4-Methylumbelliferone Inhibits Cancer Stem Cell Activation and Overcomes Chemoresistance in Ovarian Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
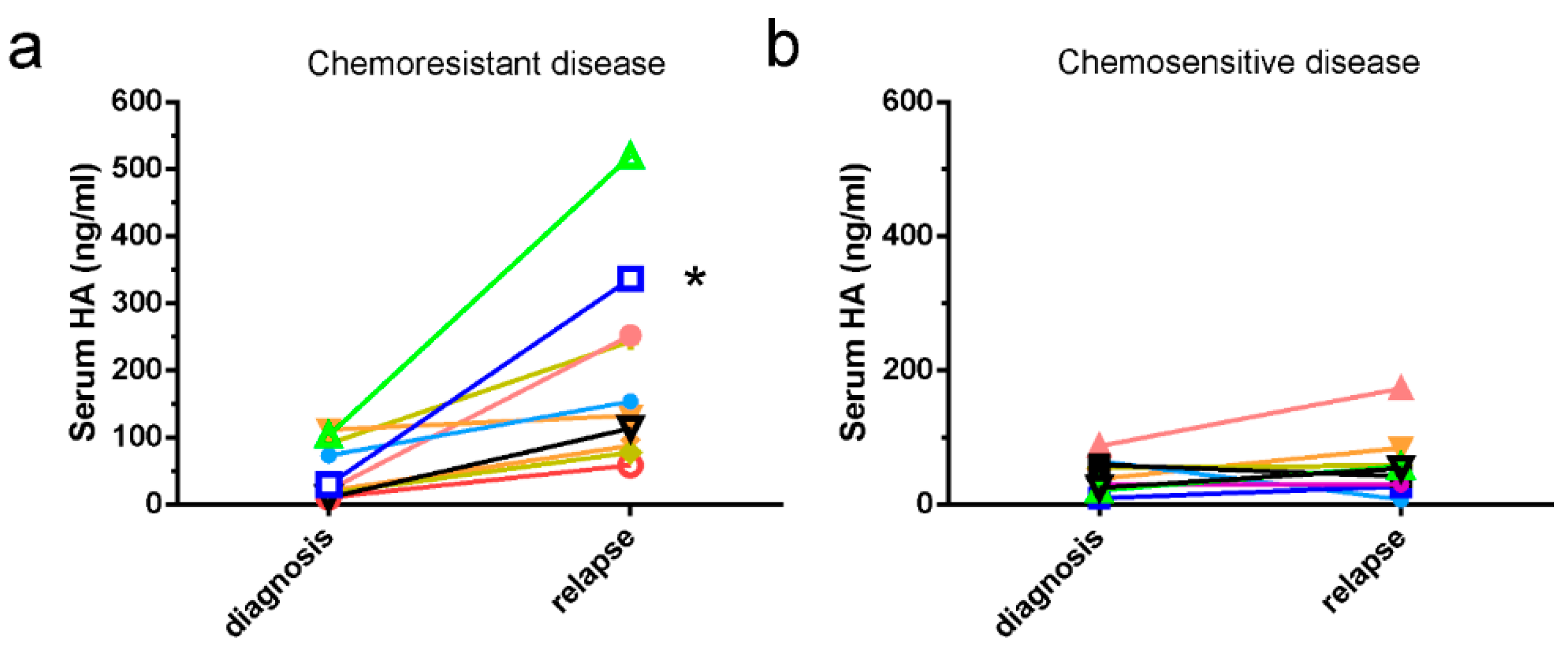
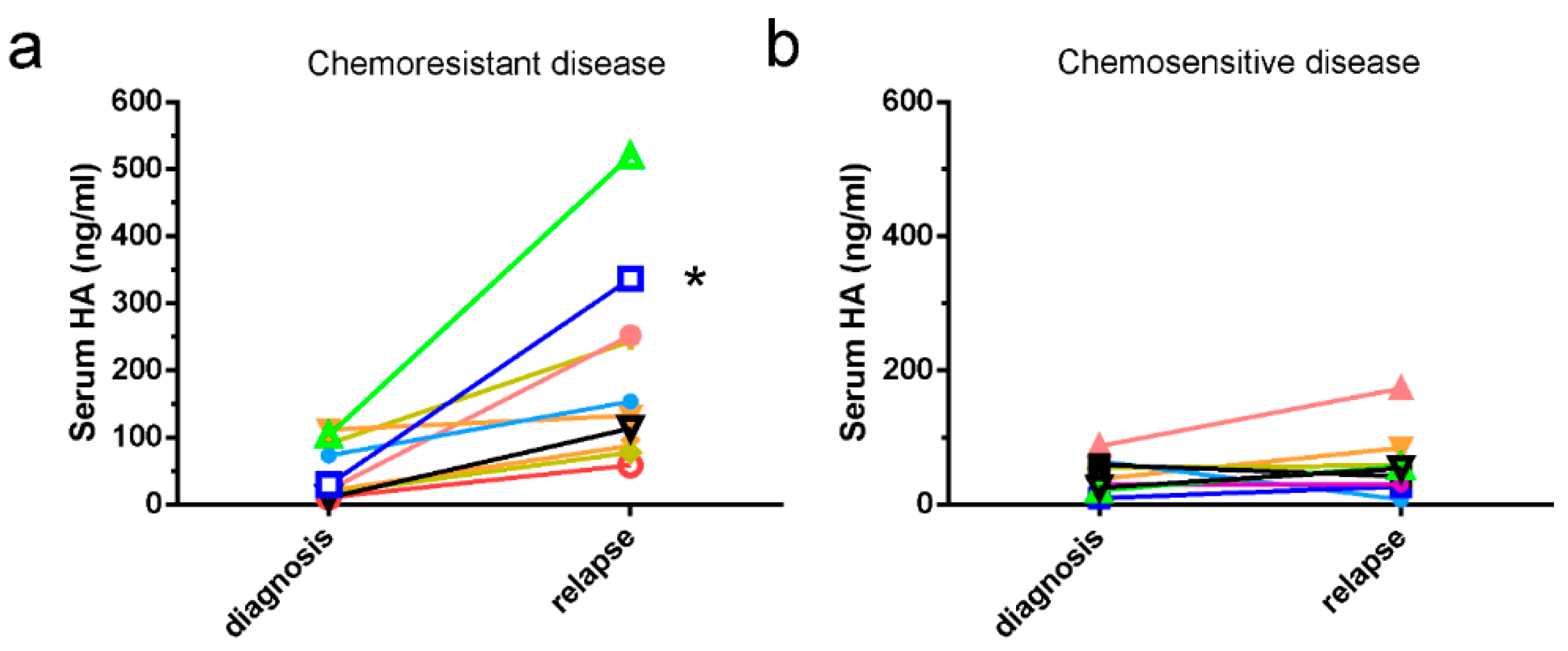
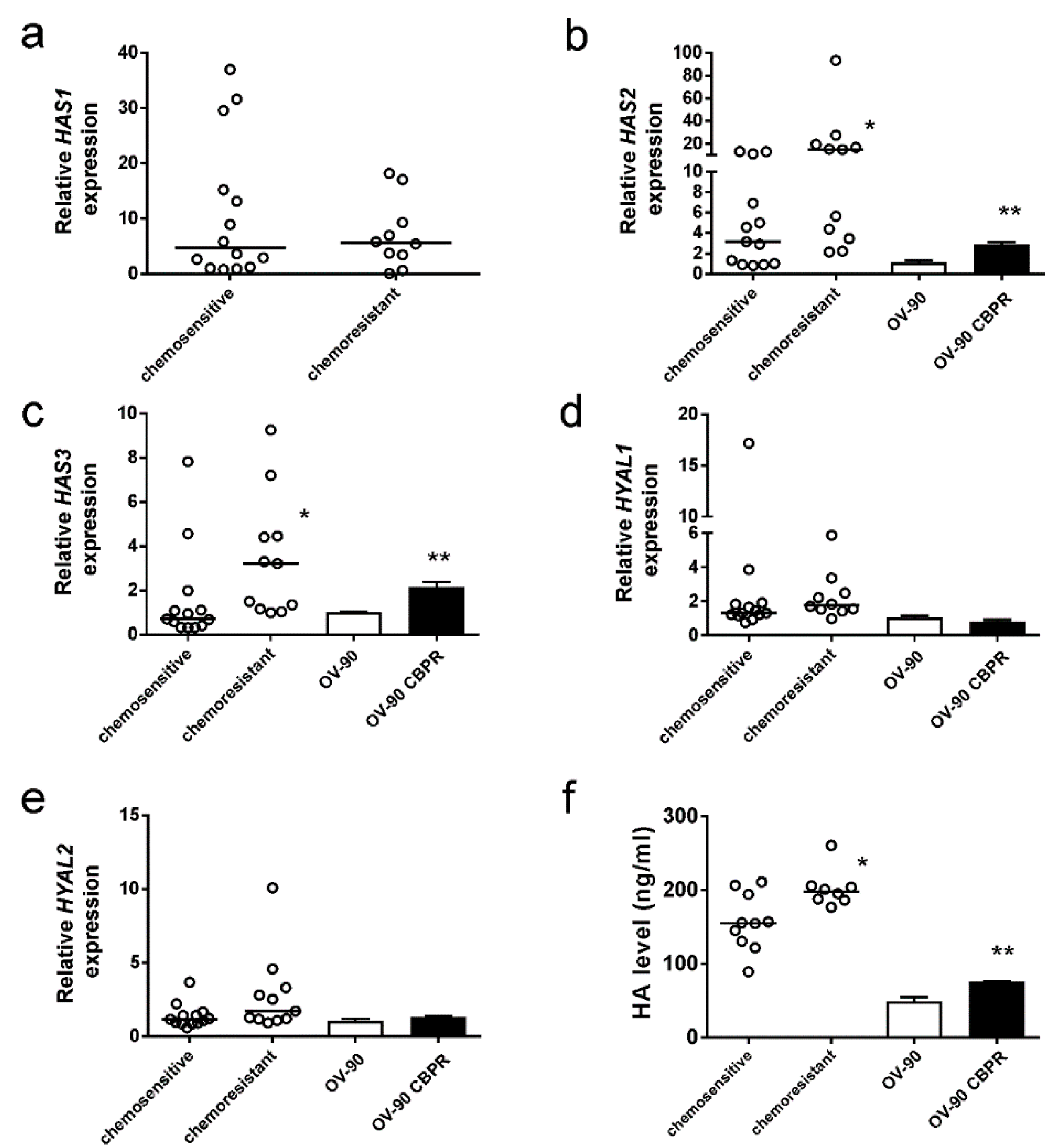
