Progesterone Receptor B signaling Reduces Breast Cancer Cell Aggressiveness: Role of Cyclin-D1/Cdk4 Mediating Paxillin Phosphorylation



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
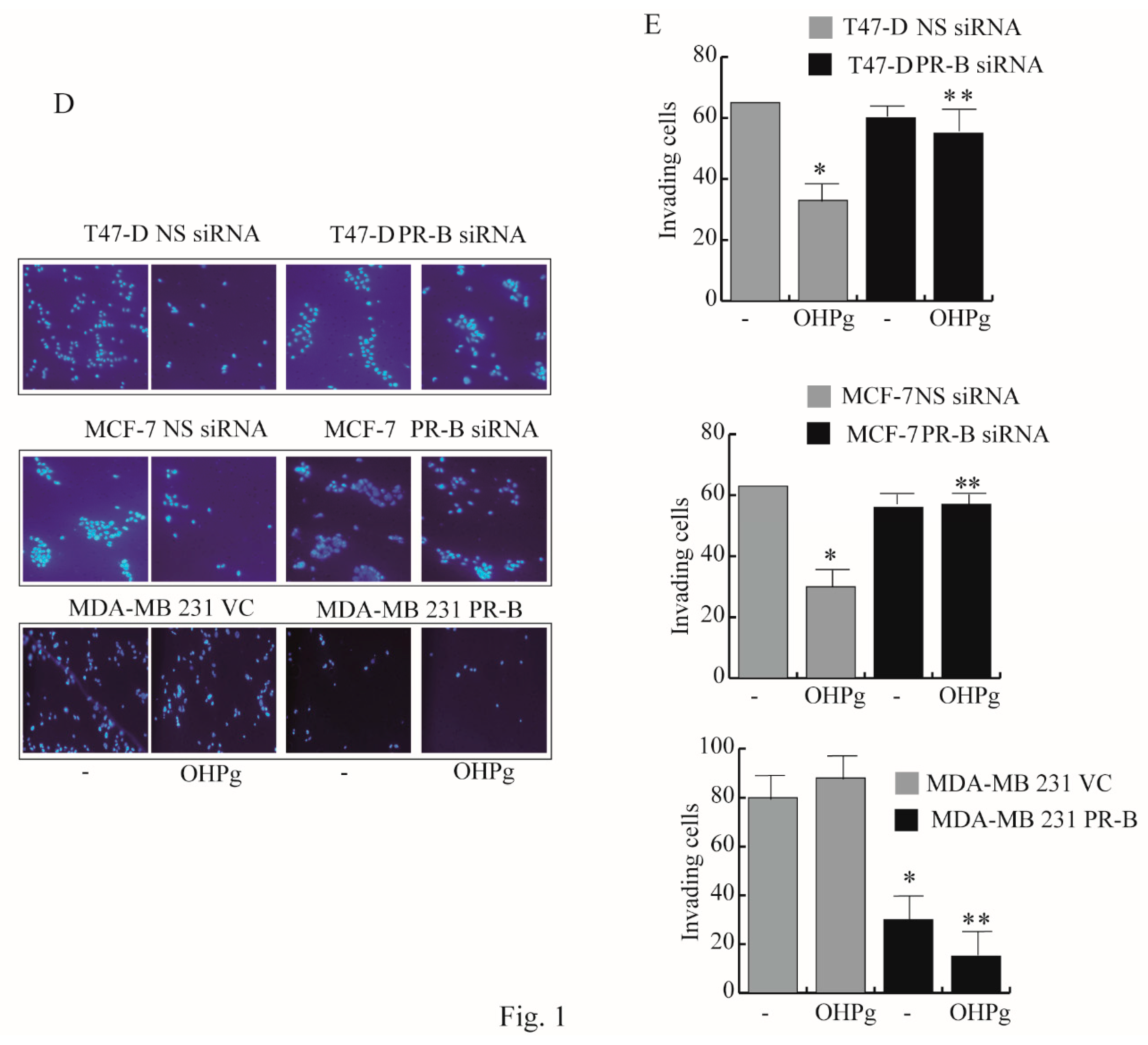
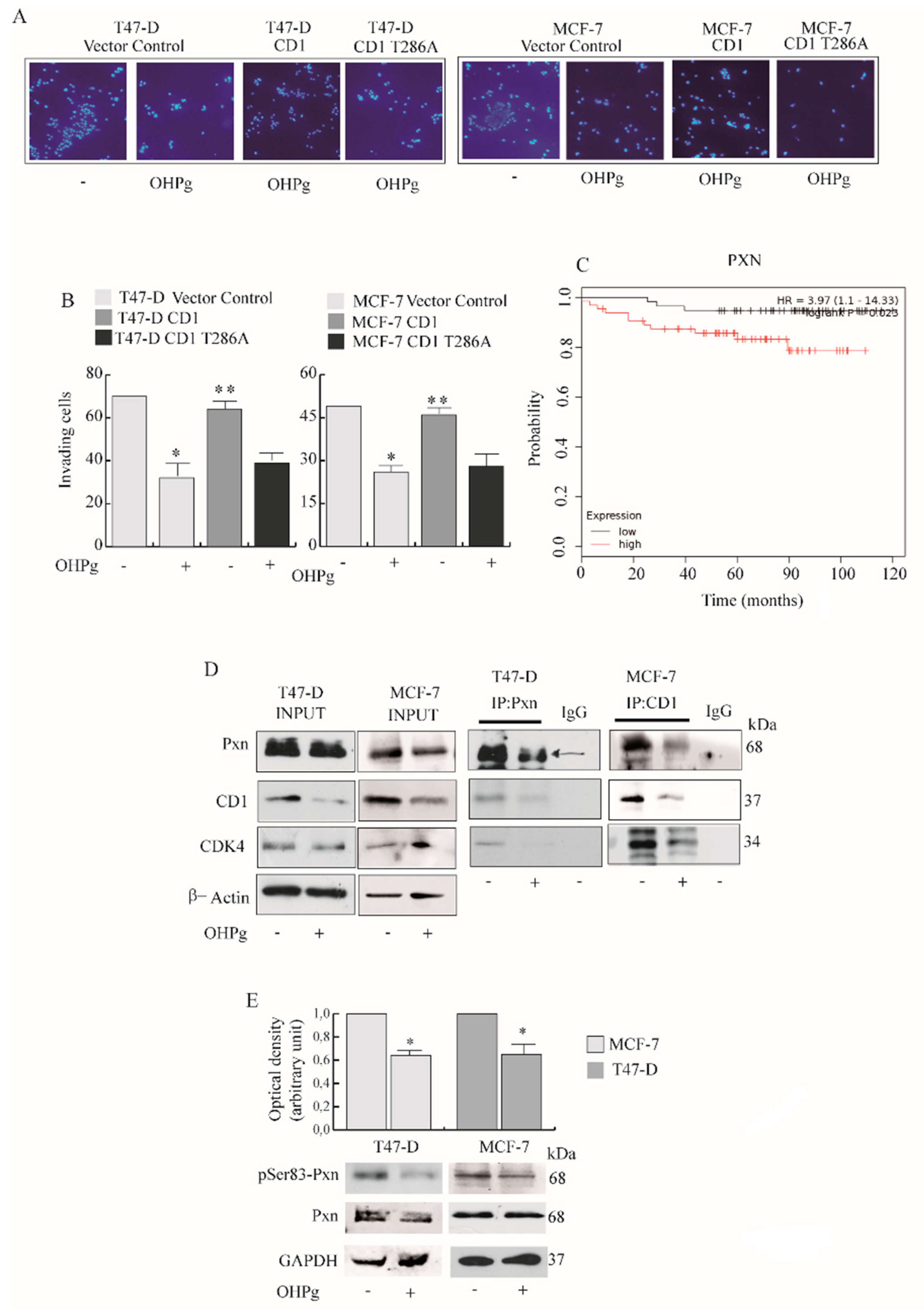
2.1. OHPg/PR-B Impair Aggressiveness of Breast Cancer Cells
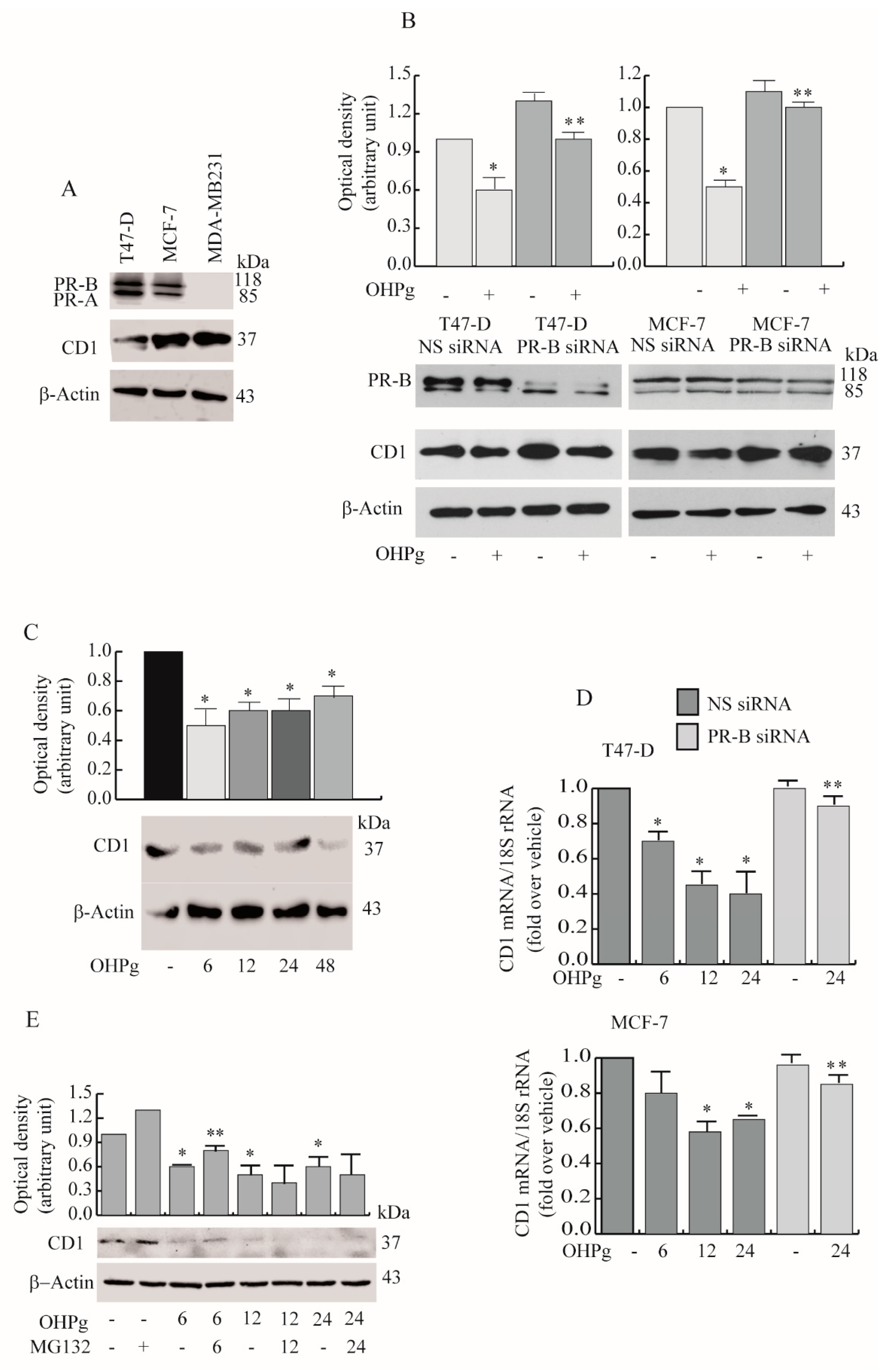
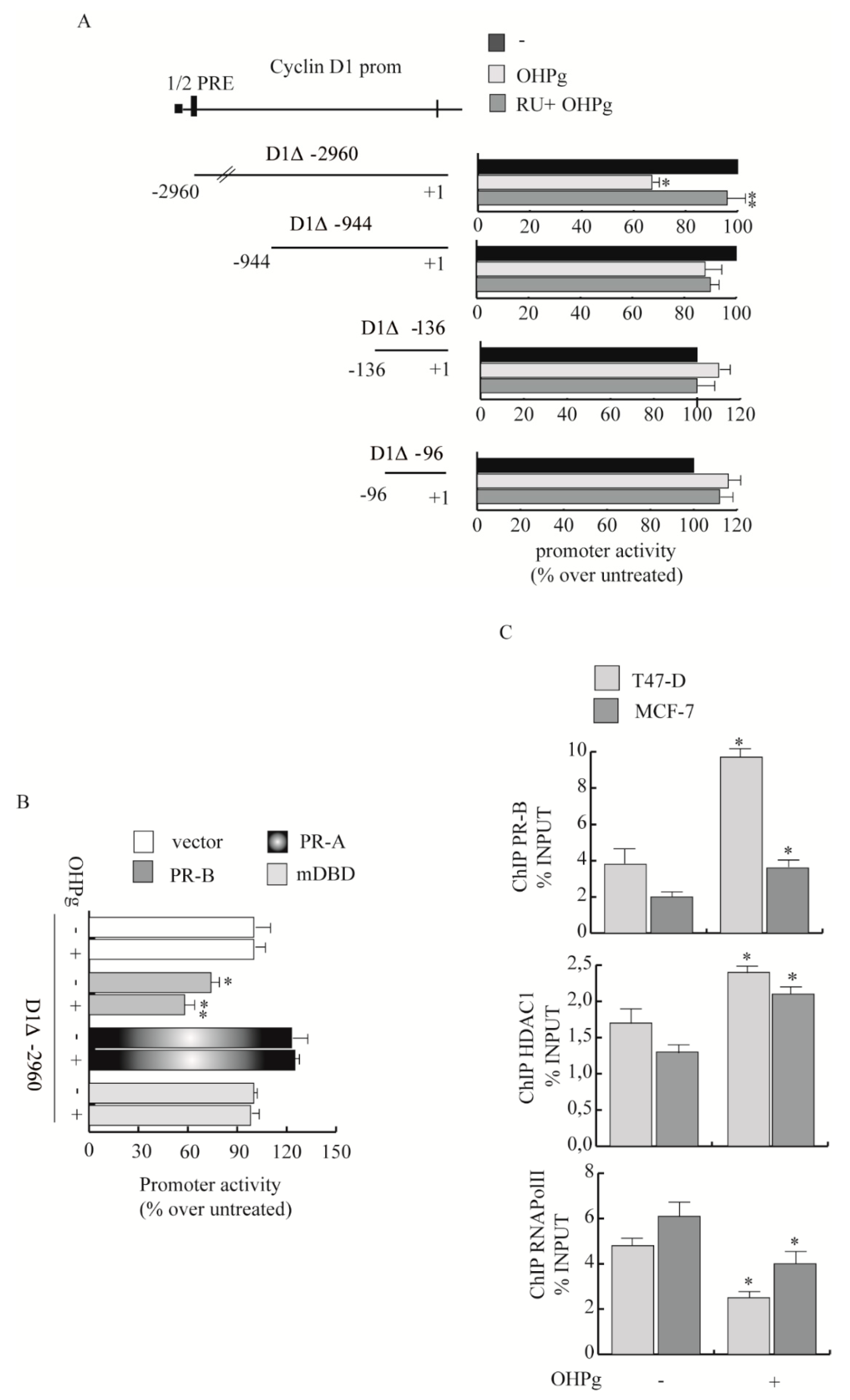
2.2. OHPg Decreases CD1 Expression Levels Through a Genomic Mechanism
2.3. OHPg Reduces the Interaction between CD1/Cdk4 and Paxillin in Vitro
2.4. OHPg/PR-B Impairs Breast Cancer Cell Migration through Pxn Phosphorylation Status
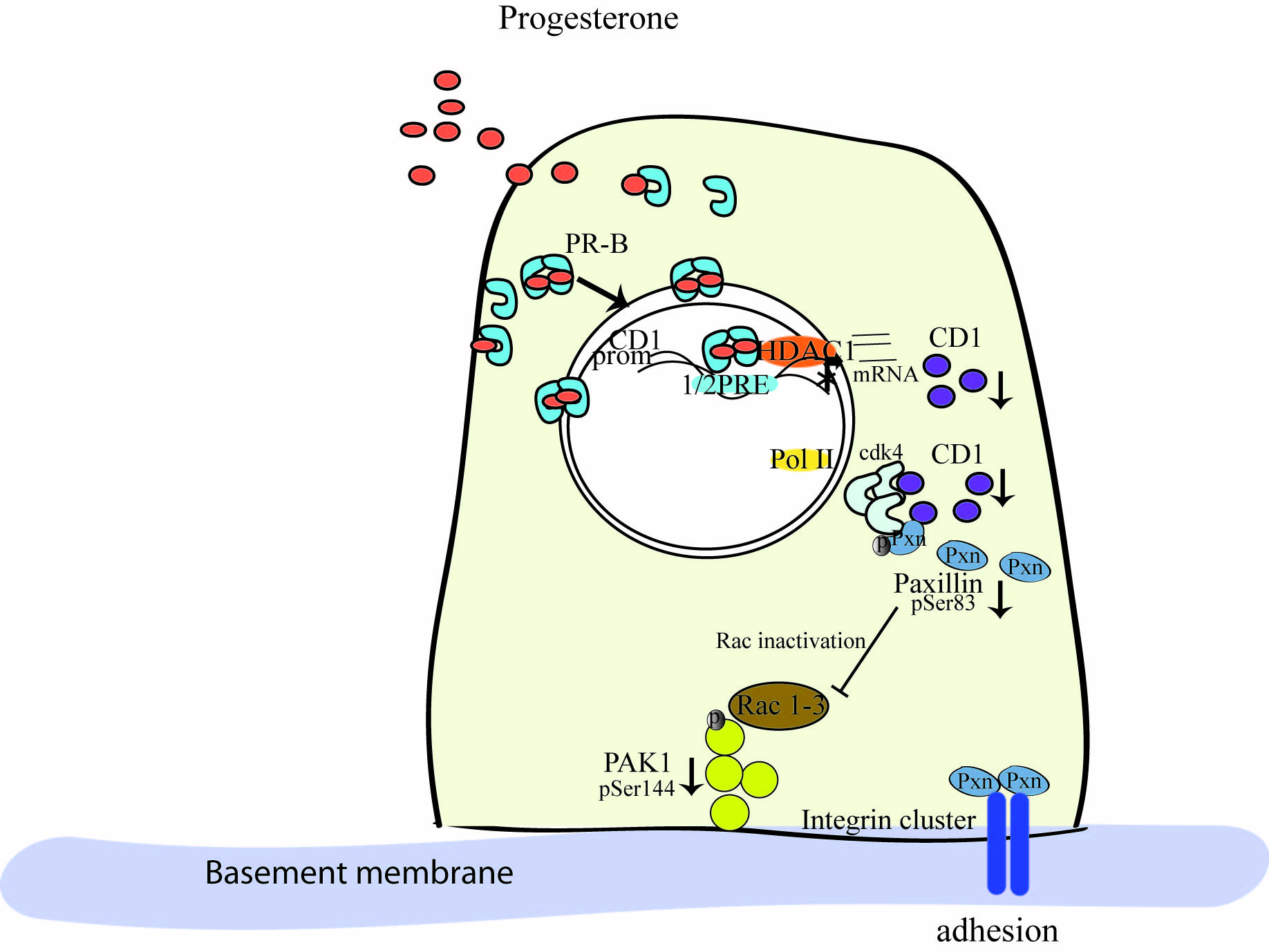
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Plasmids
4.3. Cell Culture
4.4. Total RNA Extraction, Reverse Transcription PCR and Real-Time RT-PCR Assay
4.5. Immunoprecipitation and Western Blot
4.6. Transient Transfection and Luciferase Assays
4.7. Lipid-Mediated Transfection of siRNA Duplexes
4.8. Chromatin Immunoprecipitation (ChIP) Assays and Realtime ChIP
4.9. Wound-Healing Assays
4.10. Transmigration Assays
4.11. Invasion Assays
4.12. Phalloidin Staining
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Velloso, F.J.; Bianco, A.F.; Farias, J.O.; Torres, N.E.; Ferruzo, P.Y.; Anschau, V.; Jesus-Ferreira, H.C.; Chang, T.H.; Sogayar, M.C.; Zerbini, L.F.; et al. The crossroads of breast cancer progression: Insights into the modulation of major signaling pathways. Oncol. Targets Ther. 2017, 10, 5491–5524. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, K.; Amini, R.M.; Landberg, G.; Eerola, H.; Aittomaki, K.; Heikkila, P.; Nevanlinna, H.; Blomqvist, C. Cyclin D1 expression is associated with poor prognostic features in estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2009, 113, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Drobnjak, M.; Osman, I.; Scher, H.I.; Fazzari, M.; Cordon-Cardo, C. Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin. Cancer Res. 2000, 6, 1891–1895. [Google Scholar] [PubMed]
