The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer



Abstract
1. Introduction
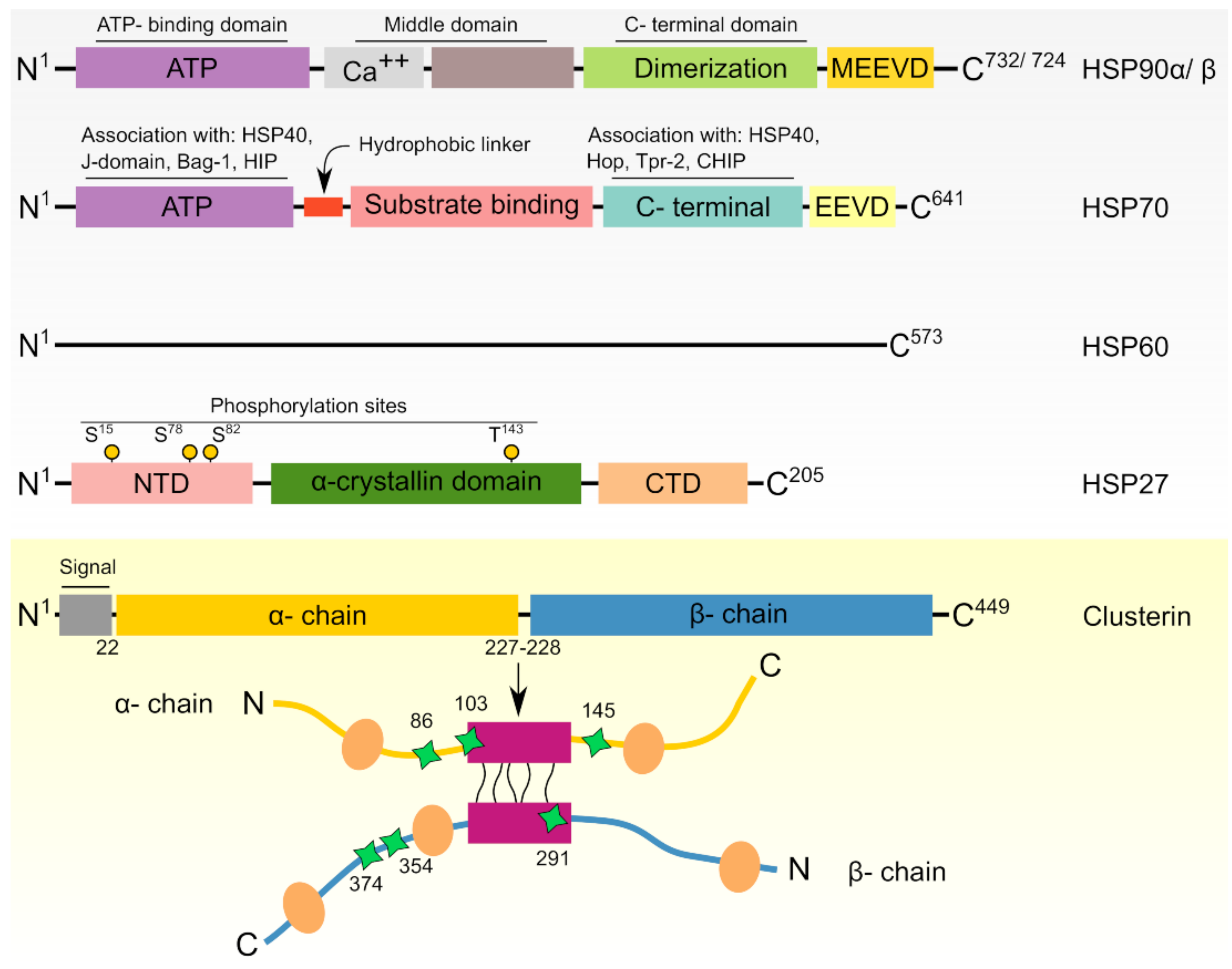
2. Heat Shock Proteins: Functional and Regulatory Overview
3. HSPs in Cancer Cells
4. HSPs in Prostate Cancer
4.1. HSP90
4.1.1. Biological Functions of HSP90 Isoforms in Prostate Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSP | Mechanism | Used cell Line/Model | Reference |
|---|---|---|---|
| Anti-apoptosis | |||
| HSP90 | Involved in several signaling and proliferative pathways via AR, ERBB2, Akt, c-RAF, survivin, EGFR, IGFR–1, STAT3, ERK, CDK-4, and CDK-6 | PC3-MM2, LNCaP-LN3, VCaP, 22Rv1, DU145 and PC3, CWR22 and CWR22R xenografts | [79,80,81,82,83,84,85,86] |
| HSP70 | Suppresses the pro-apoptotic protein MST-1, resulting in cisplatin resistance | DU145 | [87] |
| Stimulates overexpression of Bcl2-L-3, BCL2, and Bcl2-L-1, thus making PCa cells resistant to ionizing radiation and etoposide. | PC3 and LNCaP | [88] | |
| HSP27 | Hampers STAT3-regulated apoptosis, leading to resistance to androgen withdrawal | LNCaP | [89] |
| Increases the expression of eIF4E, making PCA cells refractory to androgen withdrawal and paclitaxel | LNCaP | [90] | |
| Upregulates TCTP, which diminishes docetaxel-mediated apoptosis in LNCaP cells | LNCaP | [91] | |
| Promotes IGF-1-induced phosphorylation of ERK, Akt and RPS6KA, thus inactivating the BAD-14-3-3 protein complex and inhibiting apoptosis | PC3 | [92] | |
| Hinders Fas-mediated apoptosis by allowing PEA–15 to bind FADD in an Akt-dependent mechanism | LNCaP | [93] | |
| Clusterin | Counteracts Bcl2–L–4–mediated caspase activation, resulting in apoptosis inhibition in PCa cells treated with camptothecin and etoposide | PC3 and DU145 | [94] |
| Inhibits apoptosis in rat prostatic cells treated with actinomycin D through phosphorylation of Akt, subsequent phosphorylation of BAD and reduced cytochrome c release | MLL Dunning rat prostatic adenocarcinoma cell line | [95] | |
| After stimulation by Akt, clusterin causes resistance to docetaxel | DU145 and PC3 | [96] | |
| Its overexpression upon enzalutamide treatment of PCa cells confers resistance and inhibition of apoptosis. The process occurs through the RPS6KA–YB–1 signaling pathway and involves clusterin mediated activation of Akt and MAPK | LNCaP | [97] | |
| Inhibits paclitaxel-induced apoptosis after GRP78 mediated translocation to other cellular compartments including cytosol and mitochondria | LNCaP | [98] | |
| AR, trafficking, stability, and transcription regulation | |||
| HSP90 | Protects against AR degradation | LNCaP, PC3-MM2, LNCaP-LN3, VCaP, 22Rv1, DU145 and PC3, CWR22 and CWR22R xenografts | [79,80,81,84,86,99] |
| Preserves high-affinity ligand-binding conformation of AR | LNCaP, Yeast cells | [65,99] | |
| Aids nuclear trafficking of AR, thus facilitating transcription of AR-regulated genes such as PSA and expansion of castration resistance | LNCaP cells | [100] | |
| HSP70 | Facilitates binding of BAG-1 to N-terminus of AR thus stimulating enhanced transcription of AR-regulated genes like KLK3 | PC3. LNCaP, 22Rv1 and CWR22R xenografts | [101] |
| Invasion and metastasis | |||
| HSP90 | Together with its client proteins, HSP90 is implicated in enhanced lymph node metastases | PC3LN3 orthotopic lymph node mPCa model | [102] |
| Activates NF-κB and p60-Src leading to RANKL-induced osteoclast differentiation | LNCaP xenograft model | [86] | |
| Enhances PCa cells motility through ERK and MMP-2-MMP-9 | DU145 and LNCaP, ARCaP | [103] | |
| HSP70 | Besides HSP90, HSP70 is involved in WASF3 metastasis-promoting protein stability and activity | PC3 cells | [104] |
| HSP27 | Implicated in TGF-β-mediated MMP–2 activation and invasion | PC3 cells | [105] |
| Augments EMT via IL-6-STAT3-Twist signaling resulting in increased cell migration and invasion and metastases | PC3M model | [106] | |
| Clusterin | Implicated in TGF-β-mediated invasion | PC-3 | [107] |
| Initial signals coming from Twist1 and TGF-β activates clusterin to promote EMT and increase metastasis | PC3M model | [108] | |
| Angiogenesis | |||
| HSP90 | Protects HIF-1α from proteasomal degradation, leading to increased expression of VEGF and angiogenesis | DU145, PC3 and LNCaP | [109,110] |
| Clusterin | IL-24 reduces secretory clusterin levels, thus diminishing angiogenesis | DU145 xenografts | [111] |
| Cell cycle regulation | |||
| HSP90 | Modulation of HSP90 interactors like AR, ERBB2, and Akt is associated with enforcement of G1 cell-cycle checkpoint | DU145 and LNCaP | [84] |
| Modulation of HSP90 client proteins including AR, ERBB2, Akt, c-RAF, and CDK-4 is critical for enforcement of G2–M cell-cycle checkpoint | PC3 and LNCaP | [82] | |
| HSP27 | Induces TCTP overexpression resulting in maintaining prostatic cells in S-phase of the cell cycle | PC3 and LNCaP | [91] |
| Induces PEA-15 phosphorylation, thus hindering its association to ERK. This allows for ERK nuclear translocation and promoting the cells to stay in S-phase of the cell cycle | LNCaP | [93] | |
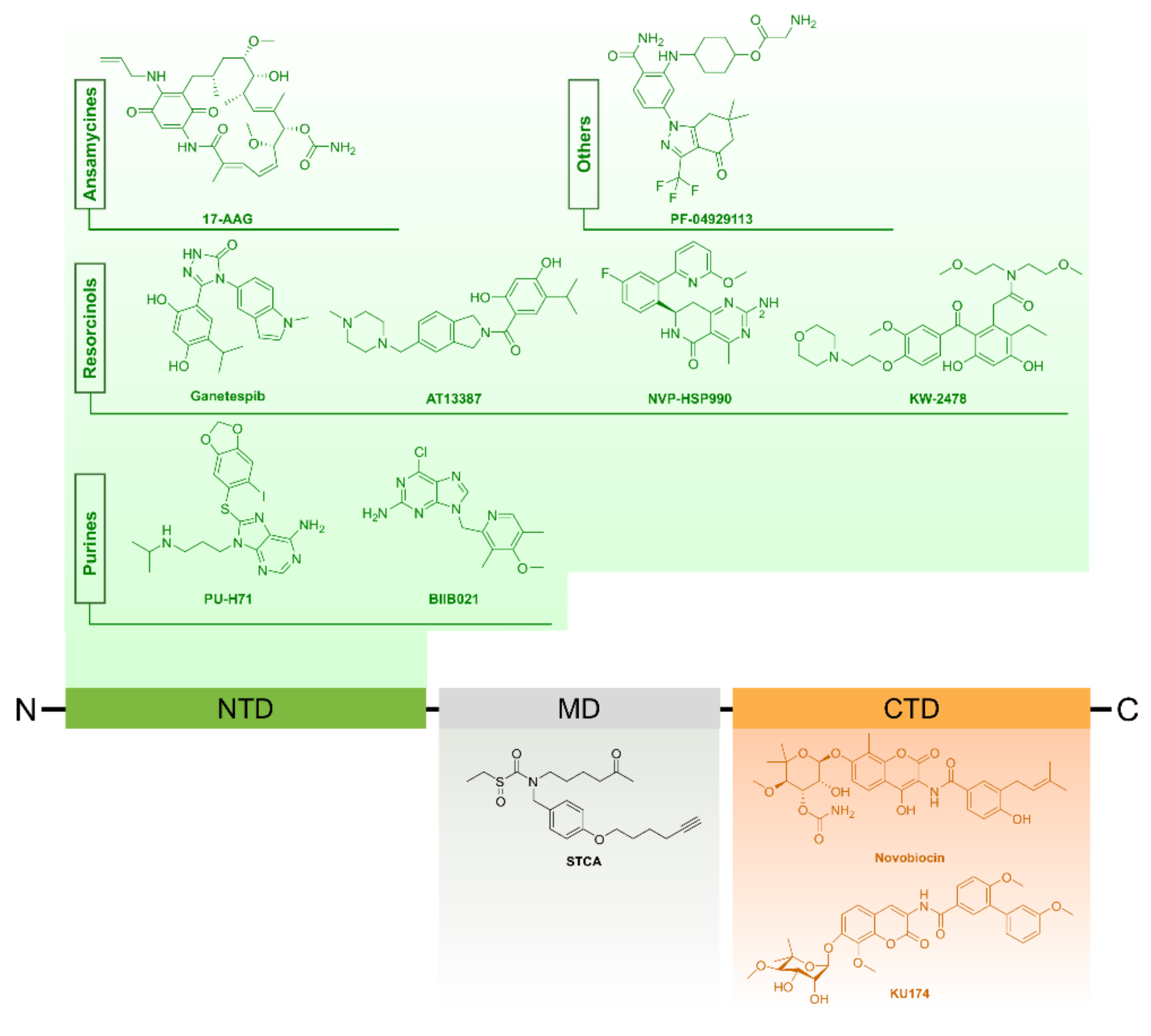
4.1.2. Targeting HSP90 in Prostate Cancer
Small Molecule Inhibitors of HSP90
HSP90 Inhibitors in Pre-clinical Prostate Cancer Studies
- N-terminal domain inhibitors
- C-terminal domain inhibitors
Clinical Assessment of HSP90 Inhibitors in CRPC
HSP90 Inhibitors in Combination Therapies for Prostate Cancer
4.1.3. Extracellular HSP90 (eHSP90) Is an Attractive Target in Metastatic Prostate Tumors
4.1.4. HSP90 Regulation as Potential Target in Prostate Cancer
Posttranslational Modification of HSP90
Co-Chaperones
4.2. HSP70
4.2.1. Biological Functions of HSP70 in Prostate Cancer
4.2.2. Targeting HSP70 in Prostate Cancer
N-terminal Domain or ATPase Domain Inhibitors
C-Terminal Domain or Peptide Binding Domain (PBD)
Inhibitors of HSP70 Co-Chaperones
HSP70 Inhibitors in Combination Therapies
4.3. HSP60
4.3.1. Biological Functions of HSP60 in Prostate Cancer
4.3.2. HSP60 as Potential Target in Cancer Therapy
4.4. HSP27
4.4.1. Biological Functions of HSP27 in Prostate Cancer
4.4.2. Prognostic Value of HSP27 in Prostate Cancer
4.4.3. Targeting HSP27 in Prostate Cancer
4.5. Clusterin
4.5.1. Biological Functions of Clusterin in Prostate Cancer
4.5.2. Prognostic Value of Clusterin in Prostate Cancer
4.5.3. Targeting Clusterin in Prostate Cancer
4.5.4. Targeting Clusterin within Combination Therapies for mCRPC
5. Conclusions, Challenges, and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 17-AAG | 17-allylamino-17-desmethoxygeldanamycin |
| Akt | Protein kinase B |
| AR | Androgen receptor |
| ARE | Androgen receptor element |
| BAD | Bcl2-associated agonist of cell death |
| BAG-1 | BAG family molecular chaperone regulator 1 |
| BCL2 | Apoptosis regulator Bcl-2 |
| Bcl2-L-1 | Bcl-2-like protein 1 |
| Bcl2-L-3 | Induced myeloid leukemia cell differentiation protein-1 |
| Bcl2-L-4 | Apoptosis regulator BAX |
| CDK | Cyclin-dependent kinase |
| c-RAF | RAF proto-oncogene serine/threonine–protein kinase |
| CRPC | Castration-resistant prostate cancer |
| EGFR | Epidermal growth factor receptor 4E |
| EMT | Epithelial–mesenchymal transition |
| EMT | Epithelial to mesenchymal transition |
| ERBB | Receptor tyrosine–protein kinase erbB |
| ERBB2 | Receptor tyrosine–protein kinase erbB-2 |
| ERK | Extracellular-signal-regulated kinase |
| FADD | Fas-associating death domain-containing protein |
| GA | Geldanamycin |
| GRP 94 | Glucose-regulated protein 94 |
| GRP-78 | 78 kDa glucose-regulated protein |
| HIF-1α | Hypoxia-inducible factor 1α |
| HSE | Heat shock element |
| HSPs | Heat shock proteins |
| IGF-1 | Insulin-like growth factor 1 |
| IGFR-1 | Insulin-like growth factor receptor 1 |
| MAPK | Mitogen-activated protein kinase; |
| MMP | Matrix metalloproteinase |
| mPCa | Metastatic prostate cancer |
| MST-1 | Mammalian STE20-like protein kinase 1 |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| p60-Src | Proto-oncogene tyrosine-protein kinase Src |
| PCa | Prostate cancer |
| PEA-15 | 15 kDa phosphoprotein enriched in astrocytes |
| PSA | Prostate specific antigen |
| PSMA | Prostate specific membrane antigen |
| RANKL | Receptor activator of nuclear factor κB ligand |
| RPS6KA | Ribosomal protein S6 kinase α |
| SGTA | Small glutamine-rich tetratricopeptide repeat-containing protein α |
| STAT3 | Signal transducer and activator of transcription 3 |
| STCA | Sulfoxythiocarbamate alkyne |
| TCTP | Translationally controlled tumor protein |
