Etiology-Specific Analysis of Hepatocellular Carcinoma Transcriptome Reveals Genetic Dysregulation in Pathways Implicated in Immunotherapy Efficacy

Abstract
:1. Introduction
2. Results
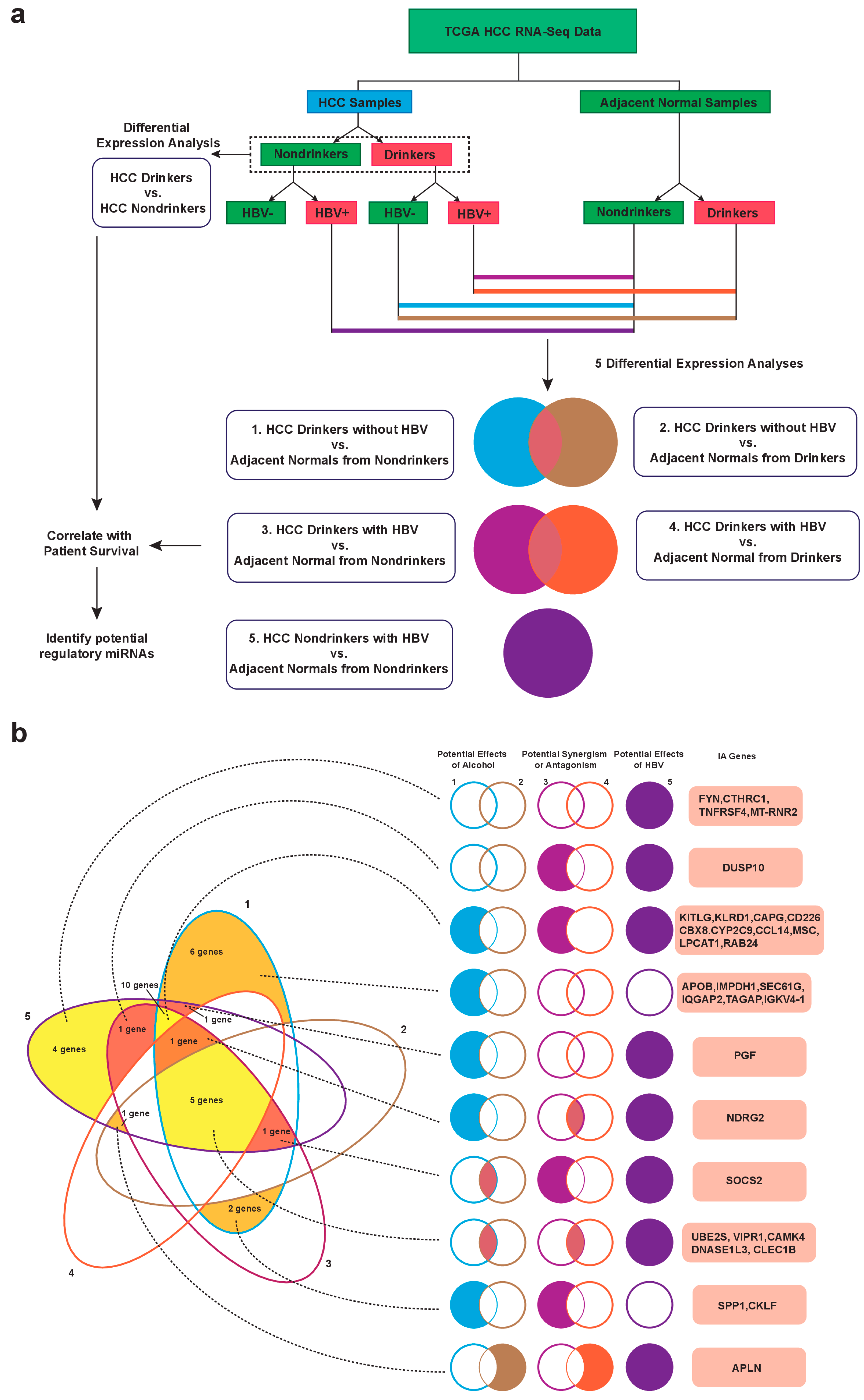
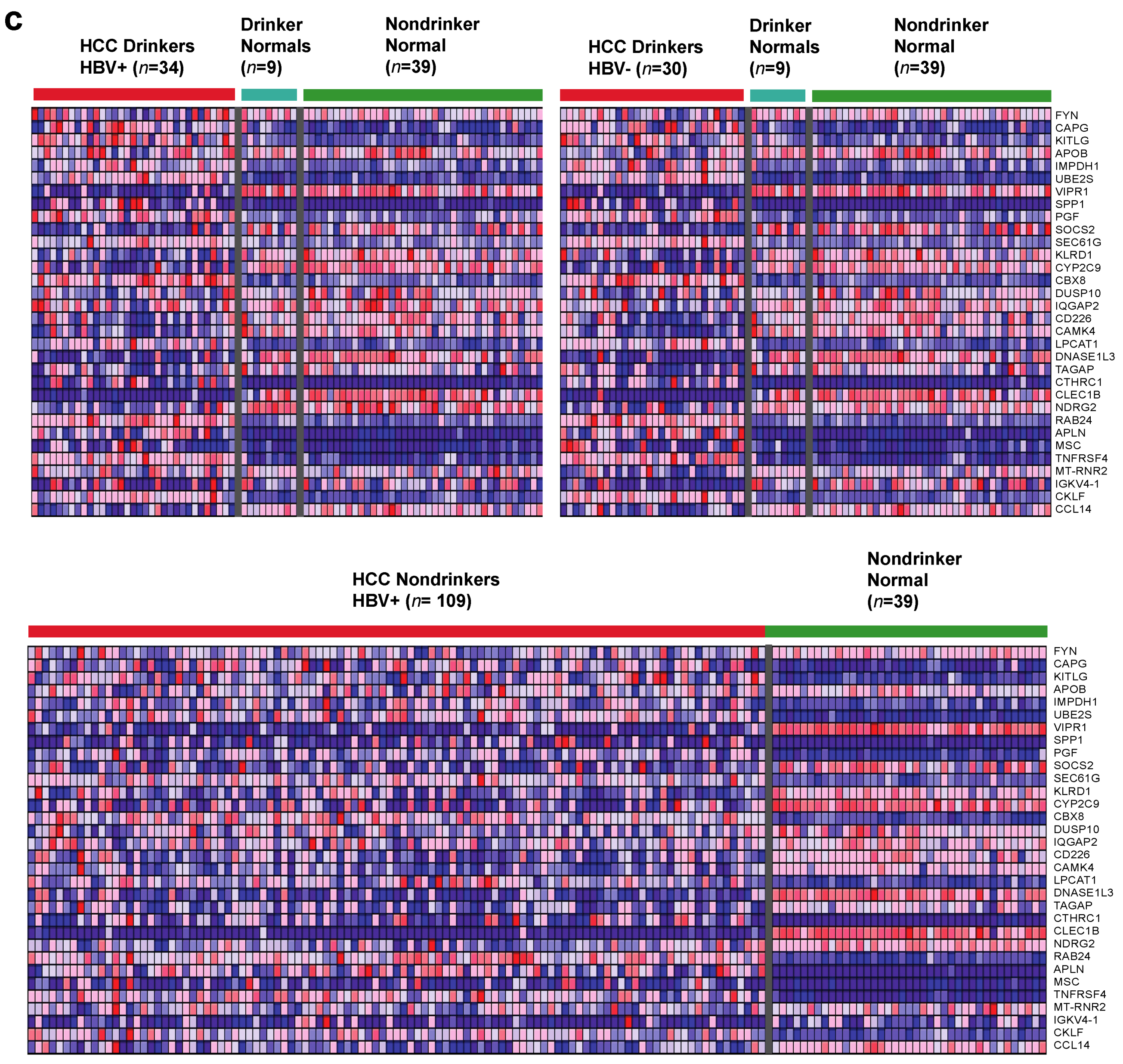
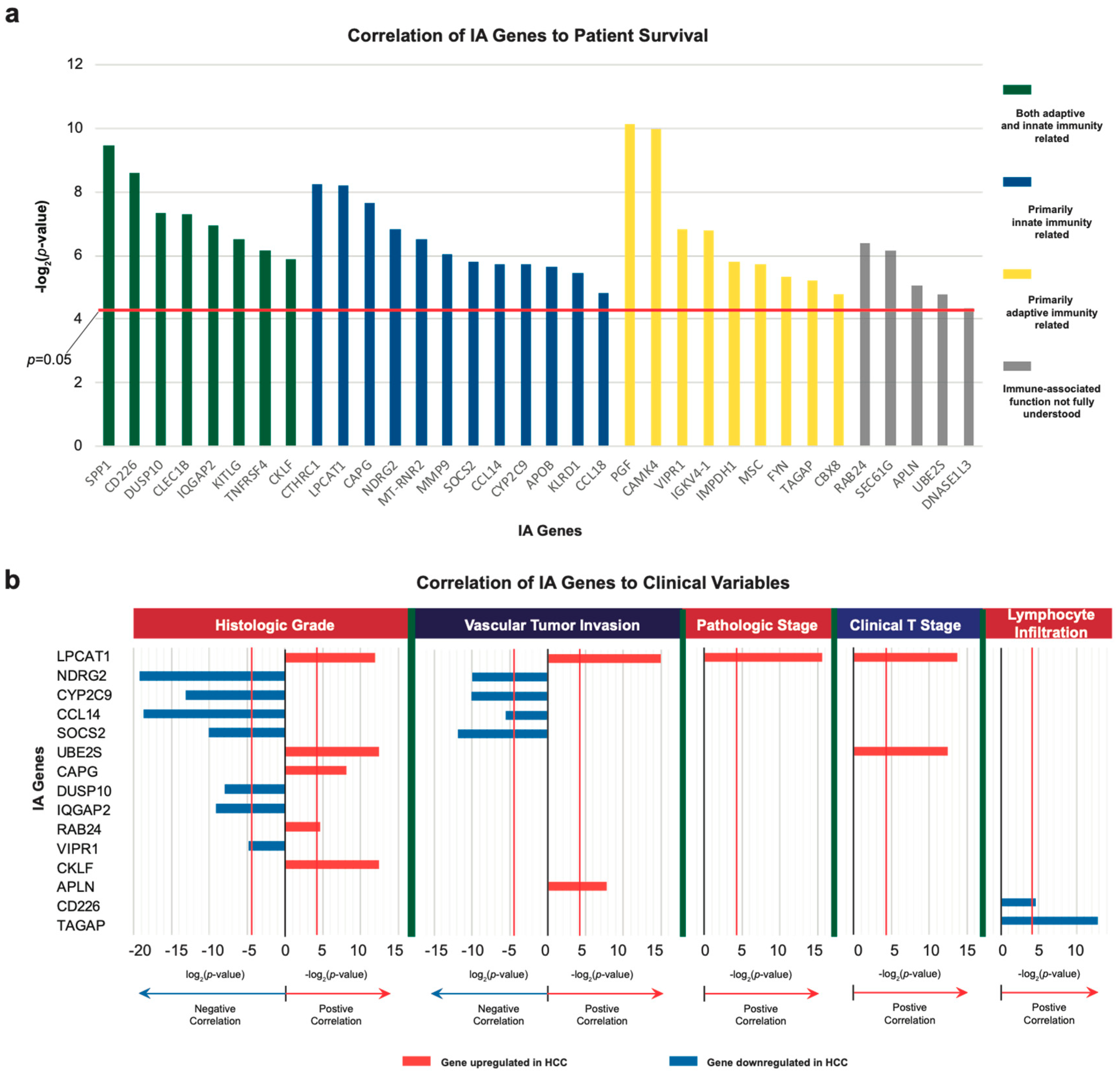
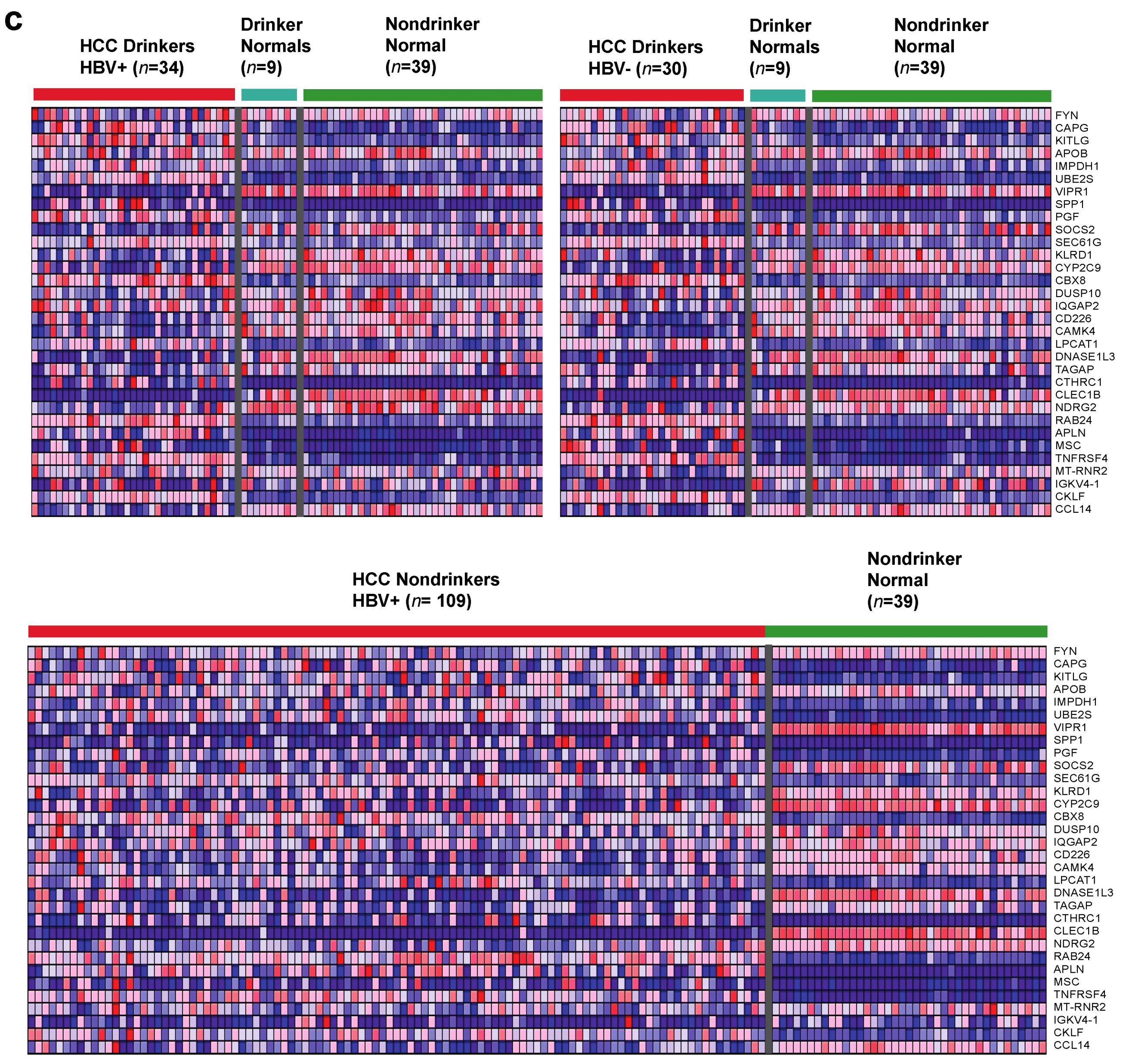
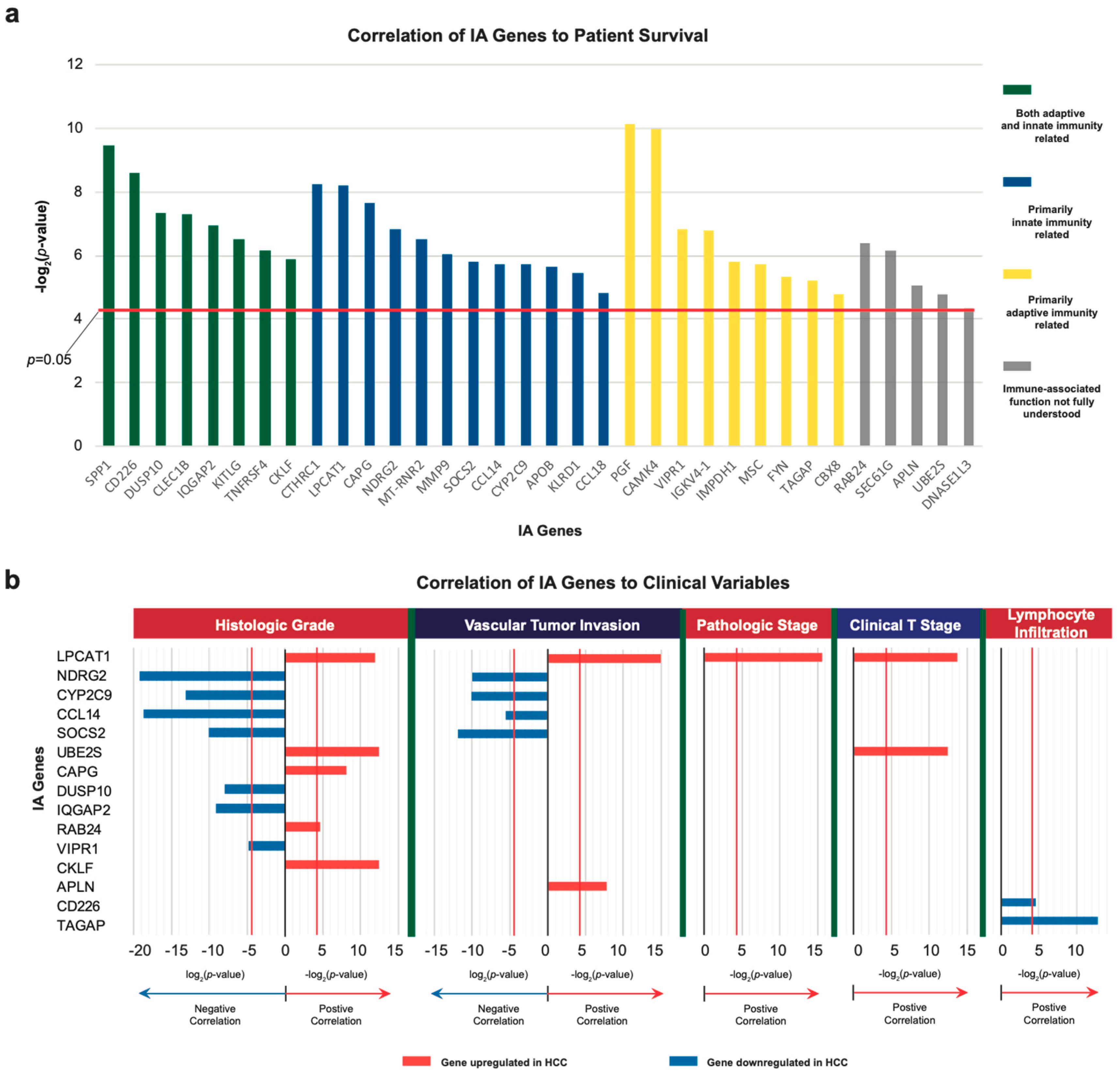
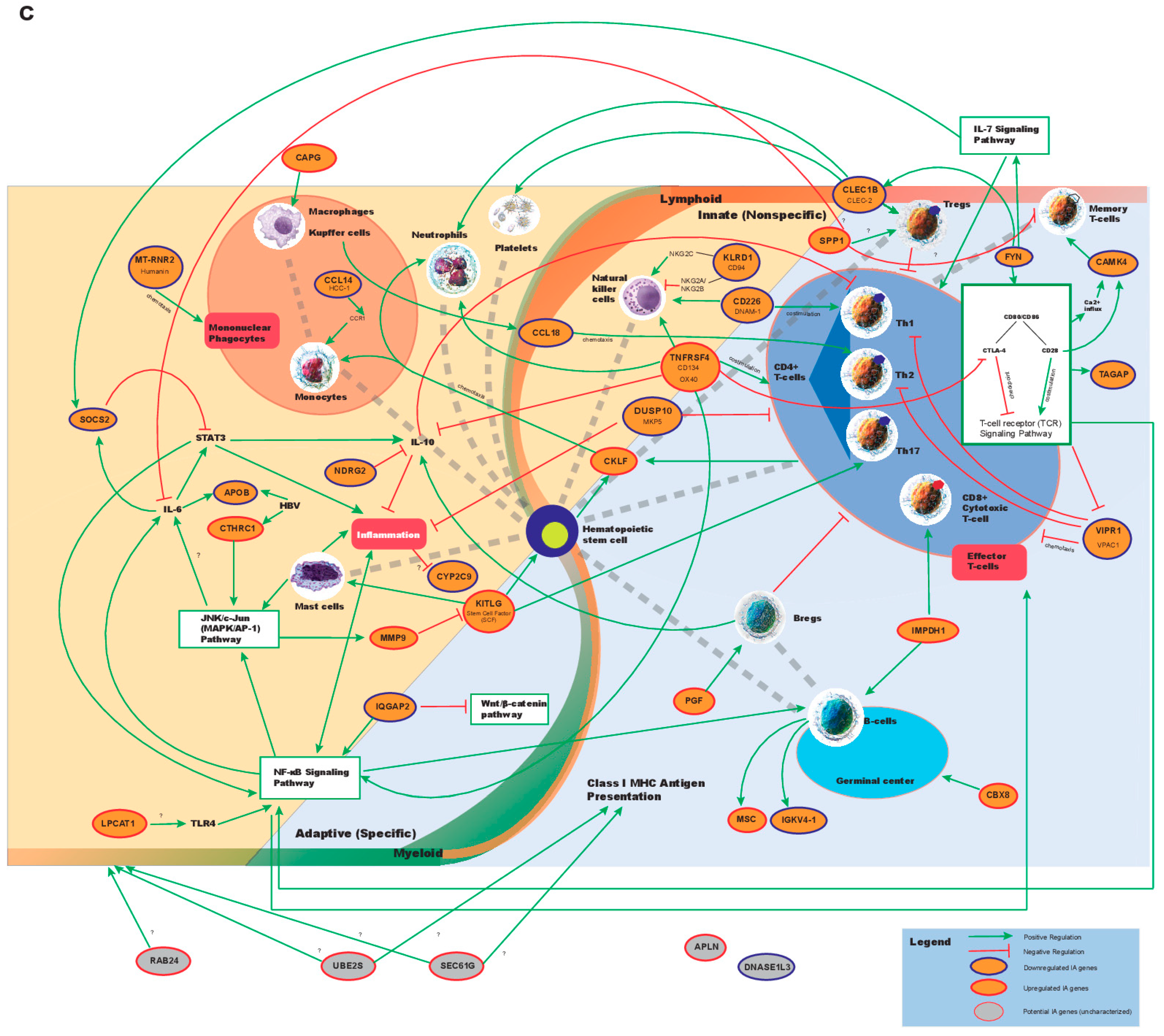
2.1. Identification of Dysregulated Immune-Associated Genes in Etiology-Specific HCC and Correlation with Patient Survival
2.2. Correlation of IA Gene Expressions with Clinical Variables
2.3. Correlation of IA Gene Expressions with Copy Number Variations and Mutation Events Using REVEALER
2.4. Identification of Frequently Disrupted Immune-Associated Pathways in HCC through Differential Mutational Load
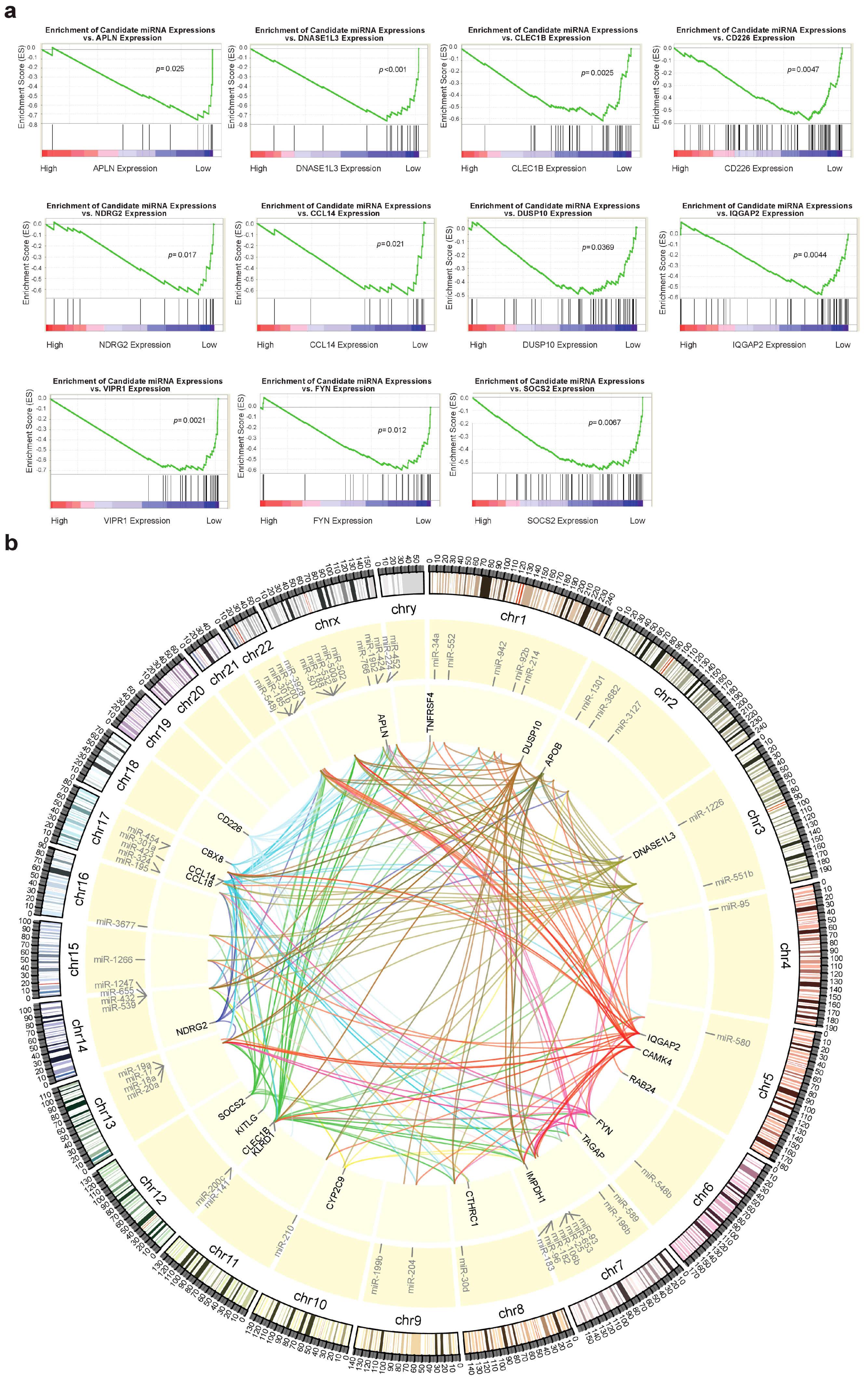
2.5. Identification of Potential miRNAs Involved in Regulation of IA Genes
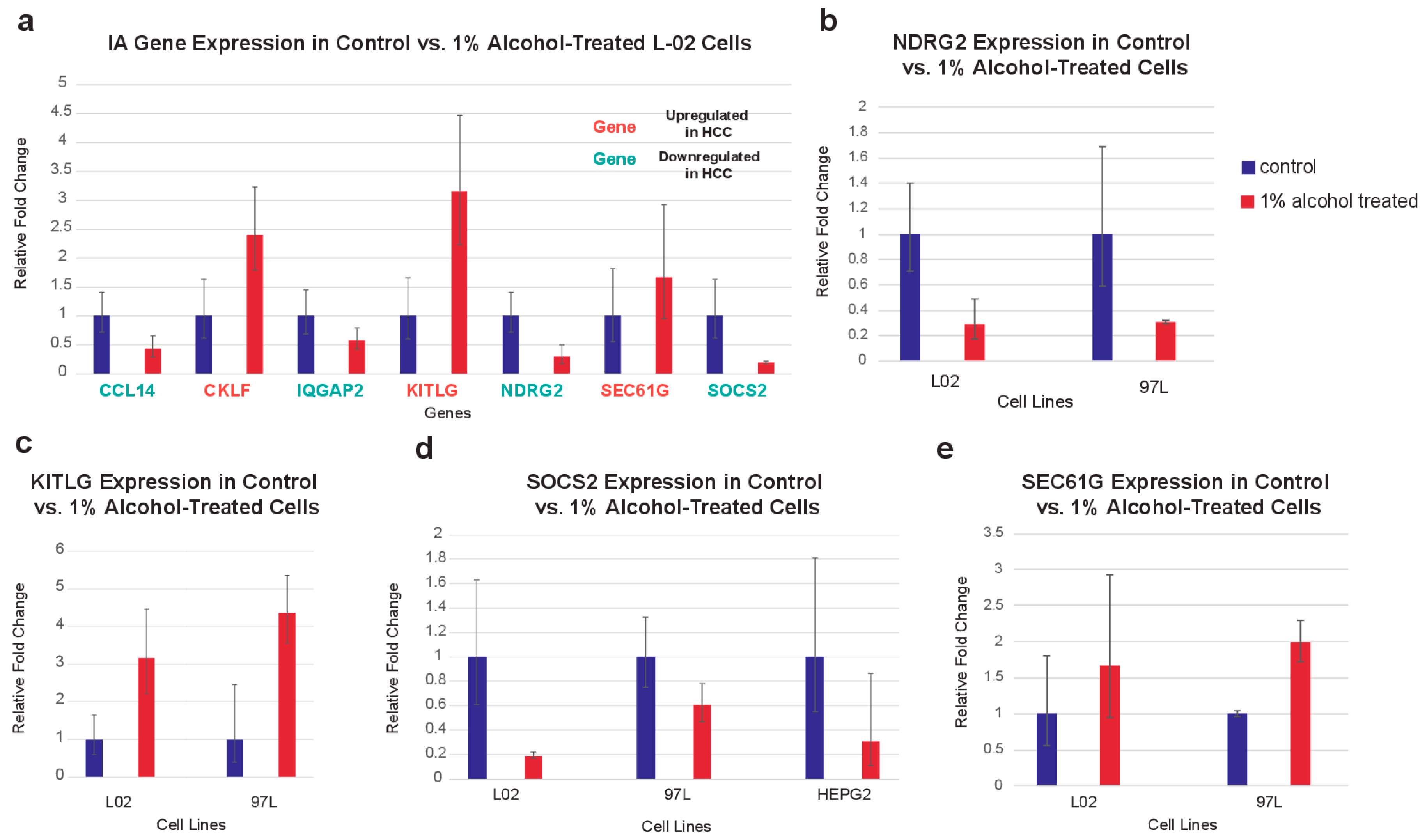
2.6. In Vitro Validation of IA Gene Expression in Cell Lines after Alcohol Exposure
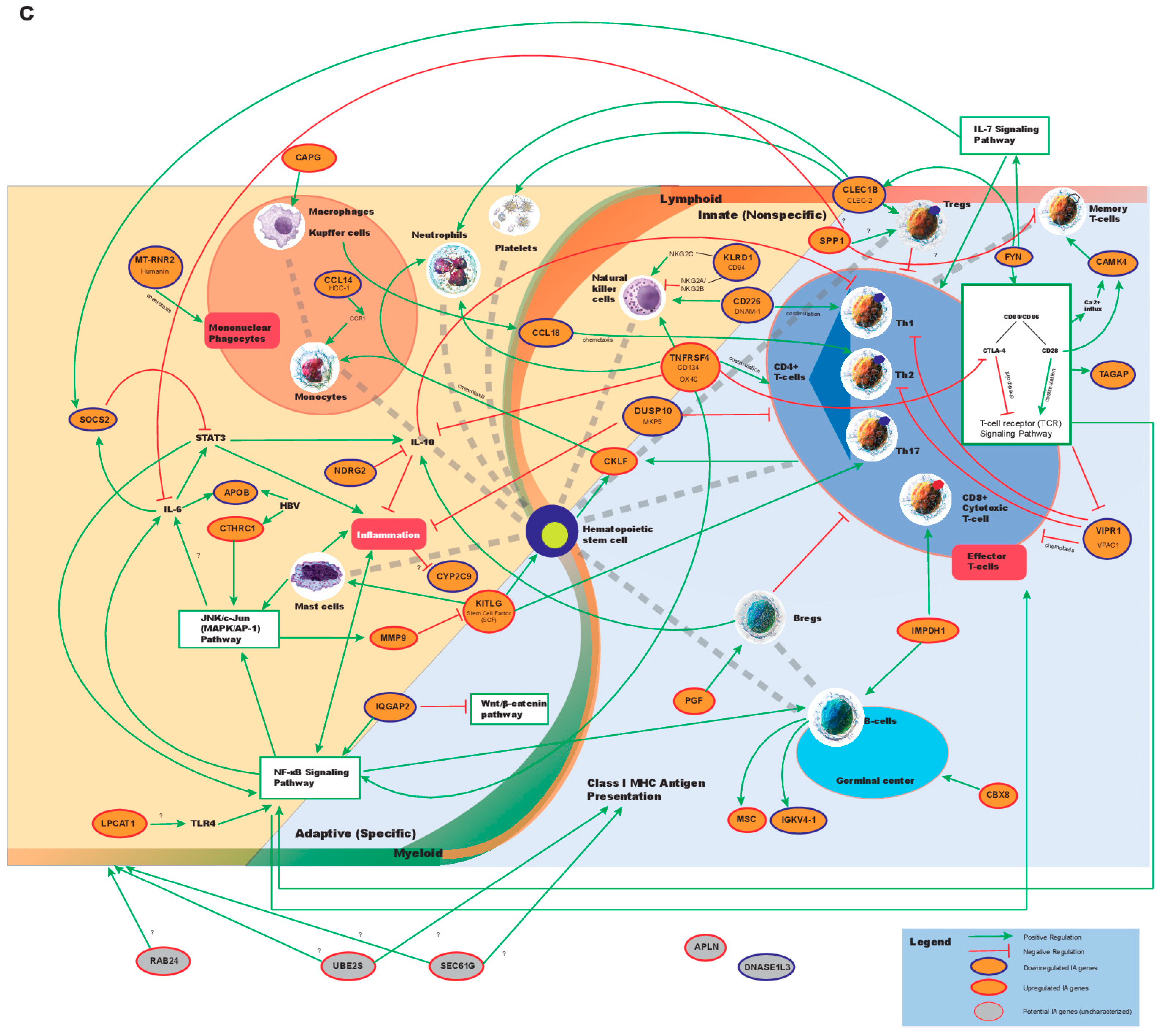
3. Discussion
4. Materials and Methods
4.1. RNA-Sequencing Datasets and Clinical Data
4.2. mRNA Differential Expression Analyses
4.3. Association of Candidate Genes’ Expressions with Patient Survival and Clinical Variables
4.4. Information-Coefficient Based Correlation of IA Gene Expression with Genomic Alterations
4.5. Identification of Differential Mutational Load in Immune-Associated Pathways
4.6. Assessing Potential Involvement of miRNAs in Regulating IA Genes
4.7. Validation of IA Gene Expression with Quantitative PCR (qPCR)
- CCL14 forward: AATACAGCTAAAGTTGGTGGGG
- CCL14 reverse: TCAAAGCAGGGAAGCTCCAA
- CKLF forward: GGCACTAACTGTGACATCTATGA
- CKLF reverse: TCACAAGTGCAAACACAAGCA
- IQGAP2 forward: TCAAGTGTAGGAAGGAGTTGTGG
- IQGAP2 reverse: CTGGATCTGGGGTGCTATTCC
- KITLG forward: TATGTCCCCGGGATGGATGT
- KITLG reverse: TTTGGCCTTCCCTTTCTCAGG
- NDRG2 forward: GGGACAGGGATGGAAAATGGT
- NDRG2 reverse: CCACATGAACCCGCACAAAG
- SEC61G forward: TTTAGGTGTCGGTTGGGTAGG
- SEC61G reverse: CTCACACCCTCACACTTGTTC
- SOCS2 forward: AGAGCCGGAGAGTCTGGTTT
- SOCS2 reverse: ATAGCGATCCTTGGCCCTTG