- Arnold, A.; Papanikolaou, A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. 2005, 23, 4215–4224. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Ladha, M.H.; McMahon, C.; Sutherland, R.L.; Ewen, M.E. Regulation of the functional interaction between cyclin D1 and the estrogen receptor. Mol. Cell. Biol. 2000, 20, 8667–8675. [Google Scholar] [CrossRef]
- Zwijsen, R.M.; Wientjens, E.; Klompmaker, R.; van der Sman, J.; Bernards, R.; Michalides, R.J. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997, 88, 405–415. [Google Scholar] [CrossRef]
- Mauro, L.; Pellegrino, M.; Giordano, F.; Ricchio, E.; Rizza, P.; De Amicis, F.; Catalano, S.; Bonofiglio, D.; Panno, M.L.; Ando, S. Estrogen receptor-alpha drives adiponectin effects on cyclin D1 expression in breast cancer cells. FASEB J. 2015, 29, 2150–2160. [Google Scholar] [CrossRef]
- Pestell, R.G. New roles of cyclin D1. Am. J. Pathol. 2013, 183, 3–9. [Google Scholar] [CrossRef]
- Body, S.; Esteve-Arenys, A.; Miloudi, H.; Recasens-Zorzo, C.; Tchakarska, G.; Moros, A.; Bustany, S.; Vidal-Crespo, A.; Rodriguez, V.; Lavigne, R.; et al. Cytoplasmic cyclin D1 controls the migration and invasiveness of mantle lymphoma cells. Sci. Rep. 2017, 7, 13946. [Google Scholar] [CrossRef]
- Fernandez, R.M.; Ruiz-Miro, M.; Dolcet, X.; Aldea, M.; Gari, E. Cyclin D1 interacts and collaborates with Ral GTPases enhancing cell detachment and motility. Oncogene 2011, 30, 1936–1946. [Google Scholar] [CrossRef]
- Zhong, Z.; Yeow, W.S.; Zou, C.; Wassell, R.; Wang, C.; Pestell, R.G.; Quong, J.N.; Quong, A.A. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res. 2010, 70, 2105–2114. [Google Scholar] [CrossRef]
- Fuste, N.P.; Fernandez-Hernandez, R.; Cemeli, T.; Mirantes, C.; Pedraza, N.; Rafel, M.; Torres-Rosell, J.; Colomina, N.; Ferrezuelo, F.; Dolcet, X.; et al. Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat. Commun. 2016, 7, 11581. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Lin, V.C.; Eng, A.S.; Hen, N.E.; Ng, E.H.; Chowdhury, S.H. Effect of progesterone on the invasive properties and tumor growth of progesterone receptor-transfected breast cancer cells MDA-MB-231. Clin. Cancer Res. 2001, 7, 2880–2886. [Google Scholar]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature 2015, 5, 313–317. [Google Scholar] [CrossRef]