| TGF-β | Transforming growth factor β |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Vav3 | Vav 3, guanine nucleotide exchange factor |
| VEGF | Vascular endothelial growth factor |
| WASF3 | Wiskott–Aldrich syndrome protein family member 3 |
| YB-1 | Nuclease-sensitive element-binding protein 1 |
References
- Tian, J.-Y.; Guo, F.-J.; Zheng, G.-Y.; Ahmad, A. Prostate cancer: Updates on current strategies for screening, diagnosis and clinical implications of treatment modalities. Carcinogenesis 2018, 39, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Lilja, H.; Ulmert, D.; Vickers, A.J. Prostate-specific antigen and prostate cancer: Prediction, detection and monitoring. Nat. Rev. Cancer 2008, 8, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Marta, G.N.; Hanna, S.A.; Da Silva, J.L.F.; De Andrade Carvalho, H. Screening for prostate cancer: An updated review. Expert Rev. Anticancer Ther. 2013, 13, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Jemaa, A.; Bouraoui, Y.; Sallami, S.; Banasr, A.; Nouira, Y.; Oueslati, R. PSMA/PSA ratio evaluated by immunohistochemistry may improve diagnosis of prostate cancer. J. Immunoass. Immunochem. 2014, 35, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Castelletti, D.; Alfalah, M.; Heine, M.; Hein, Z.; Schmitte, R.; Fracasso, G.; Colombatti, M.; Naim, H.Y. Different glycoforms of prostate-specific membrane antigen are intracellularly transported through their association with distinct detergent-resistant membranes. Biochem. J. 2008, 409, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Gericke, B.; Fracasso, G.; Ramarli, D.; Colombatti, M.; Naim, H.Y. Discriminatory Role of Detergent-Resistant Membranes in the Dimerization and Endocytosis of Prostate-Specific Membrane Antigen. PLoS ONE 2013, 8, e66193. [Google Scholar] [CrossRef]
- Colombatti, M.; Grasso, S.; Porzia, A.; Fracasso, G.; Scupoli, M.T.; Cingarlini, S.; Poffe, O.; Naim, H.Y.; Heine, M.; Tridente, G.; et al. The prostate specific membrane antigen regulates the expression of IL-6 and CCL5 in prostate tumor cells by activating the MAPK pathways. PLoS ONE 2009, 4, e4608. [Google Scholar] [CrossRef]
- So, A.; Hadaschik, B.; Sowery, R.; Gleave, M. The role of stress proteins in prostate cancer. Curr. Genom. 2007, 8, 252–261. [Google Scholar]
- Pond, G.R.; De Wit, R.; Eisenberger, M.; Berthold, D.R.; Tannock, I.F.; Soban, F. Docetaxel Plus Prednisone or Mitoxantrone Plus Prednisone for Advanced Prostate Cancer: Updated Survival in the TAX 327 Study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef]
- Zellweger, T.; Miyake, H.; July, L.V.; Akbari, M.; Kiyama, S.; Gleave, M.E. Chemosensitization of human renal cell cancer using antisense oligonucleotides targeting the antiapoptotic gene clusterin. Neoplasia 2001, 3, 360–367. [Google Scholar] [CrossRef]
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med. 1999, 5, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Bruchovsky, N.; Rennie, P.S.; Coldman, A.J.; Goldenberg, S.L.; To, M.; Lawson, D. Effects of androgen withdrawal on the stem cell composition of the Shionogi carcinoma. Cancer Res. 1990, 50, 2275–2282. [Google Scholar] [PubMed]
- Giménez-Bonafé, P.; Fedoruk, M.N.; Whitmore, T.G.; Akbari, M.; Ralph, J.L.; Ettinger, S.; Gleave, M.E.; Nelson, C.C. YB-1 is upregulated during prostate cancer tumor progression and increases P-glycoprotein activity. Prostate 2004, 59, 337–349. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Wake, N.; Coffey, D.S.; Sandberg, A.A. Genetic instability coupled to clonal selection as a mechanism for tumor progression in the Dunning R-3327 rat prostatic adenocarcinoma system. Cancer Res. 1982, 42, 2353–2371. [Google Scholar] [PubMed]
- Thapar, R.; Titus, M.A. Recent Advances in Metabolic Profiling and Imaging of Prostate Cancer. Curr. Metab. 2014, 2, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Ward, N.P.; DeNicola, G.M. Recent advances in cancer metabolism: A technological perspective. Exp. Mol. Med. 2018, 50, 31. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.M.; Ruiz-Echevarría, M.J. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int. J. Mol. Sci. 2016, 17, 1208. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Zoubeidi, A.; Gleave, M.E.; Chi, K.N. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nat. Rev. Urol. 2015, 12, 26–36. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of heat shock proteins in apoptosis, oxidative stress, human inflammatory diseases, and cancer. Pharmaceuticals 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Glaessgen, A.; Jonmarker, S.; Lindberg, A.; Nilsson, B.; Lewensohn, R.; Ekman, P.; Valdman, A.; Egevad, L. Heat shock proteins 27, 60 and 70 as prognostic markers of prostate cancer. APMIS 2008, 116, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting heat shock proteins in cancer: A promising therapeutic approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Jia, H.; Halilou, A.I.; Hu, L.; Cai, W.; Liu, J.; Huang, B. Heat shock protein 10 (Hsp10) in immune-related diseases: One coin, two sides. Int. J. Biochem. Mol. Biol. 2011, 2, 47–57. [Google Scholar]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2018, 294, 2121–2132. [Google Scholar] [CrossRef]
- De Maio, A. Heat shock proteins: Facts, thoughts, and dreams. Shock 1999, 11, 1–12. [Google Scholar] [CrossRef]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large potentials of small heat shock proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef]
- Li, J.; Qian, X.; Sha, B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef]
- Lu, B.; Garrido, N.; Spelbrink, J.N.; Suzuki, C.K. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J. Biol. Chem. 2006, 281, 13150–13158. [Google Scholar] [CrossRef]
- Meng, Q.; Li, B.X.; Xiao, X. Toward Developing Chemical Modulators of Hsp60 as Potential Therapeutics. Front. Mol. Biosci. 2018, 5, 35. [Google Scholar] [CrossRef]
- Gupta, R.S. Evolution of the chaperonin families (Hsp60, Hsp10 and Tcp-1) of proteins and the origin of eukaryotic cells. Mol. Microbiol. 1995, 15, 1–11. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2506. [Google Scholar] [CrossRef]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohászka, Z.; Nardai, G. The 90-kDa Molecular Chaperone Family: Structure, Function, and Clinical Applications. A Comprehensive Review. Pharm. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Kalmár, É.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Wang, H.; Pezeshki, A.M.; Yu, X.; Guo, C.; Subjeck, J.R.; Wang, X.-Y. The Endoplasmic Reticulum Chaperone GRP170: From Immunobiology to Cancer Therapeutics. Front. Oncol. 2014, 4, 377. [Google Scholar] [CrossRef]
- Chen, X.; Easton, D.; Oh, H.J.; Lee-Yoon, D.S.; Liu, X.; Subjeck, J. The 170 kDa glucose regulated stress protein is a large HSP70-, HSP110-like protein of the endoplasmic reticulum. FEBS Lett. 1996, 380, 68–72. [Google Scholar] [CrossRef]
- Dragovic, Z.; Broadley, S.A.; Shomura, Y.; Bracher, A.; Hartl, F.U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006, 25, 2519–2528. [Google Scholar] [CrossRef]
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar] [CrossRef]
- Fujimoto, M.; Nakai, A. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 2010, 277, 4112–4125. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef]
- Sistonen, L.; Sarge, K.D.; Morimoto, R.I. Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol. Cell. Biol. 1994, 14, 2087–2099. [Google Scholar] [CrossRef]
- Nakai, A.; Tanabe, M.; Kawazoe, Y.; Inazawa, J.; Morimoto, R.I.; Nagata, K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol. Cell. Biol. 1997, 17, 469–481. [Google Scholar] [CrossRef]
- Green, M.; Schuetz, T.J.; Sullivan, E.K.; Kingston, R.E. A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Mol. Cell. Biol. 1995, 15, 3354–3362. [Google Scholar] [CrossRef]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Fan, G.C. Role of Heat Shock Proteins in Stem Cell Behavior, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 111, ISBN 9780123984593. [Google Scholar]
- Becker, J.; Craig, E.A. Heat-shock proteins as molecular chaperones. Eur. J. Biochem. 1994, 219, 11–23. [Google Scholar] [CrossRef]
- Buchner, J. Molecular chaperones and protein quality control: An introduction to the JBC Reviews thematic series. J. Biol. Chem. 2019, 294, 2074–2075. [Google Scholar] [CrossRef]