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Qu, Z.; Yuan, C.-H.; Yin, C.-Q.; Guan, Q.; Chen, H.; Wang, F.-B. Meta-analysis of the prognostic value of abnormally expressed lncRNAs in hepatocellular carcinoma. OncoTargets Ther. 2016, 9, 5143–5152. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, J.F.; Greten, T.F.; Raymond, E.; Roskams, T.; De, T.; Ducreux, M.; Mazzaferro, V.; Governing, E. Clinical Practice Guidelines EASL—EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma European Organisation for Research and Treatment of Cancer. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef]
- Asghar, U.; Meyer, T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012, 56, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, V.; Abdo, J.; Alsuwaidan, A.N.; Agrawal, S.; Sharma, P.; Agrawal, D.K. Cellular and molecular targets for the immunotherapy of hepatocellular carcinoma. Mol. Cell. Biochem. 2017, 437, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Fujii, S.; Mitsunaga, S.; Nobuoka, D.; Mizuno, S.; Takahashi, M.; Yamauchi, C.; Endo, I.; Nakatsura, T. Remarkable tumor lysis in a hepatocellular carcinoma patient immediately following glypican-3-derived peptide vaccination. Hum. Vaccines Immunother. 2013, 9, 1228–1233. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Zheng, H.; Zou, A.E.; Saad, M.A.; Wang, X.Q.; Kwok, J.G.; Korrapati, A.; Li, P.; Kisseleva, T.; Wang-Rodriguez, J.; Ongkeko, W.M. Alcohol-dysregulated microRNAs in hepatitis B virus-related hepatocellular carcinoma. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Shafritz, D.A.; Kew, M.C. Identification of integrated hepatitis B virus DNA sequences in human hepatocellular carcinomas. Hepatology 1981, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P.; Ameres, S.; Martinez, J.; Schroeder, R.; Anders, G.; Mackowiak, S.; Jens, M.; et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Chan, S.L.; Mok, T.; Ma, B.B.Y. Management of hepatocellular carcinoma: Beyond sorafenib. Curr. Oncol. Rep. 2012, 14, 257–266. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Melero, I.; Crocenzi, T.S.; Welling, T.H.; Yau, T.C.; Yeo, W.; Chopra, A.; Grosso, J.; Lang, L.; Anderson, J.; et al. Phase I/II safety and antitumor activity of nivolumab in patients with advanced hepatocellular carcinoma (HCC): CA209-040. J. Clin. Oncol. 2015, 33, LBA101. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017. [Google Scholar] [CrossRef] [PubMed]