- Pichon, M.F.; Pallud, C.; Brunet, M.; Milgrom, E. Relationship of presence of progesterone receptors to prognosis in early breast cancer. Cancer Res. 1980, 40, 3357–3360. [Google Scholar]
- Creighton, C.J.; Kent Osborne, C.; van de Vijver, M.J.; Foekens, J.A.; Klijn, J.G.; Horlings, H.M.; Nuyten, D.; Wang, Y.; Zhang, Y.; Chamness, G.C.; et al. Molecular profiles of progesterone receptor loss in human breast tumors. Breast Cancer Res. Treat. 2009, 114, 287–299. [Google Scholar] [CrossRef]
- Bardou, V.J.; Arpino, G.; Elledge, R.M.; Osborne, C.K.; Clark, G.M. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J. Clin. Oncol. 2003, 21, 1973–1979. [Google Scholar] [CrossRef]
- Kurebayashi, J.; Kanomata, N.; Shimo, T.; Yamashita, T.; Aogi, K.; Nishimura, R.; Shimizu, C.; Tsuda, H.; Moriya, T.; Sonoo, H. Marked lymphovascular invasion, progesterone receptor negativity, and high Ki67 labeling index predict poor outcome in breast cancer patients treated with endocrine therapy alone. Breast Cancer 2014, 21, 214–222. [Google Scholar] [CrossRef]
- Knutson, T.P.; Daniel, A.R.; Fan, D.; Silverstein, K.A.; Covington, K.R.; Fuqua, S.A.; Lange, C.A. Phosphorylated and sumoylation-deficient progesterone receptors drive proliferative gene signatures during breast cancer progression. Breast Cancer Res. 2012, 14, R95. [Google Scholar] [CrossRef]
- Wang, H.C.; Lee, W.S. Molecular mechanisms underlying progesterone-enhanced breast cancer cell migration. Sci. Rep. 2016, 6, 31509. [Google Scholar] [CrossRef]
- Godbole, M.; Tiwary, K.; Badwe, R.; Gupta, S.; Dutt, A. Progesterone suppresses the invasion and migration of breast cancer cells irrespective of their progesterone receptor status-a short report. Cell. Oncol. 2017, 40, 411–417. [Google Scholar] [CrossRef]
- Kariagina, A.; Xie, J.; Langohr, I.M.; Opreanu, R.C.; Basson, M.D.; Haslam, S.Z. Progesterone decreases levels of the adhesion protein E-cadherin and promotes invasiveness of steroid receptor positive breast cancers. Horm. Cancer 2013, 4, 371–380. [Google Scholar] [CrossRef]