- Mogk, A.; Bukau, B. Role of sHsps in organizing cytosolic protein aggregation and disaggregation. Cell Stress Chaperones 2017, 22, 493–502. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Streicher, J.M. The Role of Heat Shock Proteins in Regulating Receptor Signal Transduction. Mol. Pharmacol. 2019, 95, 468–474. [Google Scholar] [CrossRef]
- Hartl, F. Ulrich Molecular chaperones in protein folding and translocation. Curr. Opin. Struct. Biol. 1996, 6, 43–50. [Google Scholar] [CrossRef]
- Basha, E.; O’Neill, H.; Vierling, E. Small heat shock proteins and α-crystallins: Dynamic proteins with flexible functions. Trends Biochem. Sci. 2012, 37, 106–117. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2018. [Google Scholar] [CrossRef]
- Cano, L.Q.; Lavery, D.N.; Bevan, C.L. Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Mol. Cell. Endocrinol. 2013, 369, 52–62. [Google Scholar] [CrossRef]
- Fang, Y.; Fliss, A.E.; Robins, D.M.; Caplan, A.J. Hsp90 regulates androgen receptor hormone binding affinity in vivo. J. Biol. Chem. 1996, 271, 28697–28702. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Zardan, A.; Beraldi, E.; Fazli, L.; Sowery, R.; Rennie, P.; Nelson, C.; Gleave, M. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 2007, 67, 10455–10465. [Google Scholar] [CrossRef]
- Li, J.; Buchner, J. Structure, function and regulation of the hsp90 machinery. Biomed. J. 2012, 36, 106–117. [Google Scholar] [CrossRef]
- Flynn, J.M.; Mishra, P.; Bolon, D.N.A. Mechanistic Asymmetry in Hsp90 Dimers. J. Mol. Biol. 2015, 427, 2904–2911. [Google Scholar] [CrossRef]
- Wayne, N.; Bolon, D.N. Dimerization of Hsp90 is required for in vivo function. Design and analysis of monomers and dimers. J. Biol. Chem. 2007, 282, 35386–35395. [Google Scholar] [CrossRef]
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 2010, 37, 344–354. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- DaSilva, J.; Gioeli, D.; Weber, M.J.; Parsons, S.J. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res. 2009, 69, 7402–7411. [Google Scholar] [CrossRef]
- Masgras, I.; Sanchez-Martin, C.; Colombo, G.; Rasola, A. The Chaperone TRAP1 As a Modulator of the Mitochondrial Adaptations in Cancer Cells. Front. Oncol. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef]
- Lisanti, S.; Garlick, D.S.; Bryant, K.G.; Tavecchio, M.; Mills, G.B.; Lu, Y.; Kossenkov, A.V.; Showe, L.C.; Languino, L.R.; Altieri, D.C. Transgenic expression of the mitochondrial chaperone TNFR-associated protein 1 (TRAP1) accelerates prostate cancer development. J. Biol. Chem. 2016, 291, 25247–25254. [Google Scholar] [CrossRef]
- Ghosh, S.; Shinogle, H.E.; Galeva, N.A.; Dobrowsky, R.T.; Blagg, B.S.J. Endoplasmic Reticulum-resident Heat Shock Protein 90 (HSP90) Isoform Glucose-regulated Protein 94 (GRP94) Regulates Cell Polarity and Cancer Cell Migration by Affecting Intracellular Transport. J. Biol. Chem. 2016, 291, 8309–8323. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Hedayat, S.; Smyth, T.; Wallis, N.; Lyons, J.; Riisnaes, R.; Crespo, M.; Nava Rodrigues, D.; Miranda, S.; Sharp, S.; et al. In vitro and in vivo antitumor activity of the next generation HSP90 inhibitor, AT13387, in both hormone-sensitive and castration-resistant prostate cancer models [Abstract 2433]. Cancer Res. 2013, 73, 2433. [Google Scholar] [CrossRef]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef]
- He, S.; Zhang, C.; Shafi, A.A.; Sequeira, M.; Acquaviva, J.; Friedland, J.C.; Sang, J.; Smith, D.L.; Weigel, N.L.; Wada, Y.; et al. Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. Int. J. Oncol. 2013, 42, 35–43. [Google Scholar] [CrossRef]
- Centenera, M.M.; Gillis, J.L.; Hanson, A.R.; Jindal, S.; Taylor, R.A.; Risbridger, G.P.; Sutherland, P.D.; Scher, H.I.; Raj, G.V.; Knudsen, K.E.; et al. Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin. Cancer Res. 2012, 18, 3562–3570. [Google Scholar] [CrossRef]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef]
- Solit, D.B.; Zheng, F.F.; Drobnjak, M.; Münster, P.N.; Higgins, B.; Verbel, D.; Heller, G.; Tong, W.; Cordon-Cardo, C.; Agus, D.B.; et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin. Cancer Res. 2002, 8, 986–993. [Google Scholar]
- Matthews, S.B.; Vielhauer, G.A.; Manthe, C.A.; Chaguturu, V.K.; Szabla, K.; Matts, R.L.; Donnelly, A.C.; Blagg, B.S.J.; Holzbeierlein, J.M. Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate 2010, 70, 27–36. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Yin, M.-J.; Kuruma, H.; Fazli, L.; Gleave, M.E.; Zoubeidi, A. A novel HSP90 inhibitor delays castrate-resistant prostate cancer without altering serum PSA levels and inhibits osteoclastogenesis. Clin. Cancer Res. 2011, 17, 2301–2313. [Google Scholar] [CrossRef]
- Ren, A.; Yan, G.; You, B.; Sun, J. Down-regulation of mammalian sterile 20-like kinase 1 by heat shock protein 70 mediates cisplatin resistance in prostate cancer cells. Cancer Res. 2008, 68, 2266–2274. [Google Scholar] [CrossRef]
- Gibbons, N.B.; Watson, R.W.G.; Coffey, R.N.T.; Brady, H.P.; Fitzpatrick, J.M. Heat-shock proteins inhibit induction of prostate cancer cell apoptosis. Prostate 2000, 45, 58–65. [Google Scholar] [CrossRef]
- Rocchi, P.; Beraldi, E.; Ettinger, S.; Fazli, L.; Vessella, R.L.; Nelson, C.; Gleave, M. Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer Res. 2005, 65, 11083–11093. [Google Scholar] [CrossRef]
- Andrieu, C.; Taieb, D.; Baylot, V.; Ettinger, S.; Soubeyran, P.; De-Thonel, A.; Nelson, C.; Garrido, C.; So, A.; Fazli, L.; et al. Heat shock protein 27 confers resistance to androgen ablation and chemotherapy in prostate cancer cells through eIF4E. Oncogene 2010, 29, 1883–1896. [Google Scholar] [CrossRef]
- Baylot, V.; Katsogiannou, M.; Andrieu, C.; Taieb, D.; Acunzo, J.; Giusiano, S.; Fazli, L.; Gleave, M.; Garrido, C.; Rocchi, P. Targeting TCTP as a new therapeutic strategy in castration-resistant prostate cancer. Mol. Ther. 2012, 20, 2244–2256. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Zardan, A.; Wiedmann, R.M.; Locke, J.; Beraldi, E.; Fazli, L.; Gleave, M.E. Hsp27 promotes insulin-like growth factor-I survival signaling in prostate cancer via p90Rsk-dependent phosphorylation and inactivation of BAD. Cancer Res. 2010, 70, 2307–2317. [Google Scholar] [CrossRef]
- Hayashi, N.; Peacock, J.W.; Beraldi, E.; Zoubeidi, A.; Gleave, M.E.; Ong, C.J. Hsp27 silencing coordinately inhibits proliferation and promotes Fas-induced apoptosis by regulating the PEA-15 molecular switch. Cell Death Differ. 2012, 19, 990–1002. [Google Scholar] [CrossRef]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.-Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [Google Scholar] [CrossRef]
- Ammar, H.; Closset, J.L. Clusterin activates survival through the phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 2008, 283, 12851–12861. [Google Scholar] [CrossRef]
- Zhong, B.; Sallman, D.A.; Gilvary, D.L.; Pernazza, D.; Sahakian, E.; Fritz, D.; Cheng, J.Q.; Trougakos, I.; Wei, S.; Djeu, J.Y. Induction of clusterin by AKT--role in cytoprotection against docetaxel in prostate tumor cells. Mol. Cancer Ther. 2010, 9, 1831–1841. [Google Scholar] [CrossRef]
- Matsumoto, H.; Yamamoto, Y.; Shiota, M.; Kuruma, H.; Beraldi, E.; Matsuyama, H.; Zoubeidi, A.; Gleave, M. Cotargeting Androgen Receptor and Clusterin Delays Castrate-Resistant Prostate Cancer Progression by Inhibiting Adaptive Stress Response and AR Stability. Cancer Res. 2013, 73, 5206–5217. [Google Scholar] [CrossRef]
- Li, N.; Zoubeidi, A.; Beraldi, E.; Gleave, M.E. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 2013, 32, 1933–1942. [Google Scholar] [CrossRef]
- Vanaja, D.K.; Mitchell, S.H.; Toft, D.O.; Young, C.Y.F. Effect of geldanamycin on androgen receptor function and stability. Cell Stress Chaperones 2002, 7, 55–64. [Google Scholar] [CrossRef]
- Saporita, A.J.; Ai, J.; Wang, Z. The Hsp90 inhibitor, 17-AAG, prevents the ligand-independent nuclear localization of androgen receptor in refractory prostate cancer cells. Prostate 2007, 67, 509–520. [Google Scholar] [CrossRef]
- Shatkina, L.; Mink, S.; Rogatsch, H.; Klocker, H.; Langer, G.; Nestl, A.; Cato, A.C.B. The cochaperone Bag-1L enhances androgen receptor action via interaction with the NH2-terminal region of the receptor. Mol. Cell. Biol. 2003, 23, 7189–7197. [Google Scholar] [CrossRef]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef]
- Hance, M.W.; Dole, K.; Gopal, U.; Bohonowych, J.E.; Jezierska-Drutel, A.; Neumann, C.A.; Liu, H.; Garraway, I.P.; Isaacs, J.S. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem. 2012, 287, 37732–37744. [Google Scholar] [CrossRef]