- Gurtsevitch, V.E. Human oncogenic viruses: Hepatitis B and hepatitis C viruses and their role in hepatocarcinogenesis. Biochemistry 2008, 73, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127, S87–S96. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.W.; Chen, W.; Young, R.L.; Longmore, G.D.; Shaw, A.S. The conserved box 1 motif of cytokine receptors is required for association with JAK kinases. J. Biol. Chem. 1995, 270, 6523–6530. [Google Scholar] [CrossRef]
- Ghazawi, F.M.; Faller, E.M.; Parmar, P.; El-Salfiti, A.; MacPherson, P.A. Suppressor of cytokine signaling (SOCS) proteins are induced by IL-7 and target surface CD127 protein for degradation in human CD8 T cells. Cell. Immunol. 2016, 306–307, 41–52. [Google Scholar] [CrossRef]
- Carrette, F.; Surh, C.D. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin. Immunol. 2012, 24, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; So, T.; Croft, M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J. Immunol. 2008, 180, 7240–7248. [Google Scholar] [CrossRef]
- Redmond, W.L.; Linch, S.N.; Kasiewicz, M.J. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol. Res. 2014, 2, 142–153. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.J.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Gnoni, A.; Casadei Gardini, A.; Pisconti, S.; Licchetta, A.; Scartozzi, M.; Memeo, R.; Palmieri, V.O.; Aprile, G.; Santini, D.; et al. Immunotherapeutic approaches for hepatocellular carcinoma. Oncotarget 2017, 8, 33897–33910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hato, T.; Goyal, L.; Greten, T.F.; Duda, D.G.; Zhu, A.X. Immune checkpoint blockade in hepatocellular carcinoma: Current progress and future directions. Hepatology 2014, 60, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Severin, S.; Pollitt, A.Y.; Navarro-Nunez, L.; Nash, C.A.; Mourao-Sa, D.; Eble, J.A.; Senis, Y.A.; Watson, S.P. Syk-dependent phosphorylation of CLEC-2: A novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J. Biol. Chem. 