2.1. HA Serum Levels Are Elevated in Patients with Chemoresistant Disease
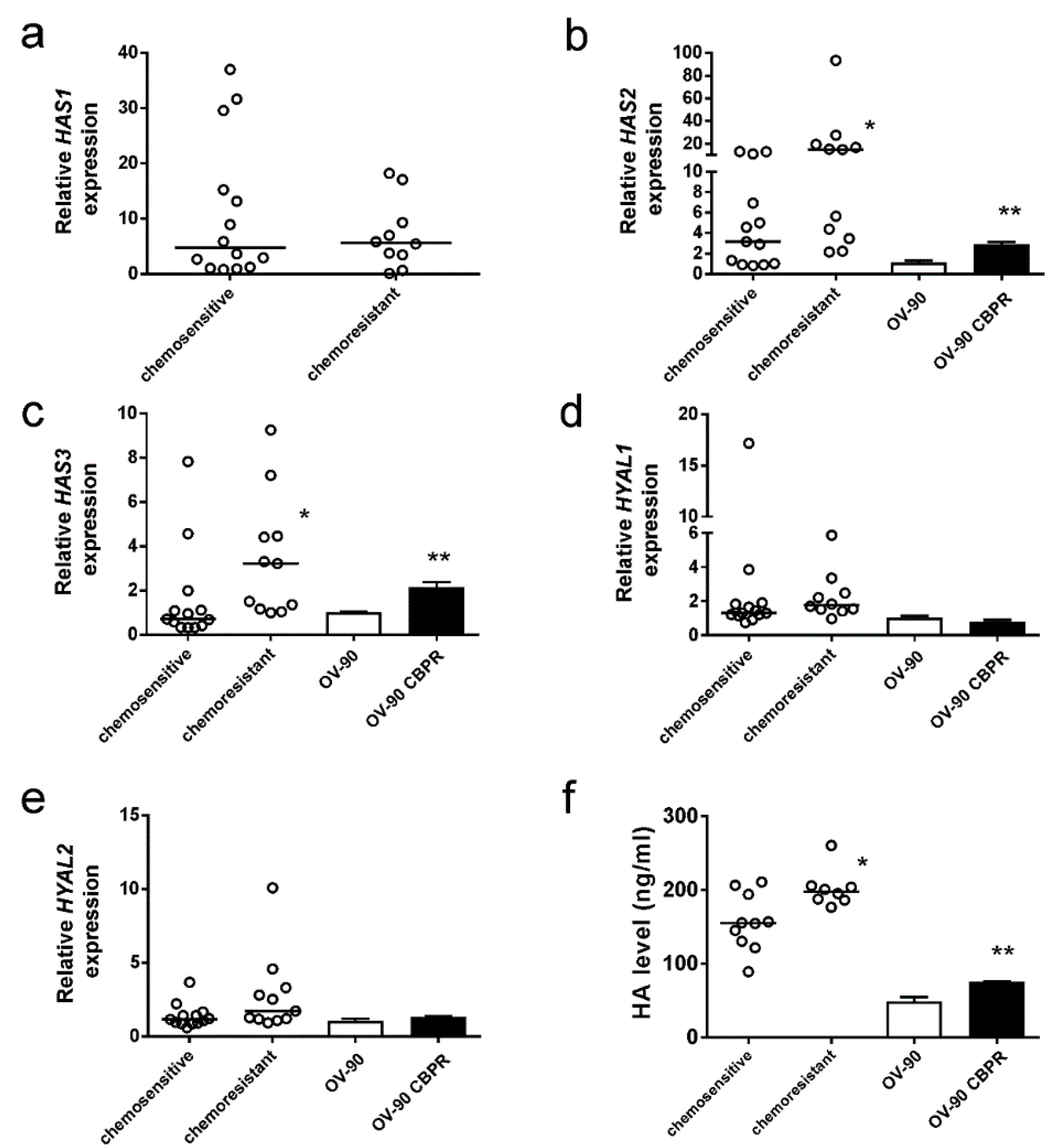
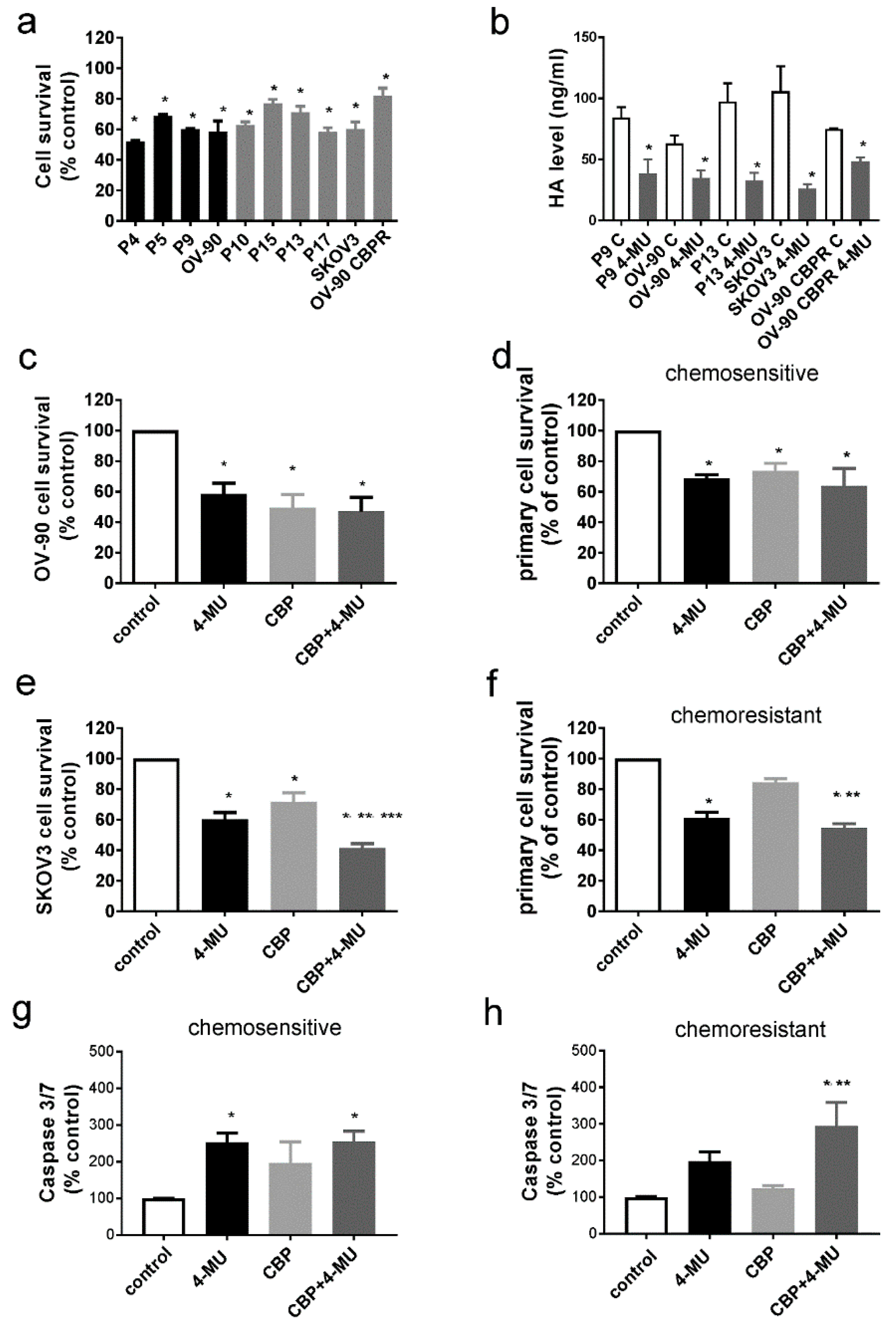
2.2. HA Production Is Increased in Serous Ovarian Cancer Cells Following Development of Chemotherapy Resistance
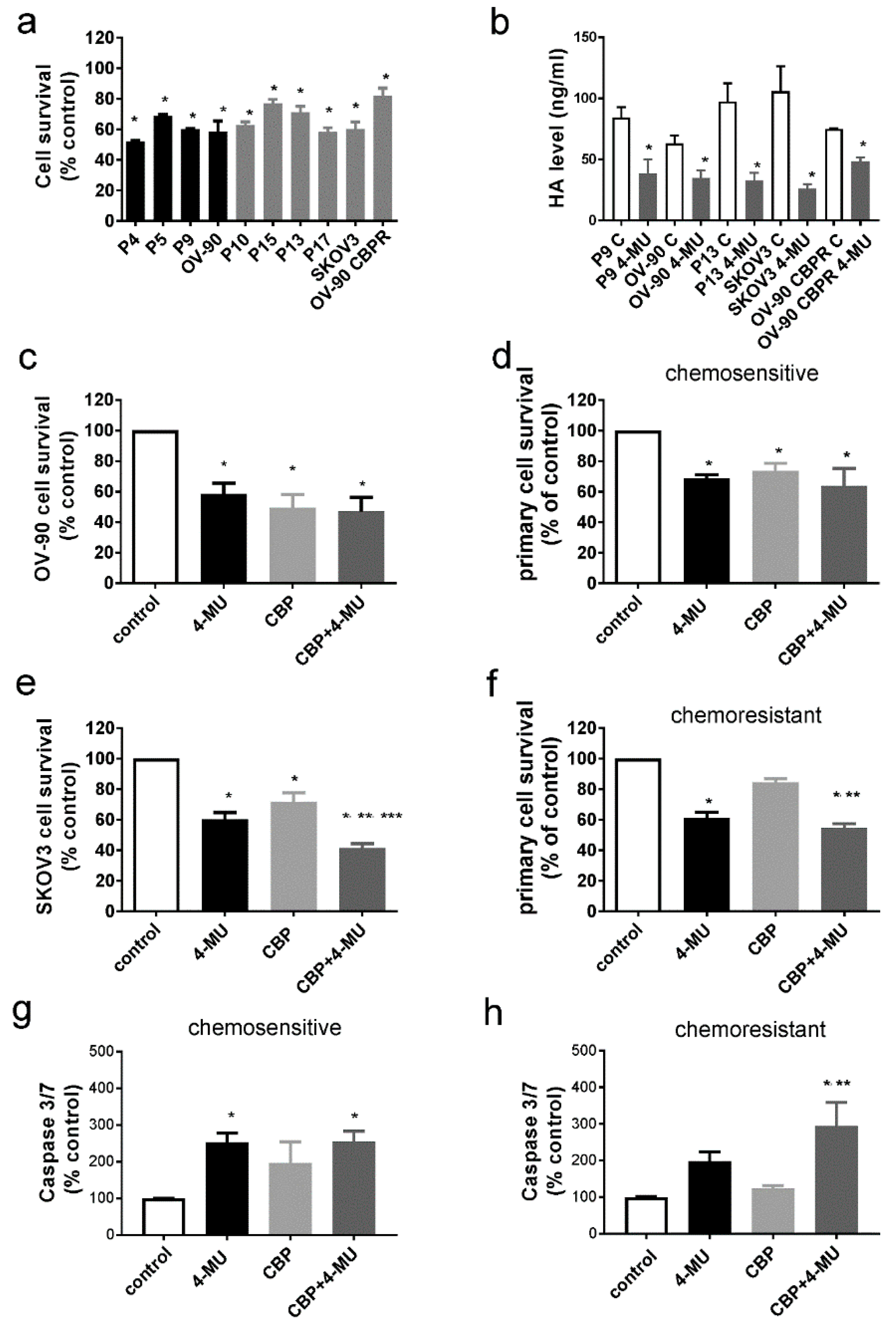