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J.K. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J. Biol. Chem. 2012, 287, 10051–10059. [Google Scholar] [CrossRef]
- Xu, L.; Chen, S.; Bergan, R.C. MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006, 25, 2987–2998. [Google Scholar] [CrossRef]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef]
- Lenferink, A.E.G.; Cantin, C.; Nantel, A.; Wang, E.; Durocher, Y.; Banville, M.; Paul-Roc, B.; Marcil, A.; Wilson, M.R.; O’Connor-McCourt, M.D. Transcriptome profiling of a TGF-beta-induced epithelial-to-mesenchymal transition reveals extracellular clusterin as a target for therapeutic antibodies. Oncogene 2010, 29, 831–844. [Google Scholar] [CrossRef]
- Shiota, M.; Zardan, A.; Takeuchi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Zoubeidi, A.; Gleave, M.E. Clusterin mediates TGF-β-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res. 2012, 72, 5261–5272. [Google Scholar] [CrossRef]
- Alqawi, O.; Moghaddas, M.; Singh, G. Effects of geldanamycin on HIF-1alpha mediated angiogenesis and invasion in prostate cancer cells. Prostate Cancer Prostatic Dis. 2006, 9, 126–135. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Post, D.E.; Willard, M.T.; Kaur, B.; Van Meir, E.G.; Simons, J.W.; Zhong, H. Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002, 62, 2478–2482. [Google Scholar]
- Bhutia, S.K.; Das, S.K.; Kegelman, T.P.; Azab, B.; Dash, R.; Su, Z.-Z.; Wang, X.-Y.; Rizzi, F.; Bettuzzi, S.; Lee, S.-G.; et al. mda-7/IL-24 differentially regulates soluble and nuclear clusterin in prostate cancer. J. Cell. Physiol. 2012, 227, 1805–1813. [Google Scholar] [CrossRef]
- Jiang, F.; Guo, A.-P.; Xu, J.-C.; You, Q.-D.; Xu, X.-L. Discovery of a Potent Grp94 Selective Inhibitor with Anti-Inflammatory Efficacy in a Mouse Model of Ulcerative Colitis. J. Med. Chem. 2018, 61, 9513–9533. [Google Scholar] [CrossRef]
- Stothert, A.R.; Suntharalingam, A.; Tang, X.; Crowley, V.M.; Mishra, S.J.; Webster, J.M.; Nordhues, B.A.; Huard, D.J.E.; Passaglia, C.L.; Lieberman, R.L.; et al. Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci. Rep. 2017, 7, 17951. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumor selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Taldone, T.; Sun, W.; Chiosis, G. Discovery and development of heat shock protein 90 inhibitors. Bioorg. Med. Chem. 2009, 17, 2225–2235. [Google Scholar] [CrossRef]
- Beebe, K.; Mollapour, M.; Scroggins, B.; Prodromou, C.; Xu, W.; Tokita, M.; Taldone, T.; Pullen, L.; Zierer, B.K.; Lee, M.-J.; et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 2013, 4, 1065–1074. [Google Scholar] [CrossRef]
- Mark, R.; Woodford, M.; Schulman, J.; Nsouli, T.; Mollapour, M. Efficacy of the Hsp90 Inhibitors in Prostate Cancer Therapy. J. Urol. Nephrol. 2014, 1, 1–9. [Google Scholar] [CrossRef]
- Patwardhan, C.A.; Fauq, A.; Peterson, L.B.; Miller, C.; Blagg, B.S.J.; Chadli, A. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J. Biol. Chem. 2013, 288, 7313–7325. [Google Scholar] [CrossRef]
- Zhao, H.; Blagg, B.S.J. Novobiocin analogues with second-generation noviose surrogates. Bioorg. Med. Chem. Lett. 2013, 23, 552–557. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef]
- Zhang, Y.; Dayalan Naidu, S.; Samarasinghe, K.; Van Hecke, G.C.; Pheely, A.; Boronina, T.N.; Cole, R.N.; Benjamin, I.J.; Cole, P.A.; Ahn, Y.-H.; et al. Sulphoxythiocarbamates modify cysteine residues in HSP90 causing degradation of client proteins and inhibition of cancer cell proliferation. Br. J. Cancer 2014, 110, 71–82. [Google Scholar] [CrossRef]
- Solit, D.B.; Scher, H.I.; Rosen, N. Hsp90 as a therapeutic target in prostate cancer. Semin. Oncol. 2003, 30, 709–716. [Google Scholar] [CrossRef]
- O’Malley, K.J.; Langmann, G.; Ai, J.; Ramos-Garcia, R.; Vessella, R.L.; Wang, Z. Hsp90 inhibitor 17-AAG inhibits progression of LuCaP35 xenograft prostate tumors to castration resistance. Prostate 2012, 72, 1117–1123. [Google Scholar] [CrossRef]
- Proia, D.A.; Bates, R.C. Ganetespib and HSP90: Translating preclinical hypotheses into clinical promise. Cancer Res. 2014, 74, 1294–1300. [Google Scholar] [CrossRef]
- Shafi, A.A.; Cox, M.B.; Weigel, N.L. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids 2013, 78, 548–554. [Google Scholar] [CrossRef]
- Shelton, S.N.; Shawgo, M.E.; Matthews, S.B.; Lu, Y.; Donnelly, A.C.; Szabla, K.; Tanol, M.; Vielhauer, G.A.; Rajewski, R.A.; Matts, R.L.; et al. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 2009, 76, 1314–1322. [Google Scholar] [CrossRef]
- Kang, B.H.; Siegelin, M.D.; Plescia, J.; Raskett, C.M.; Garlick, D.S.; Dohi, T.; Lian, J.B.; Stein, G.S.; Languino, L.R.; Altieri, D.C. Preclinical characterization of mitochondria-targeted small molecule hsp90 inhibitors, gamitrinibs, in advanced prostate cancer. Clin. Cancer Res. 2010, 16, 4779–4788. [Google Scholar] [CrossRef]
- Koya, K.; Li, Y.; Wang, H.; Ukai, T.; Tatsuta, N.; Kawakami, M.; Shishido, T.; Chen, L.B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [Google Scholar]
- Lamoureux, F.; Thomas, C.; Yin, M.-J.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Suppression of heat shock protein 27 using OGX-427 induces endoplasmic reticulum stress and potentiates heat shock protein 90 inhibitors to delay castrate-resistant prostate cancer. Eur. Urol. 2014, 66, 145–155. [Google Scholar] [CrossRef]
- Miyake, H.; Nelson, C.; Rennie, P.S.; Gleave, M.E. Testosterone-repressed prostate message-2 is an antiapoptotic gene involved in progression to androgen independence in prostate cancer. Cancer Res. 2000, 60, 170–176. [Google Scholar]
- Gleave, M.E.; Miyake, H.; Zellweger, T.; Chi, K.; July, L.; Nelson, C.; Rennie, P. Use of antisense oligonucleotides targeting the antiapoptotic gene, clusterin/testosterone-repressed prostate message 2, to enhance androgen sensitivity and chemosensitivity in prostate cancer. Urology 2001, 58, 39–49. [Google Scholar] [CrossRef]
- Miyake, H.; Nelson, C.; Rennie, P.S.; Gleave, M.E. Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 2000, 60, 2547–2554. [Google Scholar]
- Miyake, H.; Chi, K.N.; Gleave, M.E. Antisense TRPM-2 oligodeoxynucleotides chemosensitize human androgen-independent PC-3 prostate cancer cells both in vitro and in vivo. Clin. Cancer Res. 2000, 6, 1655–1663. [Google Scholar] [CrossRef]
- Zellweger, T.; Chi, K.; Miyake, H.; Adomat, H.; Kiyama, S.; Skov, K.; Gleave, M.E. Enhanced radiation sensitivity in prostate cancer by inhibition of the cell survival protein clusterin. Clin. Cancer Res. 2002, 8, 3276–3284. [Google Scholar]
- Sowery, R.D.; Hadaschik, B.A.; So, A.I.; Zoubeidi, A.; Fazli, L.; Hurtado-Coll, A.; Gleave, M.E. Clusterin knockdown using the antisense oligonucleotide OGX-011 re-sensitizes docetaxel-refractory prostate cancer PC-3 cells to chemotherapy. BJU Int. 2008, 102, 389–397. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Yin, M.-J.; Kuruma, H.; Beraldi, E.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Clusterin inhibition using OGX-011 synergistically enhances Hsp90 inhibitor activity by suppressing the heat shock response in castrate-resistant prostate cancer. Cancer Res. 2011, 71, 5838–5849. [Google Scholar] [CrossRef]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 2012, 83, 995–1004. [Google Scholar] [CrossRef]
- Ischia, J.; Saad, F.; Gleave, M. The promise of heat shock protein inhibitors in the treatment of castration resistant prostate cancer. Curr. Opin. Urol. 2013, 23, 194–200. [Google Scholar] [CrossRef]
- Banerji, U. Heat shock protein 90 as a drug target: Some like it hot. Clin. Cancer Res. 2009, 15, 9–14. [Google Scholar] [CrossRef]
- Gandhi, N.; Wild, A.T.; Chettiar, S.T.; Aziz, K.; Kato, Y.; Gajula, R.P.; Williams, R.D.; Cades, J.A.; Annadanam, A.; Song, D.; et al. Novel Hsp90 inhibitor NVP-AUY922 radiosensitizes prostate cancer cells. Cancer Biol. Ther. 2013, 14, 347–356. [Google Scholar] [CrossRef]
- Zaarur, N.; Gabai, V.L.; Porco, J.A.; Calderwood, S.; Sherman, M.Y. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res. 2006, 66, 1783–1791. [Google Scholar] [CrossRef]
- Ma, Y.; Lakshmikanthan, V.; Lewis, R.W.; Kumar, M.V. Sensitization of TRAIL-resistant cells by inhibition of heat shock protein 90 with low-dose geldanamycin. Mol. Cancer Ther. 2006, 5, 170–178. [Google Scholar] [CrossRef]
- Ding, G.; Feng, C.; Jiang, H.; Ding, Q.; Zhang, L.; Na, R.; Xu, H.; Liu, J. Combination of rapamycin, CI-1040, and 17-AAG inhibits metastatic capacity of prostate cancer via Slug inhibition. PLoS ONE 2013, 8, e77400. [Google Scholar] [CrossRef]