2011, 286, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Bénézech, C.; Nayar, S.; Finney, B.A.; Withers, D.R.; Lowe, K.; Desanti, G.E.; Marriott, C.L.; Watson, S.P.; Caamaño, J.H.; Buckley, C.D.; et al. CLEC-2 is required for development and maintenance of lymph nodes. Blood 2014, 123, 3200–3207. [Google Scholar] [CrossRef] [Green Version]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Tarao, K.; Ohkawa, S.; Miyagi, Y.; Morinaga, S.; Ohshige, K.; Yamamoto, N.; Ueno, M.; Kobayashi, S.; Kameda, R.; Tamai, S.; et al. Inflammation in background cirrhosis evokes malignant progression in HCC development from HCV-associated liver cirrhosis. Scand. J. Gastroenterol. 2013, 48, 729–735. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-κB and STAT3-key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Pozo, D.; Anderson, P.; Gonzalez-Rey, E. Induction of alloantigen-specific human T regulatory cells by vasoactive intestinal peptide. J. Immunol. 2009, 183, 4346–4359. [Google Scholar] [CrossRef]
- Zhang, Y.; Blattman, J.N.; Kennedy, N.J.; Duong, J.; Nguyen, T.; Wang, Y.; Davis, R.J.; Greenberg, P.D.; Flavell, R.A.; Dong, C. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 2004, 430, 793–797. [Google Scholar] [CrossRef]
- Thuluvath, P.J. Vascular Invasion is the Most Important Predictor of Survival in HCC, but How Do We Find It? J. Clin. Gastroenterol. 2009, 43, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Zehner, M.; Marschall, A.L.; Bos, E.; Schloetel, J.G.; Kreer, C.; Fehrenschild, D.; Limmer, A.; Ossendorp, F.; Lang, T.; Koster, A.J.; et al. The Translocon Protein Sec61 Mediates Antigen Transport from Endosomes in the Cytosol for Cross-Presentation to CD8+ T Cells. Immunity 2015. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Fan, C.; Jiang, P.; Ma, Z.; Yan, X.; Di, S.; Jiang, S.; Li, T.; Cheng, Y.; Yang, Y. Emerging role of N-myc downstream-regulated gene 2 (NDRG2) in cancer. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Monti-Rocha, R.; Cramer, A.; Leite, P.G.; Antunes, M.M.; Pereira, R.V.S.; Barroso, A.; Queiroz-Junior, C.M.; David, B.A.; Teixeira, M.M.; Menezes, G.B.; et al. SOCS2 is critical for the balancing of immune response and oxidate stress protecting against acetaminophen-induced acute liver injury. Front. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brisac, C.; Salloum, S.; Yang, V.; Schaefer, E.A.K.; Holmes, J.A.; Chevaliez, S.; Hong, J.; Carlton-Smith, C.; Alatrakchi, N.; Kruger, A.; et al. IQGAP2 is a novel interferon-alpha antiviral effector gene acting non-conventionally through the NF-κB pathway. J. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Botvinnik, O.B.; Abudayyeh, O.; Birger, C.; Rosenbluh, J.; Shrestha, Y.; Abazeed, M.E.; Hammerman, P.S.; DiCara, D.; Konieczkowski, D.J.; et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat. Biotechnol. 2016, 34, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Raspe, E.; Decraene, C.; Berx, G. Gene expression profiling to dissect the complexity of cancer biology: Pitfalls and promise. Semin. Cancer Biol. 2012, 22, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.J.; Chau, J.; Ebert, P.J.R.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. miR-181a Is an Intrinsic Modulator of T Cell Sensitivity and Selection. Cell 2007, 129, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.; Jia, H.; Tai, Q.; Li, Y.; Chen, D. miR-106b downregulates adenomatous polyposis coli and promotes cell proliferation in human hepatocellular carcinoma. Carcinogenesis 2013, 34, 211–219. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, L.; Ding, Y.; Li, Q.; Wang, R.; Liu, T.; Sun, Q.; Yang, H.; Peng, S.; Wang, W.; et al. MiR-20a Induces Cell Radioresistance by Activating the PTEN/PI3K/Akt Signaling Pathway in Hepatocellular Carcinoma. Int. J. Radiat. Oncol. 2015, 92, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Hoshino, H.; Wang, J.; Ono, S.; Iida, Y.; Hata, K.; Huang, S.K.; Colquhoun, S.; Hoon, D.S.B. MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget 2015, 6, 3211–3224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jiang, W.; Wu, L.; Chang, R.; Wu, K.; Wang, Z. miR-301a Is a Candidate Oncogene that Targets the Homeobox Gene Gax in Human Hepatocellular Carcinoma. Dig. Dis. Sci. 2012, 57, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Liang, L.; Huang, S.; Ding, J.; Tan, N.; Zhao, Y.; Yan, M.; Ge, C.; Zhang, Z.; Chen, T.; et