.; Raghav, K.P.S.; Morris, V.K.; Oh, J.; Wu, J.; Jiang, Z.; et al. A phase II and co-clinical study of an AKT inhibitor in patients (pts) with biomarker-enriched, previously treated metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2016, 34, 3563. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Krop, I.; Akcakanat, A.; Chen, H.; Liu, S.; Li, Y.; Culotta, K.S.; Tarco, E.; Piha-Paul, S.; Moulder-Thompson, S.; et al. SU2C Phase Ib Study of Paclitaxel and MK-2206 in Advanced Solid Tumors and Metastatic Breast Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, dju493. [Google Scholar] [CrossRef]
- Hudis, C.; Swanton, C.; Janjigian, Y.Y.; Lee, R.; Sutherland, S.; Lehman, R.; Chandarlapaty, S.; Hamilton, N.; Gajria, D.; Knowles, J.; et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013, 15, R110. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.; Harb, W.; Sachdev, J.; Papadopoulos, K.; Bordoni, R.; Chai, F.; Larmar, M.; Savage, R.; Abbadessa, G.; Saleh, M. 338 Results from a phase 1 study of ARQ 092, a novel pan AKT-inhibitor, in subjects with advanced solid tumors or recurrent malignant lymphoma. Eur. J. Cancer 2015, 51, S66. [Google Scholar] [CrossRef]
- Abbadessa, G.; Yu, Y.; Eathiraj, S.; Meade, J.; Wick, M.J.; Tolcher, A.; Papadopoulos, K.; Saleh, M.; Sachdev, J.; Chai, F.; et al. Abstract B181: Association of AKT1E17K and PIK3CAH1047R mutations with efficacy of ARQ 092 in vitro, in vivo and in patients. Mol. Cancer Ther. 2015, 14, B181. [Google Scholar]
- Lakhani, N.; Tolcher, A.W.; Rasco, D.W.; Patnaik, A.; O’Rourke, T.J.; Schwartz, B.E.; Abbadessa, G.; Kazakin, J.; Savage, R.; Wang, Y.; et al. Results of a phase Ib study of ARQ 092 in combination with carboplatin (C) plus paclitaxel (P), or with P in patients (pts) with solid tumors. J. Clin. Oncol. 2017, 35, 2524. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Yourick, M.R.; Yu, Y.; Savage, R.E.; Ferrari, D.; Biesecker, L.G. Repression of AKT signaling by ARQ 092 in cells and tissues from patients with Proteus syndrome. Sci. Rep. 2015, 5, 17162. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Sapp, J.C.; Lindhurst, M.J.; Darling, T.N.; Burton-Akright, J.; Bagheri, M.; Dombi, E.; Gruber, A.; Jarosinski, P.F.; Martin, S.; et al. Pharmacodynamic Study of Miransertib in Individuals with Proteus Syndrome. Am. J. Hum. Genet. 2019, 104, 484–491. [Google Scholar] [CrossRef]
- Single, A.; Beetham, H.; Telford, B.J.; Guilford, P.; Chen, A. A Comparison of Real-Time and Endpoint Cell Viability Assays for Improved Synthetic Lethal Drug Validation. J. Biomol. Screen. 2015, 20, 1286–1293. [Google Scholar] [CrossRef]
- Montero, J.; Sarosiek, K.A.; DeAngelo, J.D.; Maertens, O.; Ryan, J.; Ercan, D.; Piao, H.; Horowitz, N.S.; Berkowitz, R.S.; Matulonis, U.; et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 2015, 160, 977–989. [Google Scholar] [CrossRef]
- Kühn, R.; Torres, R.M. Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 2002, 180, 175–204. [Google Scholar]
- Pastuła, A.; Middelhoff, M.; Brandtner, A.; Tobiasch, M.; Höhl, B.; Nuber, A.H.; Demir, I.E.; Neupert, S.; Kollmann, P.; Mazzuoli-Weber, G.; et al. Three-Dimensional Gastrointestinal Organoid Culture in Combination with Nerves or Fibroblasts: A Method to Characterize the Gastrointestinal Stem Cell Niche. Available online: https://www.hindawi.com/journals/sci/2016/3710836/ (accessed on 16 February 2019).
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. gplots: Various R Programming Tools for Plotting Data. R Package Version 3.0.1. 2016. Available online: https://rdrr.io/cran/gplots/ (accessed on 12 September 2019).





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bougen-Zhukov, N.; Nouri, Y.; Godwin, T.; Taylor, M.; Hakkaart, C.; Single, A.; Brew, T.; Permina, E.; Chen, A.; Black, M.A.; et al. Allosteric AKT Inhibitors Target Synthetic Lethal Vulnerabilities in E-Cadherin-Deficient Cells. Cancers 2019, 11, 1359. https://doi.org/10.3390/cancers11091359
Bougen-Zhukov N, Nouri Y, Godwin T, Taylor M, Hakkaart C, Single A, Brew T, Permina E, Chen A, Black MA, et al. Allosteric AKT Inhibitors Target Synthetic Lethal Vulnerabilities in E-Cadherin-Deficient Cells. Cancers. 2019; 11(9):1359. https://doi.org/10.3390/cancers11091359
Chicago/Turabian StyleBougen-Zhukov, Nicola, Yasmin Nouri, Tanis Godwin, Megan Taylor, Christopher Hakkaart, Andrew Single, Tom Brew, Elizabeth Permina, Augustine Chen, Michael A. Black, and et al. 2019. "Allosteric AKT Inhibitors Target Synthetic Lethal Vulnerabilities in E-Cadherin-Deficient Cells" Cancers 11, no. 9: 1359. https://doi.org/10.3390/cancers11091359
APA StyleBougen-Zhukov, N., Nouri, Y., Godwin, T., Taylor, M., Hakkaart, C., Single, A., Brew, T., Permina, E., Chen, A., Black, M. A., & Guilford, P. (2019). Allosteric AKT Inhibitors Target Synthetic Lethal Vulnerabilities in E-Cadherin-Deficient Cells. Cancers, 11(9), 1359. https://doi.org/10.3390/cancers11091359