- Seaton, A.; Maxwell, P.J.; Hill, A.; Gallagher, R.; Pettigrew, J.; Wilson, R.H.; Waugh, D.J.J. Inhibition of constitutive and cxc-chemokine-induced NF-kappaB activity potentiates ansamycin-based HSP90-inhibitor cytotoxicity in castrate-resistant prostate cancer cells. Br. J. Cancer 2009, 101, 1620–1629. [Google Scholar] [CrossRef]
- Iwai, A.; Bourboulia, D.; Mollapour, M.; Jensen-Taubman, S.; Lee, S.; Donnelly, A.C.; Yoshida, S.; Miyajima, N.; Tsutsumi, S.; Smith, A.K.; et al. Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle 2012, 11, 3649–3655. [Google Scholar] [CrossRef]
- Mollapour, M.; Tsutsumi, S.; Donnelly, A.C.; Beebe, K.; Tokita, M.J.; Lee, M.-J.; Lee, S.; Morra, G.; Bourboulia, D.; Scroggins, B.T.; et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell 2010, 37, 333–343. [Google Scholar] [CrossRef]
- Hotte, S.J.; Yu, E.Y.; Hirte, H.W.; Higano, C.S.; Gleave, M.E.; Chi, K.N. Phase I trial of OGX-427, a 2’methoxyethyl antisense oligonucleotide (ASO), against heat shock protein 27 (Hsp27): Final results. J. Clin. Oncol. 2010, 28, 3077. [Google Scholar] [CrossRef]
- Eustace, B.K.; Sakurai, T.; Stewart, J.K.; Yimlamai, D.; Unger, C.; Zehetmeier, C.; Lain, B.; Torella, C.; Henning, S.W.; Beste, G.; et al. Functional proteomic screens reveal an essential extracellular role for hsp90α in cancer cell invasiveness. Nat. Cell Biol. 2004, 6, 507–514. [Google Scholar] [CrossRef]
- Gopal, U.; Bohonowych, J.E.; Lema-Tome, C.; Liu, A.; Garrett-Mayer, E.; Wang, B.; Isaacs, J.S. A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLoS ONE 2011, 6, e17649. [Google Scholar] [CrossRef]
- Hoter, A.; Amiri, M.; Warda, M.; Naim, H.Y. Molecular cloning, cellular expression and characterization of Arabian camel (Camelus dromedarius) endoplasmin. Int. J. Biol. Macromol. 2018, 117, 574–585. [Google Scholar] [CrossRef]
- Evdokimovskaya, Y.; Skarga, Y.; Vrublevskaya, V.; Morenkov, O. Release of the glucose-regulated protein 94 by baby hamster kidney cells. Cell Biochem. Funct. 2012, 30, 558–562. [Google Scholar] [CrossRef]
- Weinberg, R.A.; Lander, E.S.; Gupta, P.B.; Yang, J.; Onder, T.T.; Mani, S.A. Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Bohonowych, J.E.; Hance, M.W.; Nolan, K.D.; Defee, M.; Parsons, C.H.; Isaacs, J.S. Extracellular Hsp90 mediates an NF-κB dependent inflammatory stromal program: Implications for the prostate tumor microenvironment. Prostate 2014, 74, 395–407. [Google Scholar] [CrossRef]
- Ramteke, A.; Ting, H.; Agarwal, C.; Mateen, S.; Somasagara, R.; Hussain, A.; Graner, M.; Frederick, B.; Agarwal, R.; Deep, G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol. Carcinog. 2015, 54, 554–565. [Google Scholar] [CrossRef]
- Yang, Y.; Rao, R.; Shen, J.; Tang, Y.; Fiskus, W.; Nechtman, J.; Atadja, P.; Bhalla, K. Role of Acetylation and Extracellular Location of Heat Shock Protein 90A in Tumor Cell Invasion. Cancer 2008, 68, 4833–4842. [Google Scholar]
- Walton-Diaz, A.; Khan, S.; Bourboulia, D.; Trepel, J.B.; Neckers, L.; Mollapour, M. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Med. Chem. 2013, 5, 1059–1071. [Google Scholar] [CrossRef]
- Rao, R.; Fiskus, W.; Yang, Y.; Lee, P.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG-mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008, 112, 1886–1893. [Google Scholar] [CrossRef]
- Gravina, G.L.; Marampon, F.; Muzi, P.; Mancini, A.; Piccolella, M.; Negri-Cesi, P.; Motta, M.; Lenzi, A.; Di Cesare, E.; Tombolini, V.; et al. PXD101 potentiates hormonal therapy and prevents the onset of castration-resistant phenotype modulating androgen receptor, HSP90, and CRM1 in preclinical models of prostate cancer. Endocr. Relat. Cancer 2013, 20, 321–337. [Google Scholar] [CrossRef]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 21, 109–113. [Google Scholar] [CrossRef]
- Siligardi, G.; Panaretou, B.; Meyer, P.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef]
- Abbas-Terki, T.; Briand, P.-A.; Donzé, O.; Picard, D. The Hsp90 co-chaperones Cdc37 and Sti1 interact physically and genetically. Biol. Chem. 2002, 383, 1335–1342. [Google Scholar] [CrossRef]
- Wu, F.; Peacock, S.O.; Rao, S.; Lemmon, S.K.; Burnstein, K.L. Novel interaction between the co-chaperone Cdc37 and Rho GTPase exchange factor Vav3 promotes androgen receptor activity and prostate cancer growth. J. Biol. Chem. 2013, 288, 5463–5474. [Google Scholar] [CrossRef]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.A.L.; Zhan, C.-G.; Sun, D. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmacol. 2010, 79, 542–551. [Google Scholar] [CrossRef]
- Verma, S.; Singh, A.; Mishra, A. Dual inhibition of chaperoning process by taxifolin: Molecular dynamics simulation study. J. Mol. Graph. Model. 2012, 37, 27–38. [Google Scholar] [CrossRef]
- Reebye, V.; Querol Cano, L.; Lavery, D.N.; Brooke, G.N.; Powell, S.M.; Chotai, D.; Walker, M.M.; Whitaker, H.C.; Wait, R.; Hurst, H.C.; et al. Role of the HSP90-associated cochaperone p23 in enhancing activity of the androgen receptor and significance for prostate cancer. Mol. Endocrinol. 2012, 26, 1694–1706. [Google Scholar] [CrossRef]
- De Leon, J.T.; Iwai, A.; Feau, C.; Garcia, Y.; Balsiger, H.A.; Storer, C.L.; Suro, R.M.; Garza, K.M.; Lee, S.; Kim, Y.S.; et al. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11878–11883. [Google Scholar] [CrossRef]
- Trotta, A.P.; Need, E.F.; Selth, L.A.; Chopra, S.; Pinnock, C.B.; Leach, D.A.; Coetzee, G.A.; Butler, L.M.; Tilley, W.D.; Buchanan, G. Knockdown of the cochaperone SGTA results in the suppression of androgen and PI3K/Akt signaling and inhibition of prostate cancer cell proliferation. Int. J. Cancer 2013, 133, 2812–2823. [Google Scholar] [CrossRef]
- Philp, L.K.; Butler, M.S.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Day, T.K. SGTA: A new player in the molecular co-chaperone game. Horm. Cancer 2013, 4, 343–357. [Google Scholar] [CrossRef]
- Philp, L.K.; Day, T.K.; Butler, M.S.; Laven-Law, G.; Jindal, S.; Hickey, T.E.; Scher, H.I.; Butler, L.M.; Tilley, W.D. Small Glutamine-Rich Tetratricopeptide Repeat-Containing Protein Alpha (SGTA) Ablation Limits Offspring Viability and Growth in Mice. Sci. Rep. 2016, 6, 28950. [Google Scholar] [CrossRef]
- Brocchieri, L.; Conway de Macario, E.; Macario, A.J. hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol. Biol. 2008, 8, 19. [Google Scholar] [CrossRef]
- Hageman, J.; Van Waarde, M.A.W.H.; Zylicz, A.; Walerych, D.; Kampinga, H.H. The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem. J. 2011, 435, 127–142. [Google Scholar] [CrossRef]
- Vos, M.J.; Hageman, J.; Carra, S.; Kampinga, H.H. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 2008, 47, 7001–7011. [Google Scholar] [CrossRef]
- Jäättelä, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar] [CrossRef]
- Shi, Y.; Thomas, J.O. The transport of proteins into the nucleus requires the 70-kilodalton heat shock protein or its cytosolic cognate. Mol. Cell. Biol. 1992, 12, 2186–2192. [Google Scholar] [CrossRef]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef]
- Nylandsted, J.; Brand, K.; Jäättelä, M. Heat shock protein 70 is required for the survival of cancer cells. Ann. N. Y Acad. Sci. 2000, 926, 122–125. [Google Scholar] [CrossRef]
- Watson, M.J.; Bingman, W.E.; Wells, C.; Rivera-Marquez, G.M.; Gestwicki, J.E.; Kim, Y.S.; Trepel, J.B.; Zuehlke, A.D.; Beebe, K.E.; Oshima, N.; et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef]
- Pootrakul, L.; Datar, R.H.; Shi, S.-R.; Cai, J.; Hawes, D.; Groshen, S.G.; Lee, A.S.; Cote, R.J. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin. Cancer Res. 2006, 12, 5987–5993. [Google Scholar] [CrossRef]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Kuo, L.-J.; Huang, C.-Y.; Cheng, W.-L.; Hung, C.-S.; Wu, C.-T.; Lin, F.-Y.; Chang, Y.-J.; Huang, M.-T. Glucose-regulated protein 78 mediates the anticancer efficacy of shikonin in hormone-refractory prostate cancer cells. Tumor Biol. 2015, 36, 5063–5070. [Google Scholar] [CrossRef]
- Cultrara, C.N.; Kozuch, S.D.; Ramasundaram, P.; Heller, C.J.; Shah, S.; Beck, A.E.; Sabatino, D.; Zilberberg, J. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018, 18, 1263. [Google Scholar] [CrossRef]
- Calderwood, S.K. Heat shock proteins and cancer: Intracellular chaperones or extracellular signaling ligands? Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Arrigo, A.P.; Calderwood, S.K. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 2013, 87, 19–48. [Google Scholar] [CrossRef]