al. MicroRNA-30d promotes tumor invasion and metastasis by targeting Galphai2 in hepatocellular carcinoma. Hepatology 2010, 51, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Katz, G.; Pobezinsky, L.A.; Jeurling, S.; Shinzawa, M.; Van Laethem, F.; Singer, A. T cell receptor stimulation impairs IL-7 receptor signaling by inducing expression of the microRNA miR-17 to target Janus kinase 1. Sci. Signal. 2014, 7, ra83. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCC Drinkers without HBV vs. Nondrinker Normals | HCC Drinkers without HBV vs. Drinker Normals | ||||||
| Gene Name | Fold Change | FDR | p-Value | Gene Name | Fold Change | FDR | p-Value |
| APOB | −2.16 | 9.0 × 10−13 | 7.0 × 10−10 | APLN | 16.65 | 1.7 × 10−6 | 3.5 × 10−4 |
| CAMK4 | −4.12 | 7.2 × 10−16 | 3.1 × 10−13 | CAMK4 | −5.90 | 2.6 × 10−8 | 1.5 × 10−6 |
| CAPG | 3.73 | 4.0 × 10−10 | 5.5 × 10−7 | CLEC1B | −22.88 | 6.8 × 10−5 | 3.7 × 10−2 |
| CBX8 | 2.73 | 1.9 × 10−15 | 9.0 × 10−13 | DNASE1L3 | −6.63 | 1.4 × 10−5 | 5.2 × 10−3 |
| CCL14 | −2.93 | 2.2 × 10−7 | 5.4 × 10−4 | SOCS2 | −7.10 | 2.0 × 10−9 | 6.9 × 10−8 |
| CD226 | −3.03 | 8.9 × 10−9 | 1.6 × 10−5 | UBE2S | 5.93 | 1.4 × 10−5 | 5.2 × 10−3 |
| CKLF | 2.71 | 1.1 × 10−11 | 1.1 × 10−8 | VIPR1 | −12.77 | 5.2 × 10−12 | 6.3 × 10−11 |
| CLEC1B | −26.29 | 7.3 × 10−12 | 6.9 × 10−9 | ||||
| CYP2C9 | −3.34 | 7.8 × 10−8 | 1.7 × 10−4 | ||||
| DNASE1L3 | −6.62 | 2.2 × 10−13 | 1.6 × 10−10 | ||||
| IGKV4-1 | −5.33 | 1.7 × 10−6 | 4.9 × 10−3 | ||||
| IMPDH1 | 2.08 | 3.6 × 10−6 | 1.2 × 10−2 | ||||
| IQGAP2 | −1.97 | 1.3 × 10−9 | 1.9 × 10−6 | ||||
| KITLG | 2.14 | 4.6 × 10−6 | 1.5 × 10−2 | ||||
| KLRD1 | −2.54 | 7.3 × 10−7 | 2.0 × 10−3 | ||||
| LPCAT1 | 3.07 | 1.2 × 10−10 | 1.4 × 10−7 | ||||
| MSC | 6.00 | 2.1 × 10−10 | 2.6 × 10−7 | ||||
| NDRG2 | −2.46 | 1.2 × 10−9 | 1.7 × 10−6 | ||||
| PGF | 2.06 | 5.2 × 10−6 | 1.7 × 10−2 | ||||
| RAB24 | 2.31 | 9.6 × 10−15 | 5.1 × 10−12 | ||||
| SEC61G | 2.14 | 5.1 × 10−9 | 8.7 × 10−6 | ||||
| SOCS2 | −6.45 | 5.3 × 10−19 | 1.3 × 10−16 | ||||
| SPP1 | 25.97 | 8.7 × 10−22 | 1.3 × 10−19 | ||||
| TAGAP | −2.49 | 9.6 × 10−6 | 3.3 × 10−2 | ||||
| UBE2S | 5.14 | 2.2 × 10−21 | 3.7 × 10−19 | ||||