2.3. 4-MU Treatment Inhibits Survival of Chemoresistant Ovarian Cancer Cells
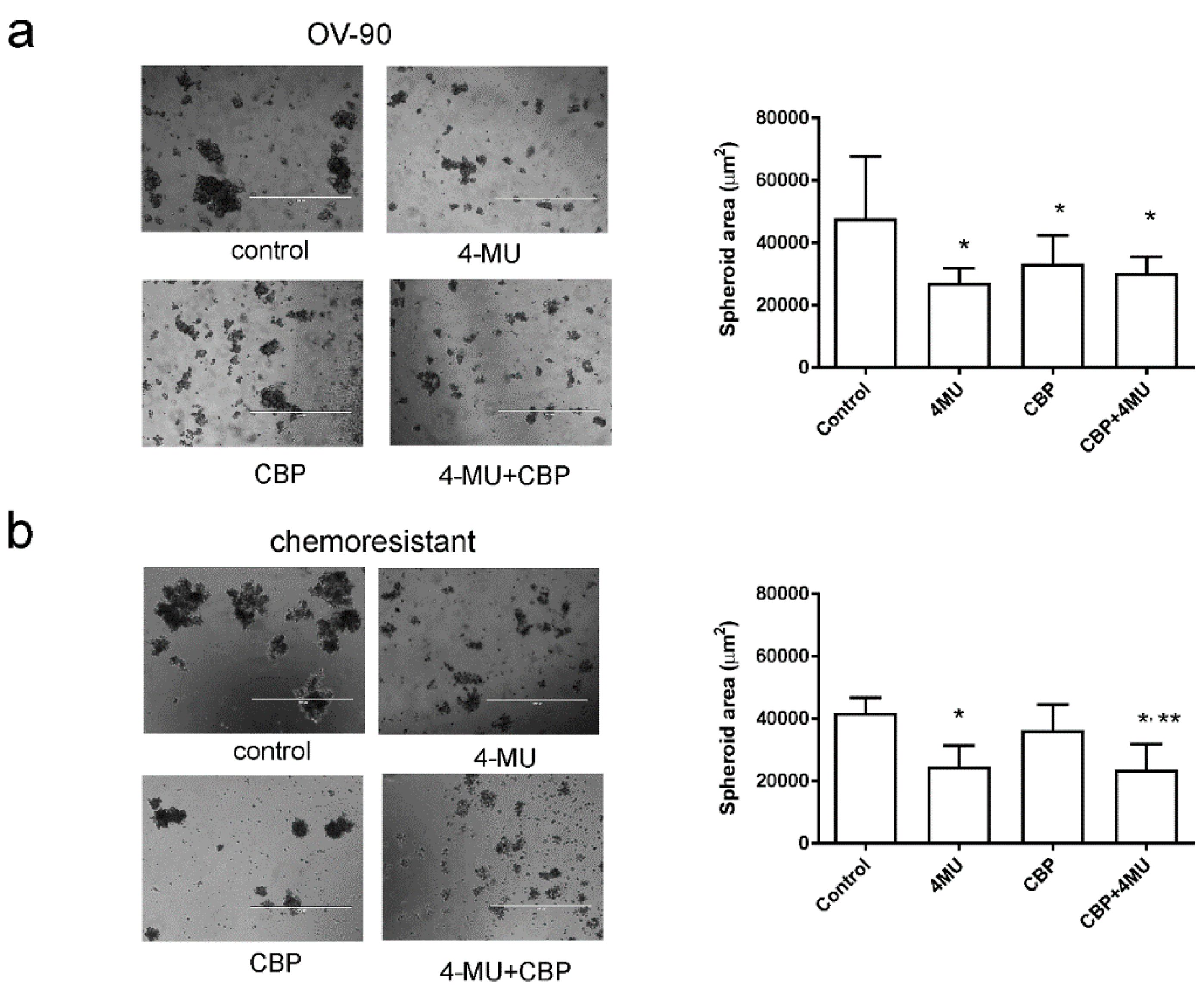
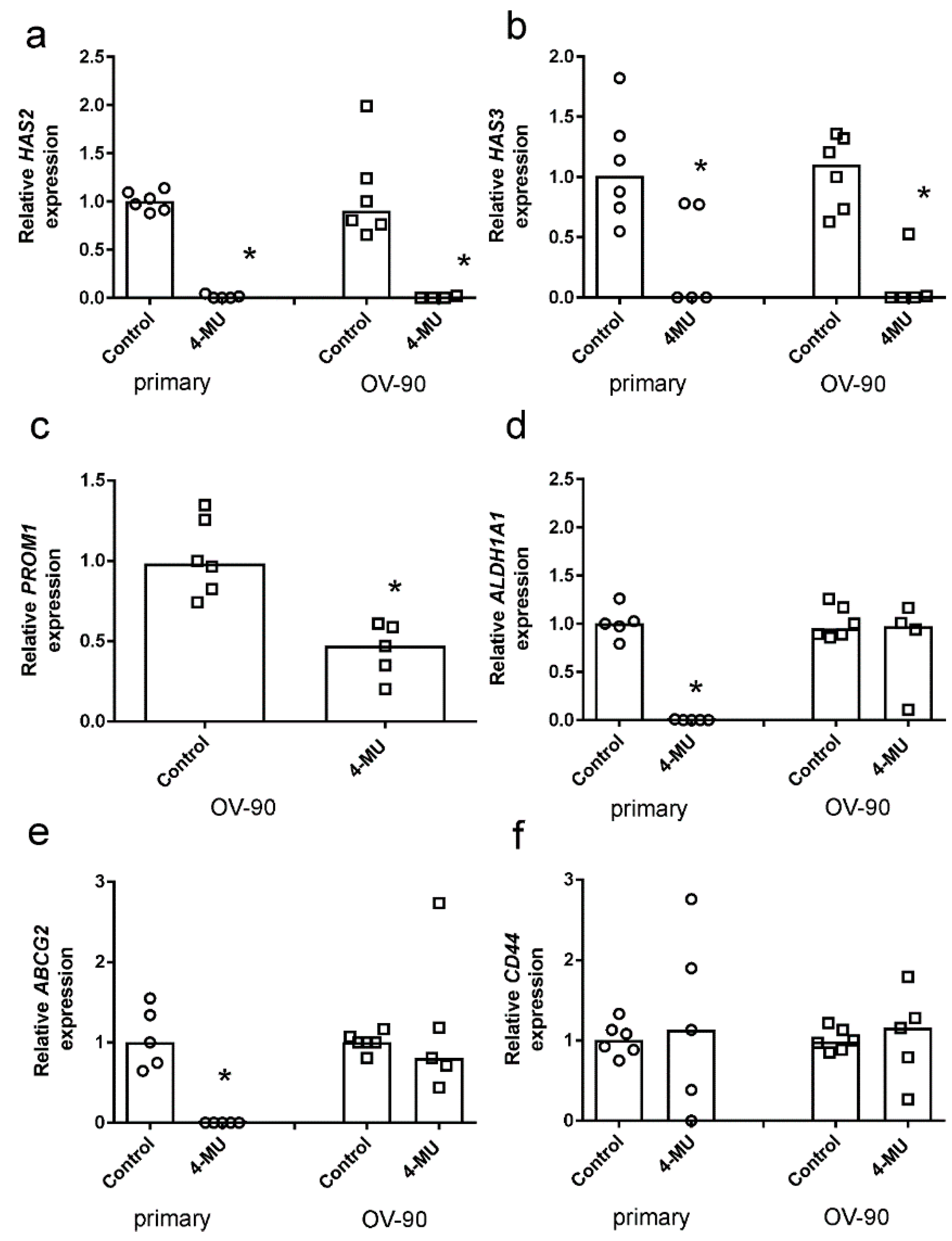
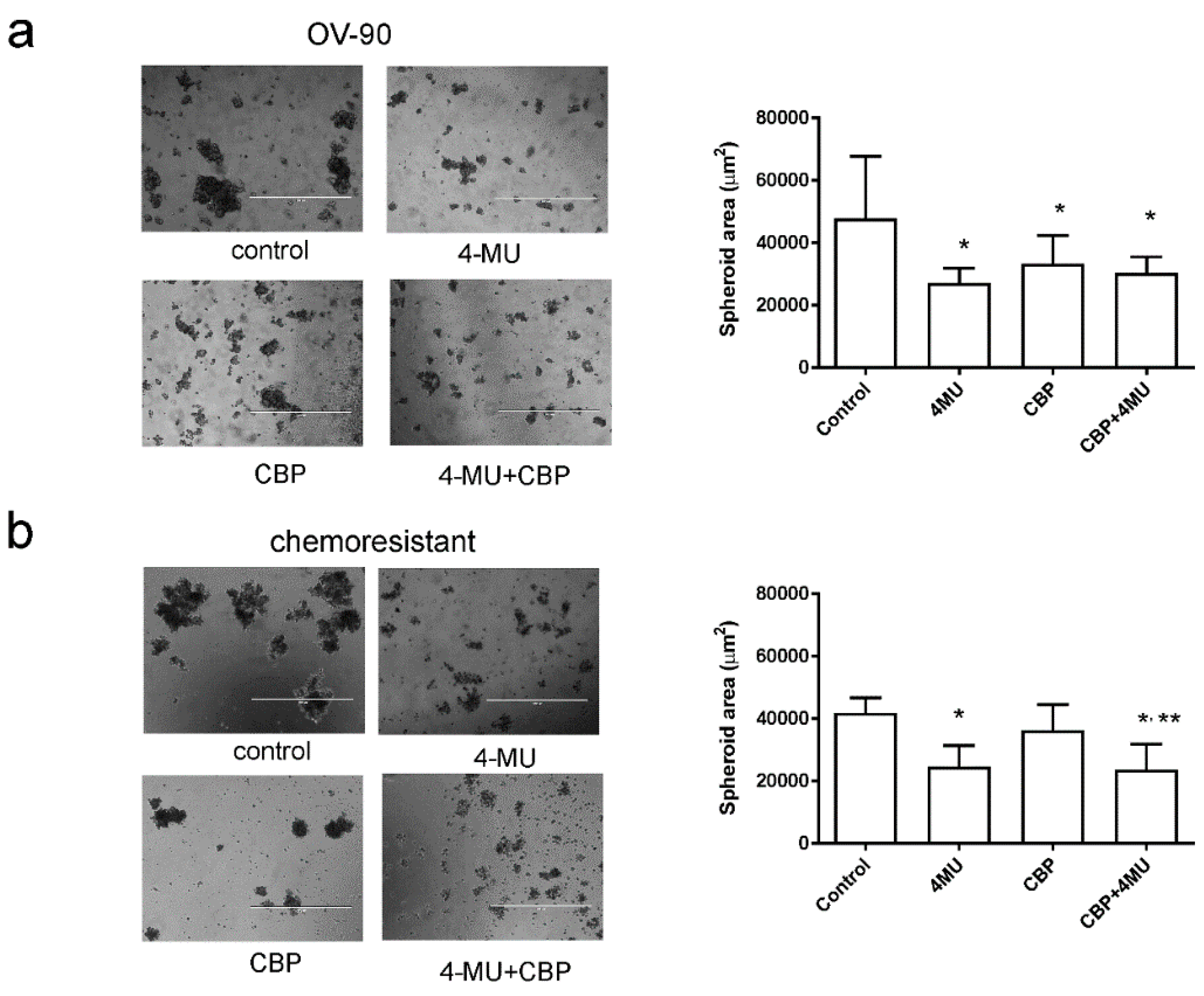
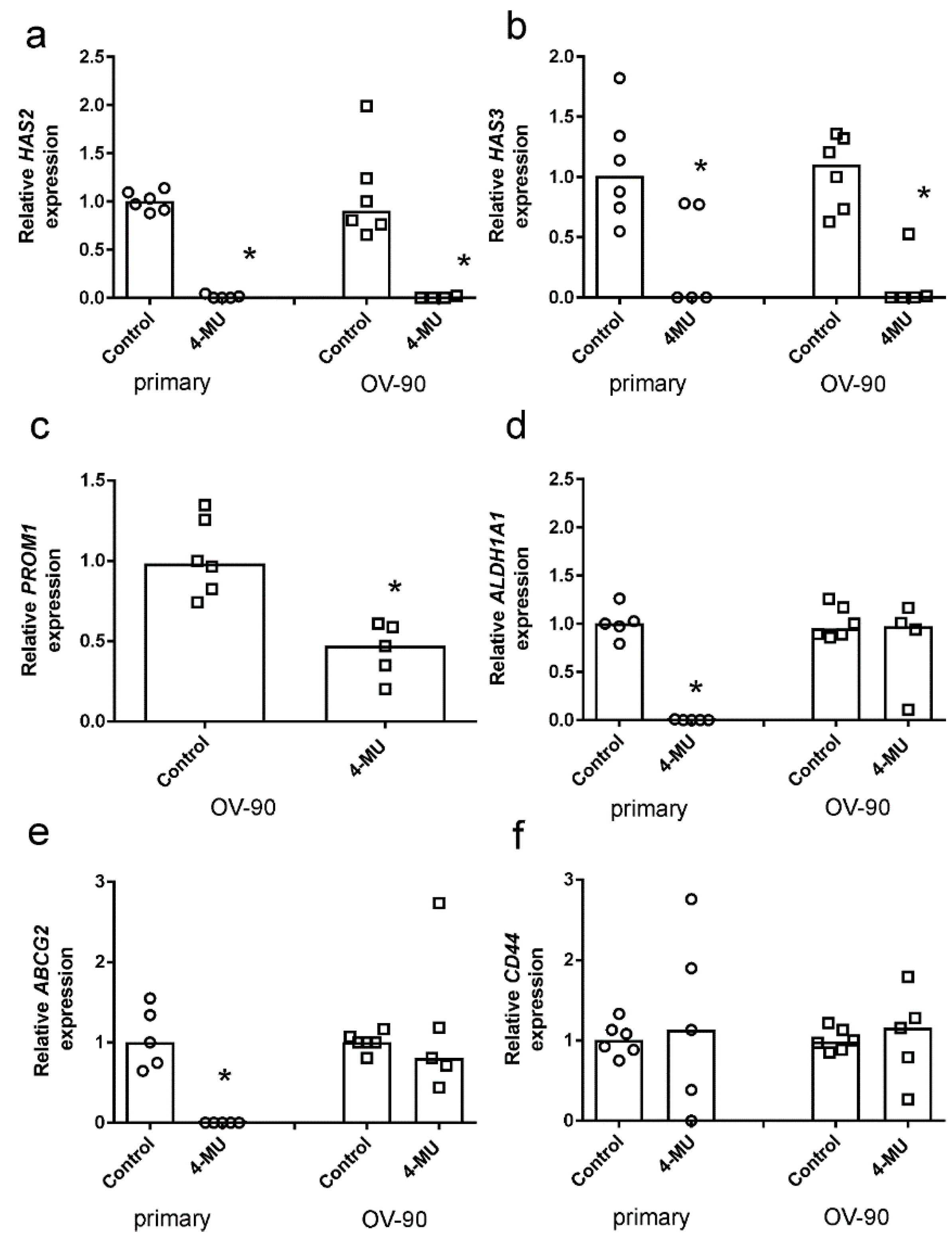
2.4. 4-MU Inhibits Spheroid Formation and Stem Cell Marker Expression