- Holley, A.K.; Kiningham, K.K.; Spitz, D.R.; Edwards, D.P.; Jenkins, J.T.; Moore, M.R. Progestin stimulation of manganese superoxide dismutase and invasive properties in T47D human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2009, 117, 23–30. [Google Scholar] [CrossRef]
- Castronovo, V.; Taraboletti, G.; Liotta, L.A.; Sobel, M.E. Modulation of laminin receptor expression by estrogen and progestins in human breast cancer cell lines. J. Natl. Cancer Inst. 1989, 81, 781–788. [Google Scholar] [CrossRef]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef]
- Velasco-Velazquez, M.A.; Li, Z.; Casimiro, M.; Loro, E.; Homsi, N.; Pestell, R.G. Examining the role of cyclin D1 in breast cancer. Future Oncol. 2011, 7, 753–765. [Google Scholar] [CrossRef]
- Shortle, B.; Dyrenfurth, I.; Ferin, M. Effects of an antiprogesterone agent, RU-486, on the menstrual cycle of the rhesus monkey. J. Clin. Endocrinol. Metab. 1985, 60, 731–735. [Google Scholar] [CrossRef]
- Deakin, N.O.; Turner, C.E. Paxillin comes of age. J. Cell Sci. 2008, 121, 2435–2444. [Google Scholar] [CrossRef]
- Brown, M.C.; Turner, C.E. Paxillin: Adapting to change. Physiol. Rev. 2004, 84, 1315–1339. [Google Scholar] [CrossRef]
- Montalto, F.I.; De Amicis, F. University of Calabria; unpublished data. 2019.
- Tang, K.; Boudreau, C.G.; Brown, C.M.; Khadra, A. Paxillin phosphorylation at serine 273 and its effects on Rac, Rho and adhesion dynamics. PLoS Comput. Biol. 2018, 14, 1006303. [Google Scholar] [CrossRef]
- Wells, C.M.; Walmsley, M.; Ooi, S.; Tybulewicz, V.; Ridley, A.J. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J. Cell Sci. 2004, 117, 1259–1268. [Google Scholar] [CrossRef]
- Chong, C.; Tan, L.; Lim, L.; Manser, E. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 2001, 276, 17347–17353. [Google Scholar] [CrossRef]
- Moshfegh, Y.; Bravo-Cordero, J.J.; Miskolci, V.; Condeelis, J.; Hodgson, L. A Trio-Rac1-Pak1 signalling axis drives invadopodia disassembly. Nat. Cell Biol. 2014, 16, 574–586. [Google Scholar] [CrossRef]