| VIPR1 | −10.80 | 6.2 × 10−27 | 4.0 × 10−25 | ||||
| HCC Drinkers with HBV vs. Drinker Normals | HCC Drinkers vs. HCC Nondrinkers | ||||||
| Gene Name | Fold Change | FDR | p-Value | Gene Name | Fold Change | FDR | p-Value |
| APLN | 18.60 | 2.6 × 10−6 | 5.0 × 10−4 | CCL18 | 1.72 | 1.0 × 10−2 | 2.6 × 10−2 |
| CAMK4 | −3.83 | 4.4 × 10−5 | 1.7 × 10−2 | CKLF | 1.36 | 3.5 × 10−3 | 5.1 × 10−3 |
| CLEC1B | −28.26 | 7.9 × 10−8 | 5.2 × 10−6 | MMP9 | 1.88 | 5.3 × 10−3 | 1.0 × 10−2 |
| DNASE1L3 | −5.45 | 9.6 × 10−6 | 2.6 × 10−3 | ||||
| NDRG2 | −2.97 | 3.9 × 10−7 | 4.2 × 10−5 | ||||
| UBE2S | 4.85 | 1.0 × 10−4 | 4.8 × 10−2 | ||||
| VIPR1 | −8.65 | 2.1 × 10−7 | 1.9 × 10−5 | ||||
| HCC Drinkers with HBV vs. Nondrinker Normals | HCC Nondrinkers with HBV vs. Nondrinker Normals | ||||||
| Gene Name | Fold Change | FDR | p-Value | Gene Name | Fold Change | FDR | p-Value |
| CAMK4 | −2.71 | 5.7 × 10−9 | 8.0 × 10−6 | APLN | 13.78 | 1.2 × 10−33 | 1.6 × 10−31 |
| CAPG | 3.47 | 3.1 × 10−9 | 4.2 × 10−6 | CAMK4 | −2.89 | 9.0 × 10−13 | 1.3 × 10−9 |
| CBX8 | 2.23 | 3.6 × 10−12 | 2.5 × 10−9 | CAPG | 3.30 | 1.1 × 10−9 | 2.7 × 10−6 |
| CCL14 | −3.01 | 2.8 × 10−9 | 3.7 × 10−6 | CBX8 | 2.14 | 7.7 × 10−14 | 9.9 × 10−11 |
| CD226 | −2.39 | 1.0 × 10−6 | 2.4 × 10−3 | CCL14 | −3.76 | 4.7 × 10−20 | 2.7 × 10−17 |
| CKLF | 3.26 | 6.9 × 10−14 | 3.3 × 10−11 | CD226 | −2.82 | 1.1 × 10−11 | 1.9 × 10−8 |
| CLEC1B | −32.63 | 7.6 × 10−20 | 1.1 × 10−17 | CLEC1B | −76.69 | 7.5 × 10−45 | 3.9 × 10−43 |
| CYP2C9 | −3.38 | 2.7 × 10−7 | 5.7 × 10−4 | CTHRC1 | 13.81 | 3.5 × 10−19 | 2.2 × 10−16 |
| DNASE1L3 | −5.40 | 4.4 × 10−14 | 2.0 × 10−11 | CYP2C9 | −3.88 | 3.3 × 10−12 | 5.3 × 10−9 |
| DUSP10 | −2.15 | 7.5 × 10−8 | 1.4 × 10−4 | DNASE1L3 | −7.82 | 1.7 × 10−37 | 1.6 × 10−35 |
| KITLG | 2.97 | 1.2 × 10−11 | 9.7 × 10−9 | DUSP10 | −2.35 | 5.0 × 10−14 | 6.2 × 10−11 |
| KLRD1 | −2.73 | 1.0 × 10−9 | 1.2 × 10−6 | FYN | −2.03 | 1.5 × 10−12 | 2.3 × 10−9 |
| LPCAT1 | 2.76 | 2.1 × 10−9 | 2.6 × 10−6 | KITLG | 2.39 | 7.4 × 10−8 | 2.3 × 10−4 |
| MSC | 7.90 | 2.4 × 10−11 | 2.1 × 10−8 | KLRD1 | −2.82 | 1.3 × 10−11 | 2.2 × 10−8 |
| NDRG2 | −2.79 | 4.8 × 10−19 | 8.3 × 10−17 | LPCAT1 | 2.16 | 7.6 × 10−8 | 2.4 × 10−4 |
| RAB24 | 2.26 | 5.0 × 10−14 | 2.3 × 10−11 | MSC | 6.65 | 8.2 × 10−9 | 2.2 × 10−5 |