- Roigas, J.; Wallen, E.S.; Loening, S.A.; Moseley, P.L. Effects of combined treatment of chemotherapeutics and hyperthermia on survival and the regulation of heat shock proteins in Dunning R3327 prostate carcinoma cells. Prostate 1998, 34, 195–202. [Google Scholar] [CrossRef]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar] [CrossRef]
- Williamson, D.S.; Borgognoni, J.; Clay, A.; Daniels, Z.; Dokurno, P.; Drysdale, M.J.; Foloppe, N.; Francis, G.L.; Graham, C.J.; Howes, R.; et al. Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J. Med. Chem. 2009, 52, 1510–1513. [Google Scholar] [CrossRef]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Miyata, Y.; Koren, J.; Jones, J.R.; Trotter, J.H.; Chang, L.; O’Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J. Neurosci. 2009, 29, 12079–12088. [Google Scholar] [CrossRef]
- Britten, C.D.; Rowinsky, E.K.; Baker, S.D.; Weiss, G.R.; Smith, L.; Stephenson, J.; Rothenberg, M.; Smetzer, L.; Cramer, J.; Collins, W.; et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin. Cancer Res. 2000, 6, 42–49. [Google Scholar]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1-42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef]
- Gurbuxani, S.; Schmitt, E.; Cande, C.; Parcellier, A.; Hammann, A.; Daugas, E.; Kouranti, I.; Spahr, C.; Pance, A.; Kroemer, G.; et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 2003, 22, 6669–6678. [Google Scholar] [CrossRef]
- Schmitt, E.; Maingret, L.; Puig, P.-E.; Rerole, A.-L.; Ghiringhelli, F.; Hammann, A.; Solary, E.; Kroemer, G.; Garrido, C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006, 66, 4191–4197. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef]
- Steele, A.J.; Prentice, A.G.; Hoffbrand, A.V.; Yogashangary, B.C.; Hart, S.M.; Lowdell, M.W.; Samuel, E.R.; North, J.M.; Nacheva, E.P.; Chanalaris, A.; et al. 2-Phenylacetylenesulfonamide (PAS) induces p53-independent apoptotic killing of B-chronic lymphocytic leukemia (CLL) cells. Blood 2009, 114, 1217–1225. [Google Scholar] [CrossRef]
- Yi, F.; Regan, L. A novel class of small molecule inhibitors of Hsp90. ACS Chem. Biol. 2008, 3, 645–654. [Google Scholar] [CrossRef]
- Roodveldt, C.; Bertoncini, C.W.; Andersson, A.; van der Goot, A.T.; Hsu, S.-T.; Fernández-Montesinos, R.; de Jong, J.; van Ham, T.J.; Nollen, E.A.; Pozo, D.; et al. Chaperone proteostasis in Parkinson’s disease: Stabilization of the Hsp70/alpha-synuclein complex by Hip. EMBO J. 2009, 28, 3758–3770. [Google Scholar] [CrossRef]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.M.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005, 65, 10536–10544. [Google Scholar] [CrossRef]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell. Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef]
- Munje, C.; Shervington, L.; Khan, Z.; Shervington, A. Could Upregulated Hsp70 Protein Compensate for the Hsp90-Silence-Induced Cell Death in Glioma Cells? Int. J. Brain Sci. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Powers, M.V.; Clarke, P.A.; Workman, P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 2008, 14, 250–262. [Google Scholar] [CrossRef]
- Welch, W.J. Mammalian stress response: Cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol. Rev. 1992, 72, 1063–1081. [Google Scholar] [CrossRef]
- Viitanen, P.V.; Lorimer, G.; Bergmeier, W.; Weiss, C.; Kessel, M.; Goloubinoff, P. Purification of mammalian mitochondrial chaperonin 60 through in vitro reconstitution of active oligomers. Methods Enzymol. 1998, 290, 203–217. [Google Scholar]
- Jindal, S.; Dudani, A.K.; Singh, B.; Harley, C.B.; Gupta, R.S. Primary structure of a human mitochondrial protein homologous to the bacterial and plant chaperonins and to the 65-kilodalton mycobacterial antigen. Mol. Cell. Biol. 1989, 9, 2279–2283. [Google Scholar] [CrossRef]
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; Conway de Macario, E.; Macario, A.J.L. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2014, 18, 185–208. [Google Scholar] [CrossRef]
- Bini, L.; Magi, B.; Marzocchi, B.; Arcuri, F.; Tripodi, S.; Cintorino, M.; Sanchez, J.C.; Frutiger, S.; Hughes, G.; Pallini, V.; et al. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis 1997, 18, 2832–2841. [Google Scholar] [CrossRef]
- He, Y.; Wu, Y.; Mou, Z.; Li, W.; Zou, L.; Fu, T.; Zhang, A.; Xiang, D.; Xiao, H.; Wang, X. Proteomics-based identification of HSP60 as a tumor-associated antigen in colorectal cancer. Proteom. Clin. Appl. 2007, 1, 336–342. [Google Scholar] [CrossRef]
- Cappello, F.; Bellafiore, M.; Palma, A.; David, S.; Marcianò, V.; Bartolotta, T.; Sciumè, C.; Modica, G.; Farina, F.; Zummo, G.; et al. 60KDa chaperonin (HSP60) is over-expressed during colorectal carcinogenesis. Eur. J. Histochem. 2003, 47, 105–110. [Google Scholar] [CrossRef]
- Xu, X.; Wang, W.; Shao, W.; Yin, W.; Chen, H.; Qiu, Y.; Mo, M.; Zhao, J.; Deng, Q.; He, J. Heat shock protein-60 expression was significantly correlated with the prognosis of lung adenocarcinoma. J. Surg. Oncol. 2011, 104, 598–603. [Google Scholar] [CrossRef]
- Hjerpe, E.; Egyhazi, S.; Carlson, J.; Stolt, M.F.; Schedvins, K.; Johansson, H.; Shoshan, M.; Avall-Lundqvist, E. HSP60 predicts survival in advanced serous ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 448–455. [Google Scholar] [CrossRef]
- Cappello, F.; Rappa, F.; David, S.; Anzalone, R.; Zummo, G. Immunohistochemical evaluation of PCNA, p53, HSP60, HSP10 and MUC-2 presence and expression in prostate carcinogenesis. Anticancer Res. 2003, 23, 1325–1331. [Google Scholar]
- Castilla, C.; Congregado, B.; Conde, J.M.; Medina, R.; Torrubia, F.J.; Japón, M.A.; Sáez, C. Immunohistochemical expression of Hsp60 correlates with tumor progression and hormone resistance in prostate cancer. Urology 2010, 76, 1017.e1–1017.e6. [Google Scholar] [CrossRef]
- Cornford, P.A.; Dodson, A.R.; Parsons, K.F.; Desmond, A.D.; Woolfenden, A.; Fordham, M.; Neoptolemos, J.P.; Ke, Y.; Foster, C.S. Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res. 2000, 60, 7099–7105. [Google Scholar]
- Johansson, B.; Pourian, M.R.; Chuan, Y.-C.; Byman, I.; Bergh, A.; Pang, S.-T.; Norstedt, G.; Bergman, T.; Pousette, A. Proteomic comparison of prostate cancer cell lines LNCaP-FGC and LNCaP-r reveals heatshock protein 60 as a marker for prostate malignancy. Prostate 2006, 66, 1235–1244. [Google Scholar] [CrossRef]
- Mizuno, K.; Tsujino, M.; Takada, M.; Hayashi, M.; Atsumi, K. Studies on bredinin. I. Isolation, characterization and biological properties. J. Antibiot. 1974, 27, 775–782. [Google Scholar] [CrossRef]
- Tanabe, M.; Ishida, R.; Izuhara, F.; Komatsuda, A.; Wakui, H.; Sawada, K.; Otaka, M.; Nakamura, N.; Itoh, H. The ATPase activity of molecular chaperone HSP60 is inhibited by immunosuppressant mizoribine. AJMB 2012, 2012, 93–102. [Google Scholar] [CrossRef]
- Honda, M.; Itoh, H.; Suzuki, T.; Hashimoto, Y. Population pharmacokinetics of higher-dose mizoribine in healthy male volunteers. Biol. Pharm. Bull. 2006, 29, 2460–2464. [Google Scholar] [CrossRef]
- Kakeya, H.; Takahashi, I.; Okada, G.; Isono, K.; Osada, H. Epolactaene, a novel neuritogenic compound in human neuroblastoma cells, produced by a marine fungus. J. Antibiot. 1995, 48, 733–735. [Google Scholar] [CrossRef]
- Nagumo, Y.; Kakeya, H.; Shoji, M.; Hayashi, Y.; Dohmae, N.; Osada, H. Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem. J. 2005, 387, 835–840. [Google Scholar] [CrossRef]
- Nagumo, Y.; Kakeya, H.; Yamaguchi, J.; Uno, T.; Shoji, M.; Hayashi, Y.; Osada, H. Structure-activity relationships of epolactaene derivatives: Structural requirements for inhibition of Hsp60 chaperone activity. Bioorg. Med. Chem. Lett. 2004, 14, 4425–4429. [Google Scholar] [CrossRef]
- Wiechmann, K.; Müller, H.; König, S.; Wielsch, N.; Svatoš, A.; Jauch, J.; Werz, O. Mitochondrial Chaperonin HSP60 Is the Apoptosis-Related Target for Myrtucommulone. Cell Chem. Biol. 2017, 24, 614–623. [Google Scholar] [CrossRef]
- Wiechmann, K.; Müller, H.; Huch, V.; Hartmann, D.; Werz, O.; Jauch, J. Synthesis and biological evaluation of novel myrtucommulones and structural analogues that target mPGES-1 and 5-lipoxygenase. Eur. J. Med. Chem. 2015, 101, 133–149. [Google Scholar] [CrossRef]
- Ban, H.S.; Shimizu, K.; Minegishi, H.; Nakamura, H. Identification of HSP60 as a primary target of o-carboranylphenoxyacetanilide, an HIF-1alpha inhibitor. J. Am. Chem. Soc. 2010, 132, 11870–11871. [Google Scholar] [CrossRef]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef]