- McFall, T.; McKnight, B.; Rosati, R.; Kim, S.; Huang, Y.; Viola-Villegas, N.; Ratnam, M. Role of the short isoform of the progesterone receptor in breast cancer cell invasiveness at estrogen and progesterone levels in the pre- and post-menopausal ranges. Oncotarget 2015, 6, 33146–33164. [Google Scholar] [CrossRef]
- McFall, T.; McKnight, B.; Rosati, R.; Kim, S.; Huang, Y.; Viola-Villegas, N.; Ratnam, M. Progesterone receptor A promotes invasiveness and metastasis of luminal breast cancer by suppressing regulation of critical microRNAs by estrogen. J. Biol. Chem. 2018, 293, 1163–1177. [Google Scholar] [CrossRef]
- Lin, V.C.; Ng, E.H.; Aw, S.E.; Tan, M.G.; Ng, E.H.; Bay, B.H. Progesterone induces focal adhesion in breast cancer cells MDA-MB-231 transfected with progesterone receptor complementary DNA. Mol. Endocrinol. 2000, 14, 348–358. [Google Scholar] [CrossRef]
- Van Mackelenbergh, M.T.; Denkert, C.; Nekljudova, V.; Karn, T.; Schem, C.; Marme, F.; Stickeler, E.; Jackisch, C.; Hanusch, C.; Huober, J.; et al. Outcome after neoadjuvant chemotherapy in estrogen receptor-positive and progesterone receptor-negative breast cancer patients: A pooled analysis of individual patient data from ten prospectively randomized controlled neoadjuvant trials. Breast Cancer Res. Treat. 2018, 167, 59–71. [Google Scholar] [CrossRef]
- Ono, M.; Tsuda, H.; Yoshida, M.; Shimizu, C.; Kinoshita, T.; Tamura, K. Prognostic Significance of Progesterone Receptor Expression in Estrogen-Receptor Positive, HER2-Negative, Node-Negative Invasive Breast Cancer With a Low Ki-67 Labeling Index. Clin. Breast Cancer 2017, 17, 41–47. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef]
- Pang, H.; Rowan, B.G.; Al-Dhaheri, M.; Faber, L.E. Epidermal growth factor suppresses induction by progestin of the adhesion protein desmoplakin in T47D breast cancer cells. Breast Cancer Res. 2004, 6, 239–245. [Google Scholar] [CrossRef]
- Shyamala, G.; Yang, X.; Silberstein, G.; Barcellos-Hoff, M.H.; Dale, E. Transgenic mice carrying an imbalance in the native ratio of A to B forms of progesterone receptor exhibit developmental abnormalities in mammary glands. Proc. Natl. Acad. Sci. USA 1998, 95, 696–701. [Google Scholar] [CrossRef]