| SOCS2 | −3.70 | 1.6 × 10−9 | 2.0 × 10−6 | MT-RNR2 | −2.21 | 1.5 × 10−11 | 2.7 × 10−8 |
| SPP1 | 15.71 | 6.9 × 10−15 | 2.7 × 10−12 | NDRG2 | −2.81 | 7.8 × 10−23 | 3.2 × 10−20 |
| UBE2S | 4.06 | 2.2 × 10−17 | 5.5 × 10−15 | PGF | 3.09 | 5.0 × 10−8 | 1.5 × 10−4 |
| VIPR1 | −7.31 | 1.2 × 10−16 | 3.5 × 10−14 | RAB24 | 2.19 | 1.3 × 10−17 | 1.0 × 10−14 |
| SOCS2 | −3.93 | 3.9 × 10−16 | 3.7 × 10−13 | ||||
| TNFRSF4 | 5.61 | 9.0 × 10−27 | 2.3 × 10−24 | ||||
| UBE2S | 3.67 | 6.8 × 10−20 | 4.0 × 10−17 | ||||
| VIPR1 | −13.83 | 9.5 × 10−55 | 1.3 × 10−53 | ||||
| Comparison: HCC Drinkers vs. HCC Nondrinkers | ||
|---|---|---|
| Pathway Name | Odds Ratio | p-Value |
| B-cell receptor pathway | 2.735 | 0.004 |
| T-cell receptor pathway | 1.901 | 0.029 |
| Classical complement system activation pathway | 2.184 | 0.047 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.T.; Zou, A.E.; Honda, C.O.; Zheng, H.; Wang, X.Q.; Kisseleva, T.; Chang, E.Y.; Ongkeko, W.M. Etiology-Specific Analysis of Hepatocellular Carcinoma Transcriptome Reveals Genetic Dysregulation in Pathways Implicated in Immunotherapy Efficacy. Cancers 2019, 11, 1273. https://doi.org/10.3390/cancers11091273
Li WT, Zou AE, Honda CO, Zheng H, Wang XQ, Kisseleva T, Chang EY, Ongkeko WM. Etiology-Specific Analysis of Hepatocellular Carcinoma Transcriptome Reveals Genetic Dysregulation in Pathways Implicated in Immunotherapy Efficacy. Cancers. 2019; 11(9):1273. https://doi.org/10.3390/cancers11091273
Chicago/Turabian StyleLi, Wei Tse, Angela E. Zou, Christine O. Honda, Hao Zheng, Xiao Qi Wang, Tatiana Kisseleva, Eric Y. Chang, and Weg M. Ongkeko. 2019. "Etiology-Specific Analysis of Hepatocellular Carcinoma Transcriptome Reveals Genetic Dysregulation in Pathways Implicated in Immunotherapy Efficacy" Cancers 11, no. 9: 1273. https://doi.org/10.3390/cancers11091273
APA StyleLi, W. T., Zou, A. E., Honda, C. O., Zheng, H., Wang, X. Q., Kisseleva, T., Chang, E. Y., & Ongkeko, W. M. (2019). Etiology-Specific Analysis of Hepatocellular Carcinoma Transcriptome Reveals Genetic Dysregulation in Pathways Implicated in Immunotherapy Efficacy. Cancers, 11(9), 1273. https://doi.org/10.3390/cancers11091273