- Kriehuber, T.; Rattei, T.; Weinmaier, T.; Bepperling, A.; Haslbeck, M.; Buchner, J. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 2010, 24, 3633–3642. [Google Scholar] [CrossRef]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.J.J.M.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef]
- Voll, E.A.; Ogden, I.M.; Pavese, J.M.; Huang, X.; Xu, L.; Jovanovic, B.D.; Bergan, R.C. Heat shock protein 27 regulates human prostate cancer cell motility and metastatic progression. Oncotarget 2014, 5, 2648–2663. [Google Scholar] [CrossRef]
- Salinthone, S.; Tyagi, M.; Gerthoffer, W.T. Small heat shock proteins in smooth muscle. Pharm. Ther. 2008, 119, 44–54. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Gleave, M. Small heat shock proteins in cancer therapy and prognosis. Int. J. Biochem. Cell Biol. 2012, 44, 1646–1656. [Google Scholar] [CrossRef]
- Kostenko, S.; Moens, U. Heat shock protein 27 phosphorylation: Kinases, phosphatases, functions and pathology. Cell. Mol. Life Sci. 2009, 66, 3289–3307. [Google Scholar] [CrossRef]
- Parcellier, A.; Schmitt, E.; Brunet, M.; Hammann, A.; Solary, E.; Garrido, C. Small heat shock proteins HSP27 and alphaB-crystallin: Cytoprotective and oncogenic functions. Antioxid. Redox Signal. 2005, 7, 404–413. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Levin, E.R. Heat shock protein 27 is required for sex steroid receptor trafficking to and functioning at the plasma membrane. Mol. Cell. Biol. 2010, 30, 3249–3261. [Google Scholar] [CrossRef]
- Cui, Y.; Sun, Y.; Hu, S.; Luo, J.; Li, L.; Li, X.; Yeh, S.; Jin, J.; Chang, C. Neuroendocrine prostate cancer (NEPCa) increased the neighboring PCa chemoresistance via altering the PTHrP/p38/Hsp27/androgen receptor (AR)/p21 signals. Oncogene 2016, 35, 6065–6076. [Google Scholar] [CrossRef]
- Garrido, C.; Fromentin, A.; Bonnotte, B.; Favre, N.; Moutet, M.; Arrigo, A.P.; Mehlen, P.; Solary, E. Heat shock protein 27 enhances the tumorigenicity of immunogenic rat colon carcinoma cell clones. Cancer Res. 1998, 58, 5495–5499. [Google Scholar]
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-Coll, A.; Yamanaka, K.; Gleave, M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res. 2004, 64, 6595–6602. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kang, S.; Kim, D.S.; Na, H.J.; Kim, Y.J.; Choi, Y.D.; Cho, N.H. HSP27, ALDH6A1 and Prohibitin Act as a Trio-biomarker to Predict Survival in Late Metastatic Prostate Cancer. Anticancer Res. 2018, 38, 6551–6560. [Google Scholar] [CrossRef]
- Foster, C.S.; Dodson, A.R.; Ambroisine, L.; Fisher, G.; Møller, H.; Clark, J.; Attard, G.; De-Bono, J.; Scardino, P.; Reuter, V.E.; et al. Hsp-27 expression at diagnosis predicts poor clinical outcome in prostate cancer independent of ETS-gene rearrangement. Br. J. Cancer 2009, 101, 1137–1144. [Google Scholar] [CrossRef]
- Bubendorf, L.; Kolmer, M.; Kononen, J.; Koivisto, P.; Mousses, S.; Chen, Y.; Mahlamäki, E.; Schraml, P.; Moch, H.; Willi, N.; et al. Hormone therapy failure in human prostate cancer: Analysis by complementary DNA and tissue microarrays. J. Natl. Cancer Inst. 1999, 91, 1758–1764. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Lin, P.J.C.; Beraldi, E.; Zhang, F.; Kawai, Y.; Leong, J.; Katsumi, H.; Fazli, L.; Fraser, R.; Cullis, P.R.; et al. siRNA Lipid Nanoparticle Potently Silences Clusterin and Delays Progression When Combined with Androgen Receptor Cotargeting in Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4845–4855. [Google Scholar] [CrossRef]
- Rocchi, P.; Jugpal, P.; So, A.; Sinneman, S.; Ettinger, S.; Fazli, L.; Nelson, C.; Gleave, M. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU Int. 2006, 98, 1082–1089. [Google Scholar] [CrossRef]
- Zielinski, R.; Chi, K.N. Custirsen (OGX-011): A second-generation antisense inhibitor of clusterin in development for the treatment of prostate cancer. Future Oncol. 2012, 8, 1239–1251. [Google Scholar] [CrossRef]
- Kumano, M.; Furukawa, J.; Shiota, M.; Zardan, A.; Zhang, F.; Beraldi, E.; Wiedmann, R.M.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol. Cancer Ther. 2012, 11, 1661–1671. [Google Scholar] [CrossRef]
- Chi, K.N.; Yu, E.Y.; Jacobs, C.; Bazov, J.; Kollmannsberger, C.; Higano, C.S.; Mukherjee, S.D.; Gleave, M.E.; Stewart, P.S.; Hotte, S.J. A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and other advanced cancers. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1116–1122. [Google Scholar] [CrossRef]
- Yu, E.Y.; Ellard, S.L.; Hotte, S.J.; Gingerich, J.R.; Joshua, A.M.; Gleave, M.E.; Chi, K.N. A randomized phase 2 study of a HSP27 targeting antisense, apatorsen with prednisone versus prednisone alone, in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 278–287. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, Y.J.; Choi, B.; Lee, N.L.; Lee, H.J.; Kwak, S.Y.; Kwon, Y.; Na, Y.; Lee, Y.-S. Overcoming HSP27-mediated resistance by altered dimerization of HSP27 using small molecules. Oncotarget 2016, 7, 53178–53190. [Google Scholar] [CrossRef]
- Choi, S.-H.; Lee, Y.-J.; Seo, W.D.; Lee, H.-J.; Nam, J.-W.; Lee, Y.J.; Kim, J.; Seo, E.-K.; Lee, Y.-S. Altered cross-linking of HSP27 by zerumbone as a novel strategy for overcoming HSP27-mediated radioresistance. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1196–1205. [Google Scholar] [CrossRef]
- Choi, B.; Choi, S.-K.; Park, Y.N.; Kwak, S.-Y.; Lee, H.J.; Kwon, Y.; Na, Y.; Lee, Y.-S. Sensitization of lung cancer cells by altered dimerization of HSP27. Oncotarget 2017, 8, 105372–105382. [Google Scholar] [CrossRef]
- Heinrich, J.C.; Donakonda, S.; Haupt, V.J.; Lennig, P. New HSP27 inhibitors efficiently down-regulate resistance development in cancer cells. Oncotarget 2016, 7, 68156–68169. [Google Scholar] [CrossRef]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef]
- Wilson, M.R.; Easterbrook-Smith, S.B. Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 2000, 25, 95–98. [Google Scholar] [CrossRef]
- Shannan, B.; Seifert, M.; Leskov, K.; Willis, J.; Boothman, D.; Tilgen, W.; Reichrath, J. Challenge and promise: Roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006, 13, 12–19. [Google Scholar] [CrossRef]
- Pucci, S.; Bonanno, E.; Pichiorri, F.; Angeloni, C.; Spagnoli, L.G. Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene 2004, 23, 2298–2304. [Google Scholar] [CrossRef]
- Trougakos, I.P.; So, A.; Jansen, B.; Gleave, M.E.; Gonos, E.S. Silencing expression of the clusterin/apolipoprotein j gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress. Cancer Res. 2004, 64, 1834–1842. [Google Scholar] [CrossRef]
- Loison, F.; Debure, L.; Nizard, P.; Le Goff, P.; Michel, D.; Le Dréan, Y. Up-regulation of the clusterin gene after proteotoxic stress: Implication of HSF1-HSF2 heterocomplexes. Biochem. J. 2006, 395, 223–231. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Wang, Z.; Muramaki, M.; Gleave, M.E.; Nelson, C.C. Differential regulation of clusterin and its isoforms by androgens in prostate cells. J. Biol. Chem. 2007, 282, 2278–2287. [Google Scholar] [CrossRef]
- Shiota, M.; Zoubeidi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Nelson, C.C.; Sorensen, P.H.B.; Gleave, M.E. Clusterin is a critical downstream mediator of stress-induced YB-1 transactivation in prostate cancer. Mol. Cancer Res. 2011, 9, 1755–1766. [Google Scholar] [CrossRef]
- Lee, K.-B.; Jeon, J.-H.; Choi, I.; Kwon, O.-Y.; Yu, K.; You, K.-H. Clusterin, a novel modulator of TGF-beta signaling, is involved in Smad2/3 stability. Biochem. Biophys. Res. Commun. 2008, 366, 905–909. [Google Scholar] [CrossRef]
- Chou, T.-Y.; Chen, W.-C.; Lee, A.-C.; Hung, S.-M.; Shih, N.-Y.; Chen, M.-Y. Clusterin silencing in human lung adenocarcinoma cells induces a mesenchymal-to-epithelial transition through modulating the ERK/Slug pathway. Cell. Signal. 2009, 21, 704–711. [Google Scholar] [CrossRef]
- Song, H.; Zhang, B.; Watson, M.A.; Humphrey, P.A.; Lim, H.; Milbrandt, J. Loss of Nkx3.1 leads to the activation of discrete downstream target genes during prostate tumorigenesis. Oncogene 2009, 28, 3307–3319. [Google Scholar] [CrossRef]
- Steinberg, J.; Oyasu, R.; Lang, S.; Sintich, S.; Rademaker, A.; Lee, C.; Kozlowski, J.M.; Sensibar, J.A. Intracellular levels of SGP-2 (Clusterin) correlate with tumor grade in prostate cancer. Clin. Cancer Res. 1997, 3, 1707–1711. [Google Scholar]