- Denner, L.A.; Weigel, N.L.; Maxwell, B.L.; Schrader, W.T.; O’Malley, B.W. Regulation of progesterone receptor-mediated transcription by phosphorylation. Science 1990, 250, 1740–1743. [Google Scholar] [CrossRef]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef]
- Radu, A.; Neubauer, V.; Akagi, T.; Hanafusa, H.; Georgescu, M.M. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol. Cell. Biol. 2003, 23, 6139–6149. [Google Scholar] [CrossRef]
- De Amicis, F.; Guido, C.; Santoro, M.; Lanzino, M.; Panza, S.; Avena, P.; Panno, M.L.; Perrotta, I.; Aquila, S.; Ando, S. A novel functional interplay between Progesterone Receptor-B and PTEN, via AKT, modulates autophagy in breast cancer cells. J. Cell. Mol. Med. 2014, 18, 2252–2265. [Google Scholar] [CrossRef]
- Shore, A.N.; Chang, C.H.; Kwon, O.J.; Weston, M.C.; Zhang, M.; Xin, L.; Rosen, J.M. PTEN is required to maintain luminal epithelial homeostasis and integrity in the adult mammary gland. Dev. Biol. 2016, 409, 202–217. [Google Scholar] [CrossRef]
- Aggarwal, P.; Vaites, L.P.; Kim, J.K.; Mellert, H.; Gurung, B.; Nakagawa, H.; Herlyn, M.; Hua, X.; Rustgi, A.K.; McMahon, S.B.; et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010, 18, 329–340. [Google Scholar] [CrossRef]
- Lazarov, M.; Kubo, Y.; Cai, T.; Dajee, M.; Tarutani, M.; Lin, Q.; Fang, M.; Tao, S.; Green, C.L.; Khavari, P.A. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat. Med. 2002, 8, 1105–1114. [Google Scholar] [CrossRef]
- De Amicis, F.; Zupo, S.; Panno, M.L.; Malivindi, R.; Giordano, F.; Barone, I.; Mauro, L.; Fuqua, S.A.; Ando, S. Progesterone receptor B recruits a repressor complex to a half-PRE site of the estrogen receptor alpha gene promoter. Mol. Endocrinol. 2009, 23, 454–465. [Google Scholar] [CrossRef]
- Yu, S.; Yang, X.; Zhu, Y.; Xie, F.; Lu, Y.; Yu, T.; Yan, C.; Shao, J.; Gao, Y.; Mo, F.; et al. Systems pharmacology of mifepristone (RU486) reveals its 47 hub targets and network: Comprehensive analysis and pharmacological focus on FAK-Src-Paxillin complex. Sci. Rep. 2015, 19, 7830. [Google Scholar] [CrossRef]