- July, L.V.; Akbari, M.; Zellweger, T.; Jones, E.C.; Goldenberg, S.L.; Gleave, M.E. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate 2002, 50, 179–188. [Google Scholar] [CrossRef]
- Pins, M.R.; Fiadjoe, J.E.; Korley, F.; Wong, M.; Rademaker, A.W.; Jovanovic, B.; Yoo, T.K.; Kozlowski, J.M.; Raji, A.; Yang, X.J.; et al. Clusterin as a possible predictor for biochemical recurrence of prostate cancer following radical prostatectomy with intermediate Gleason scores: A preliminary report. Prostate Cancer Prostatic Dis. 2004, 7, 243–248. [Google Scholar] [CrossRef]
- Girard, F.P.; Byrne, J.; Downes, M.; Fanning, D.; Desgrandchamps, F.; Fitzpatrick, J.M.; Watson, R.W. Detecting soluble clusterin in in-vitro and in-vivo models of prostate cancer. Neoplasma 2010, 57, 488–493. [Google Scholar] [CrossRef][Green Version]
- Chi, K.N.; Eisenhauer, E.; Fazli, L.; Jones, E.C.; Goldenberg, S.L.; Powers, J.; Tu, D.; Gleave, M.E. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2’-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Natl. Cancer Inst. 2005, 97, 1287–1296. [Google Scholar] [CrossRef]
- Chi, K.N.; Hotte, S.J.; Yu, E.Y.; Tu, D.; Eigl, B.J.; Tannock, I.; Saad, F.; North, S.; Powers, J.; Gleave, M.E.; et al. Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 4247–4254. [Google Scholar] [CrossRef]
- Chi, K.N.; Higano, C.S.; Blumenstein, B.; Ferrero, J.-M.; Reeves, J.; Feyerabend, S.; Gravis, G.; Merseburger, A.S.; Stenzl, A.; Bergman, A.M.; et al. Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): A phase 3, multicentre, open-label, randomised trial. Lancet. Oncol. 2017, 18, 473–485. [Google Scholar] [CrossRef]
- Beer, T.M.; Hotte, S.J.; Saad, F.; Alekseev, B.; Matveev, V.; Fléchon, A.; Gravis, G.; Joly, F.; Chi, K.N.; Malik, Z.; et al. Custirsen (OGX-011) combined with cabazitaxel and prednisone versus cabazitaxel and prednisone alone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel (AFFINITY): A randomised, open-label, international, ph. Lancet Oncol. 2017, 18, 1532–1542. [Google Scholar] [CrossRef]




| Family Name | Sample Protein Members | Gene Name/M.W (kDa) | Cellular Location | Co-Chaperones | Roles | Citation |
|---|---|---|---|---|---|---|
| Small HSPs | HSP10 | HSPE1/10 | Mitochondria | None | Molecular chaperone (co-factor for HSP60) | [27] |
| HSP27 | HSPB1/22 | Cytosol/nucleus | Molecular chaperone | [28,29,30] | ||
| HSP40/DNAJ | HSP40 | DNAJB1/38 | Cytosol | None | Molecular chaperone (co-factor for HSP70) | [31,32,33] |
| Tid1 | DNAJA3/Isoform 1/52 | Cytosol | ||||
| DNAJA3/Isoform 2/49 | Mitochondria | |||||
| HSP60 | HSP60 | HSPD1/61 | Cytosol, mitochondria, chloroplast | HSP10 | Chaperonin | [34,35] |
| HSP70 | HSP70 | HSPA1A/70 | Cytosol | HSP40, Grpe, Bag1, Bag3, Hip, Hop, CHIP | Molecular chaperone | [36,37] |
| HSP70-2 | HSPA1B/70 | Cell surface | ||||
| HSC70 | HSPA8/71 | Cytosol | ||||
| GRP75/Mortalin | HSPA9/73 | Mitochondria | ||||
| GRP78 | HSPA5/72 | ER | ||||
| HSP90 | HSP90A | HSPC1/86 | Cytosol | P23, Aha1, Cyp40, Cdc37, Hop, FKBP51, FKBP52 | Molecular chaperone | [38,39,40] |
| HSP90B | HSPC3/84 | Cytosol | ||||
| GRP94 | HSPC4/92 | ER, cytosol | ||||
| TRAP1 | HSPC5/75 | Mitochondria | ||||
| Large HSPs | HSP110 | HSP110/96 | Cytosol | None | Holdase, molecular chaperone | [41,42,43] |
| GRP170 | HYOU1/170 | ER |
| HSP Inhibitor | Molecular Mechanism | Used Cell Line/Model | Reference |
|---|---|---|---|
| Targeting HSP90 | |||
| 17-AAG | Interferes with nuclear translocation of AR, postpones castration resistance, and promotes cell viability | LuCaP35 xenograft model | [124] |
| Downregulates the levels of HSP90 clients including AR, ERBB2, ERBB3, and Akt, and inhibits PCa growth | CWR22R & CWRSA6 xenografts | [84] | |
| Ganetespib | Reduces the expression of HSP90 client proteins such as AR, Akt, ERK, IGFR-1, EGFR, and STAT3, enhances apoptosis and interferes with growth of PCa cells | PC3 and 22Rv1 xenografts | [81] |
| NVP-AUY922 | Decreases HSP90 client proteins like ERBB2, c-RAF, CDK-4, Akt, and HIF-1α, inhibits growth of PC3 xenografts, and suppresses lymphatic metastasis | PC3LN3 orthotopic lymph node mPCa model | [102] |
| Exhibits anti-proliferative and pro-apoptotic functions | ex vivo model of primary PCa | [82] | |
| AT13387 | Causes degradation of HSP90 client proteins such as AR, Akt, ERBB2, and c-RAF, hinders AR nuclear translocation and inhibits its transcriptional activity and displays anti-proliferative and growth limiting effects in vivo | VCaP, LNCaP, 22Rv1 | [79] |
| NVP-HSP990 | Performs anti-proliferative and pro-apoptotic functions | ex vivo model of primary PCa | [82] |
| PF-04929113 (SNX-5422) | Depletes HSP90 client proteins like AR, ERBB2, Akt, and ERK, suppresses RANKL-mediated osteoclast differentiation interferes with growth of PCa xenografts | LNCaP xenografts | [86] |
| Gamitrinibs | Exhibits pro-apoptotic effects, reduces growth of PCa xenografts and inhibits bone metastasis | PC3 xenografts and orthotopic model of PCa | [128] |
| Shepherdin | Decreases HSP90 client proteins levels including survivin, Akt, CDK-4, and CDK-6, displays pro-apoptotic effects and delays growth of PCa xenografts | PC3 xenografts | [83] |
| KU174 | Supports apoptosis, delays growth of PCa xenografts, and depletes HSP90 client proteins, such as AR, survivin, ERBB2, and Akt | PC3 xenografts | [80] |
| Targeting HSP70 | |||
| MKT-077 | Delays growth of PCa cells | DU145 xenografts | [129] |
| Targeting HSP27 | |||
| OGX-427 | Interferes with HSP90-AR binding, promotes AR proteasomal degradation, supports apoptosis, suppresses growth of PCa xenografts, reduces serum PSA levels | LNCaP xenografts | [66] |
| Hinders metastasis | PC3M model of mPCa | [106] | |
| Targeting HSP90 and HSP27 | |||
| Combination of PF-04929113 and OGX-427 | The anti-proliferative and pro-apoptotic actions of PF-04929113 are augmented by OGX-427 due to its synergetic effect resulting in PCa growth inhibition | LNCaP xenograft | [130] |
| Targeting clusterin | |||
| Clusterin antisense oligonucleotide | Enhances apoptosis and counteracts recurrence in castration-sensitive PCa | Shionogi rat prostate tumors | [131] |
| Potentiates paclitaxel efficacy | Shionogi rat prostate tumors | [132,133] | |
| Increases efficacy of paclitaxel or mitoxantrone | PC3 xenografts | [134] | |
| Enhances response to radiation therapy | PC3 xenografts | [135] | |
| OGX-011 | Anti-metastatic effect | PC3M model | [108] |
| Enhances efficacy of enzalutamide | LNCaP xenografts | [97] | |
| Increases sensitivity to paclitaxel or mitoxantrone | PC3 xenografts | [136] | |
| Targeting HSP90 and clusterin | |||
| Combination of 17-AAG, PF-04929113 and OGX-011 | OGX-011 enhances apoptotic and anti-proliferative actions of 17-AAG and PF-04929113 resulting in synergistic inhibition of PCa growth | PC3 and LNCaP xenograft | [137] |
| Co-Chaperone | Roles | Targeting Approach | References |
|---|---|---|---|
| Cdc37 | Controls the HSP90 ATPase cycle by assisting the recruitment of kinase client proteins to the Hsp90 machinery | siRNA and natural product inhibitors (celastrol, withaferin A and taxifolin) | [121,161,162,163,164,165] |
| Interacts with Vav3, a co-activator of AR transcriptional activity, thus, increasing prostatic cell proliferation | |||
| p32 | Activates AR transcription by facilitating the binding of AR to the androgen response element (ARE) Participates in steroid receptor folding and signaling | Gedunin (inhibitor) | [119,166] |
| Mediates the binding of AR to HSP90 by acting as a bridge between the two molecules | |||
| Immunophilin FKBP52 | Stabilizes Hsp90-AR interaction | Knockout and inhibition by MJC13 | [64,126,167] |
| SGTA | Interacts with AR and keeps it in the cytoplasm | Knockdown and knockout | [168,169,170] |
| Enhances PCa proliferation and survival by promoting Akt signaling pathway |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoter, A.; Rizk, S.; Naim, H.Y. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers 2019, 11, 1194. https://doi.org/10.3390/cancers11081194
Hoter A, Rizk S, Naim HY. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers. 2019; 11(8):1194. https://doi.org/10.3390/cancers11081194
Chicago/Turabian StyleHoter, Abdullah, Sandra Rizk, and Hassan Y. Naim. 2019. "The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer" Cancers 11, no. 8: 1194. https://doi.org/10.3390/cancers11081194
APA StyleHoter, A., Rizk, S., & Naim, H. Y. (2019). The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers, 11(8), 1194. https://doi.org/10.3390/cancers11081194