- Chen, H.Y.; Shen, C.H.; Tsai, Y.T.; Lin, F.C.; Huang, Y.P.; Chen, R.H. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol. Cell. Biol. 2004, 24, 10558–10572. [Google Scholar] [CrossRef]
- Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Lanzino, M.; De Amicis, F.; Mauro, L.; Sisci, D.; Catalano, S.; Dahlman Wright, K.; et al. Estrogen receptor beta as a novel target of androgen receptor action in breast cancer cell lines. Breast Cancer Res. 2014, 16, R21. [Google Scholar] [CrossRef]
- Sirianni, R.; Chimento, A.; Malivindi, R.; Mazzitelli, I.; Ando, S.; Pezzi, V. Insulin-like growth factor-I, regulating aromatase expression through steroidogenic factor 1, supports estrogen-dependent tumor Leydig cell proliferation. Cancer Res. 2007, 67, 8368–8377. [Google Scholar] [CrossRef]
- Abmayr, S.M.; Yao, T.; Parmely, T.; Workman, J.L. Preparation of nuclear and cytoplamic extracts from mammalian cells. Curr. Protoc. Mol. Biol. 2006, 12, 12.3.1–12.3.13. [Google Scholar]
- Panza, S.; Santoro, M.; De Amicis, F.; Morelli, C.; Passarelli, V.; D’Aquila, P.; Giordano, F.; Cione, E.; Passarino, G.; Bellizzi, D.; et al. Estradiol via estrogen receptor beta influences ROS levels through the transcriptional regulation of SIRT3 in human seminoma TCam-2 cells. Tumor Biol. 2017, 39, 1010428317701642. [Google Scholar] [CrossRef]
- De Amicis, F.; Giordano, F.; Vivacqua, A.; Pellegrino, M.; Panno, M.L.; Tramontano, D.; Fuqua, S.A.; Ando, S. Resveratrol, through NF-Y/p53/Sin3/HDAC1 complex phosphorylation, inhibits estrogen receptor alpha gene expression via p38MAPK/CK2 signaling in human breast cancer cells. FASEB J. 2011, 25, 3695–3707. [Google Scholar] [CrossRef]
- Rovito, D.; Giordano, C.; Plastina, P.; Barone, I.; De Amicis, F.; Mauro, L.; Rizza, P.; Lanzino, M.; Catalano, S.; Bonofiglio, D.; et al. Omega-3 DHA- and EPA-dopamine conjugates induce PPARgamma-dependent breast cancer cell death through autophagy and apoptosis. Biochim. Biophys. Acta 2015, 1850, 2185–2195. [Google Scholar] [CrossRef]
- De Amicis, F.; Lanzino, M.; Kisslinger, A.; Cali, G.; Chieffi, P.; Ando, S.; Mancini, F.P.; Tramontano, D. Loss of proline-rich tyrosine kinase 2 function induces spreading and motility of epithelial prostate cells. J. Cell. Physiol. 2006, 209, 74–80. [Google Scholar] [CrossRef]
- De Amicis, F.; Perri, A.; Vizza, D.; Russo, A.; Panno, M.L.; Bonofiglio, D.; Giordano, C.; Mauro, L.; Aquila, S.; Tramontano, D.; et al. Epigallocatechin gallate inhibits growth and epithelial-to-mesenchymal transition in human thyroid carcinoma cell lines. J. Cell. Physiol. 2013, 228, 2054–2062. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montalto, F.I.; Giordano, F.; Chiodo, C.; Marsico, S.; Mauro, L.; Sisci, D.; Aquila, S.; Lanzino, M.; Panno, M.L.; Andò, S.; et al. Progesterone Receptor B signaling Reduces Breast Cancer Cell Aggressiveness: Role of Cyclin-D1/Cdk4 Mediating Paxillin Phosphorylation. Cancers 2019, 11, 1201. https://doi.org/10.3390/cancers11081201
Montalto FI, Giordano F, Chiodo C, Marsico S, Mauro L, Sisci D, Aquila S, Lanzino M, Panno ML, Andò S, et al. Progesterone Receptor B signaling Reduces Breast Cancer Cell Aggressiveness: Role of Cyclin-D1/Cdk4 Mediating Paxillin Phosphorylation. Cancers. 2019; 11(8):1201. https://doi.org/10.3390/cancers11081201
Chicago/Turabian StyleMontalto, Francesca Ida, Francesca Giordano, Chiara Chiodo, Stefania Marsico, Loredana Mauro, Diego Sisci, Saveria Aquila, Marilena Lanzino, Maria Luisa Panno, Sebastiano Andò, and et al. 2019. "Progesterone Receptor B signaling Reduces Breast Cancer Cell Aggressiveness: Role of Cyclin-D1/Cdk4 Mediating Paxillin Phosphorylation" Cancers 11, no. 8: 1201. https://doi.org/10.3390/cancers11081201
APA StyleMontalto, F. I., Giordano, F., Chiodo, C., Marsico, S., Mauro, L., Sisci, D., Aquila, S., Lanzino, M., Panno, M. L., Andò, S., & De Amicis, F. (2019). Progesterone Receptor B signaling Reduces Breast Cancer Cell Aggressiveness: Role of Cyclin-D1/Cdk4 Mediating Paxillin Phosphorylation. Cancers, 11(8), 1201. https://doi.org/10.3390/cancers11081201