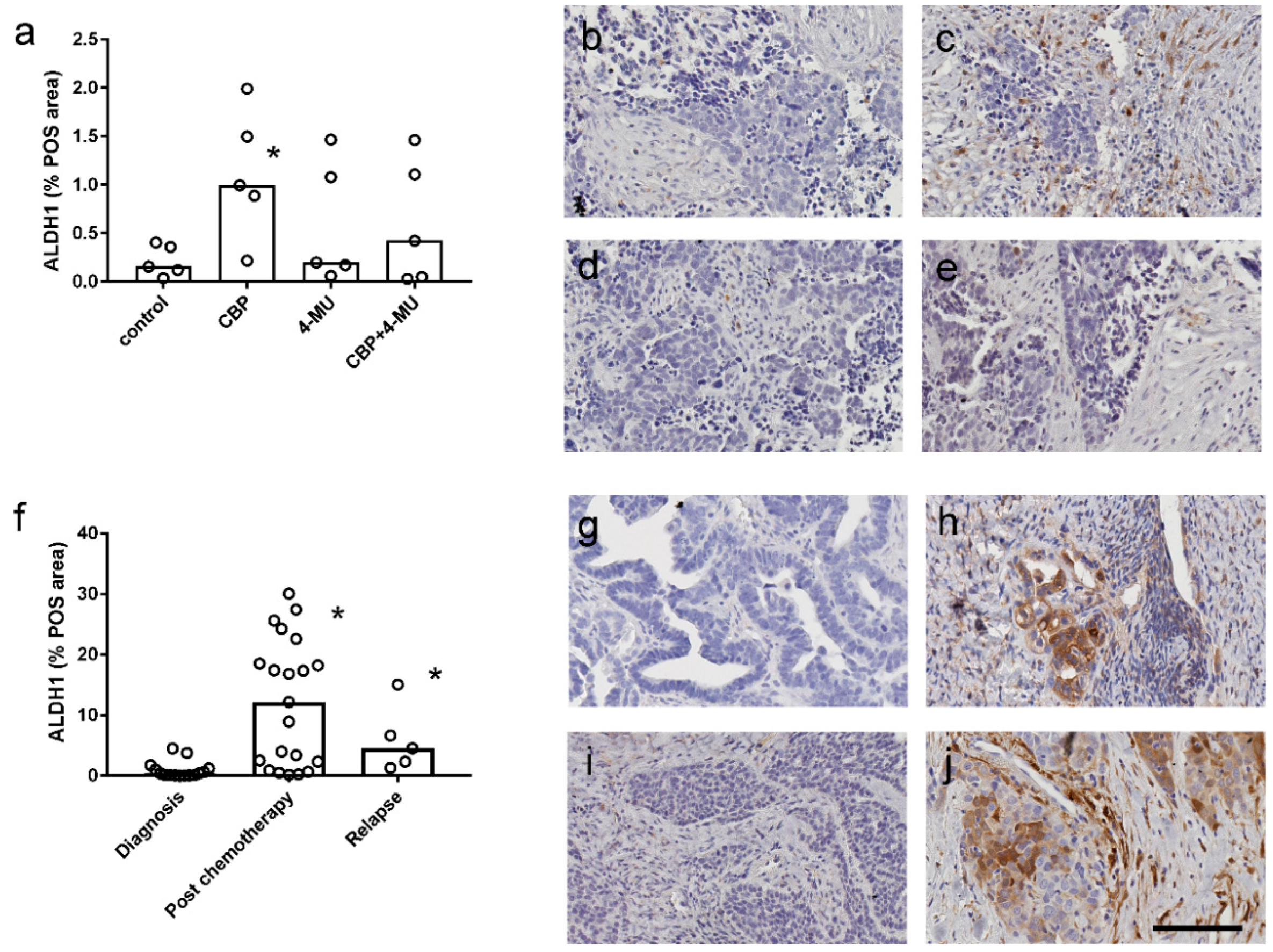
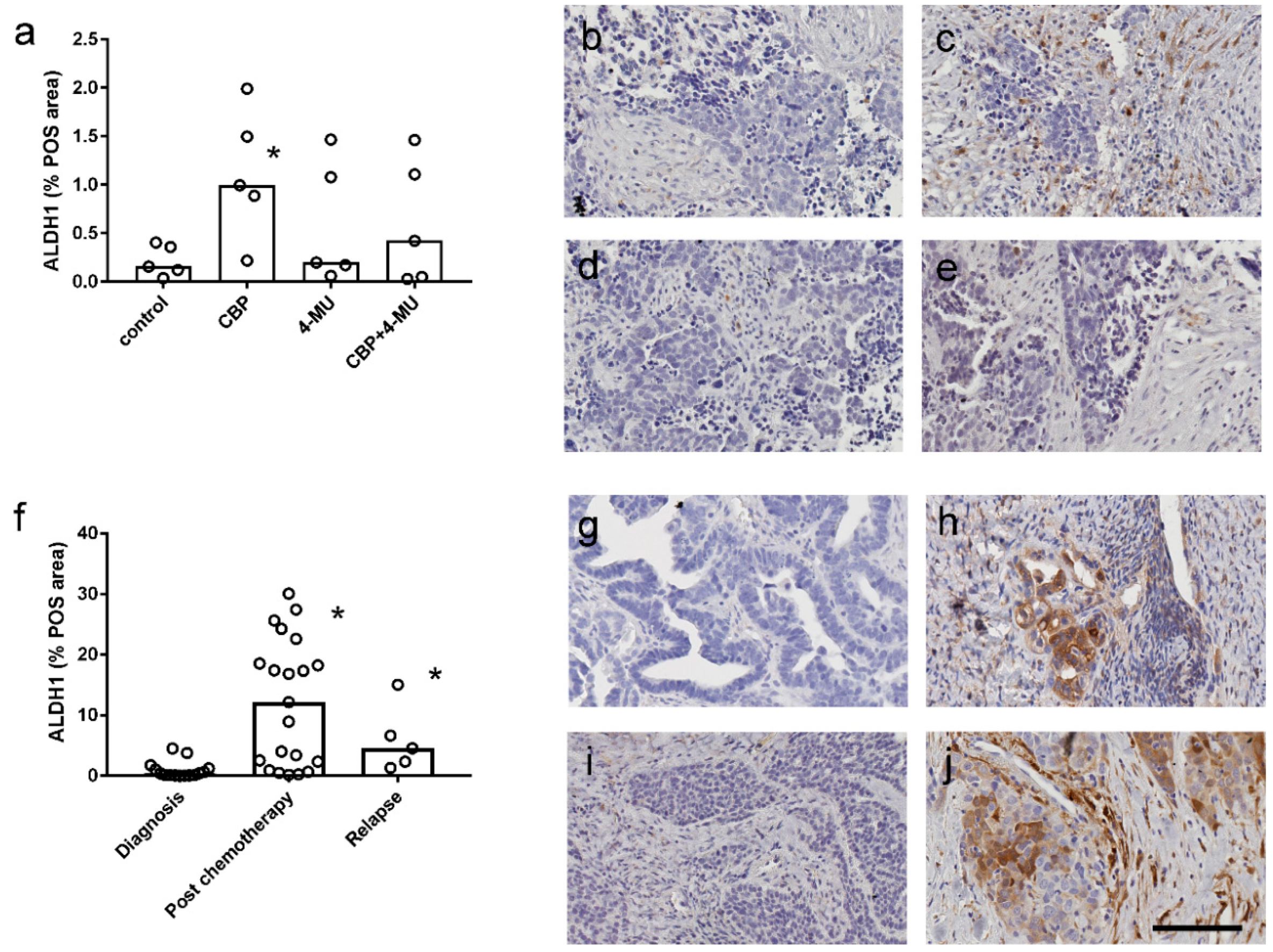
2.5. ALDH1 Protein Expression is Increased Following Chemotherapy Treatment and Reduced by 4-MU Treatment in Patient-Derived Explant Assays
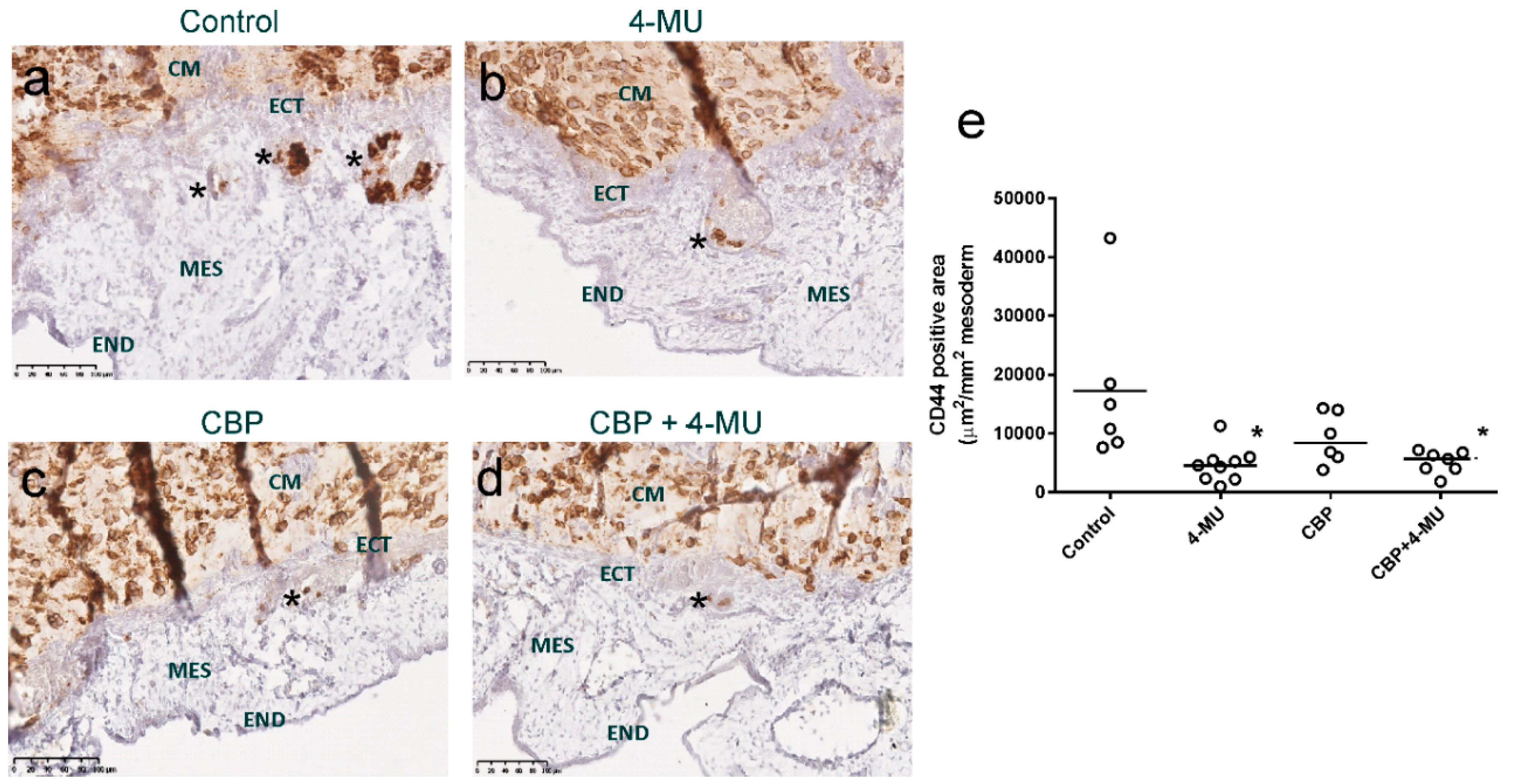
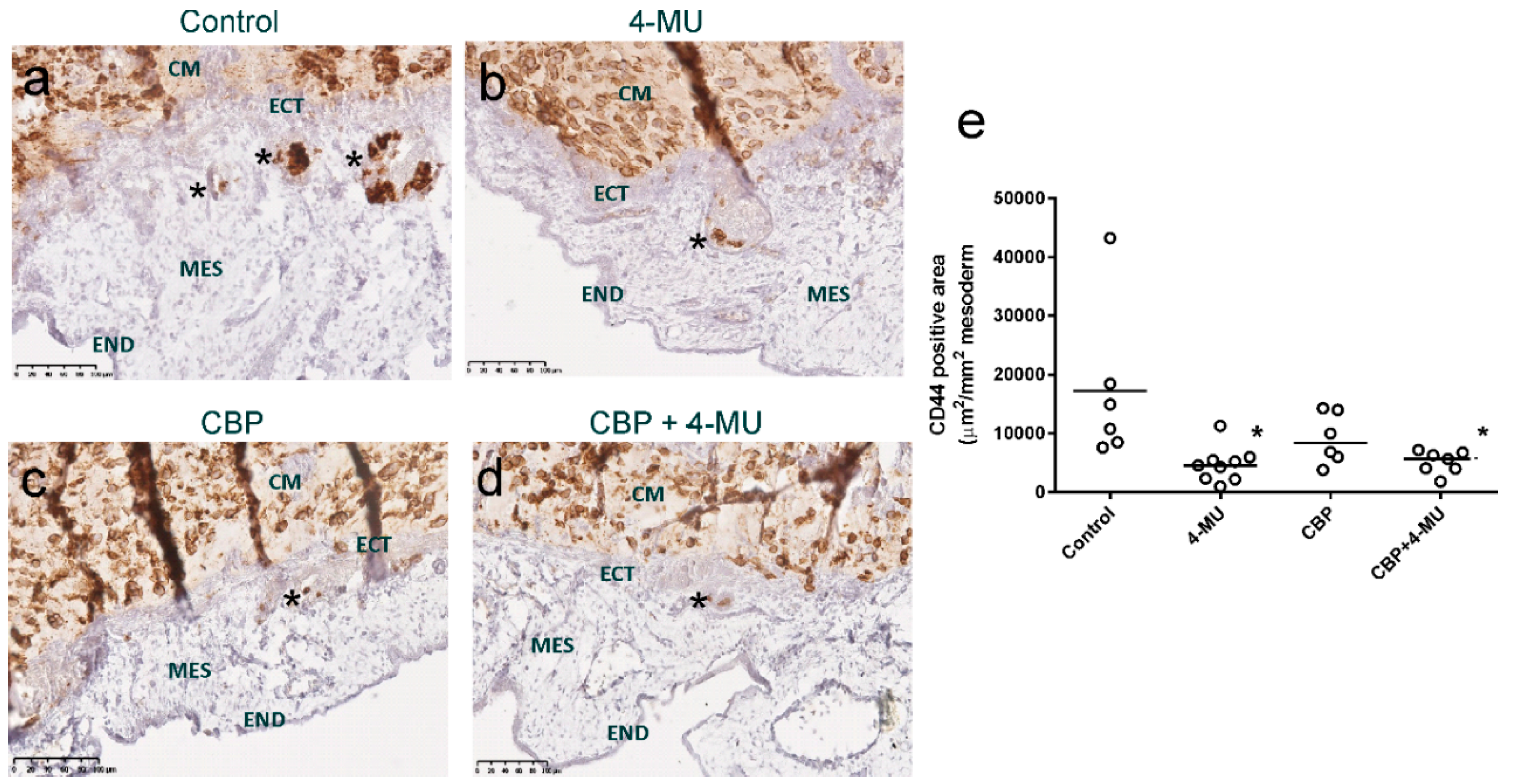
2.6. 4-MU Treatment Reduces In Vivo Invasion of Chemoresistant Ovarian Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Line Culture
4.2. Primary Cell Isolation
4.3. HA Detection
4.4. Cell Survival Assays
4.5. Apoptosis Assays
4.6. Spheroid Assays
4.7. Quantitative RT-PCR
4.8. ALDH1 Immunohistochemistry
4.9. CAM In Vivo Invasion Assays
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.E.; Fabbro, M.; Theillet, C.; Bibeau, F.; Rouanet, P.; Ray-Coquard, I. Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer. Critical Rev. Oncol./Hematol. 2014, 89, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.; Kuang, R.; Landen, C.; Shridhar, V. Platinum-sensitive recurrence in ovarian cancer: The role of tumor microenvironment. Front. Oncol. 2013, 3, 251. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Oehler, M.K.; Ricciardelli, C. Role of versican, hyaluronan and CD44 in ovarian cancer metastasis. Int. J. Mol. Sci. 2011, 12, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Hummitzsch, K.; Rodgers, R.J.; Oehler, M.K.; Ricciardelli, C. Versican induces a pro-metastatic ovarian cancer cell behavior which can be inhibited by small hyaluronan oligosaccharides. Clin. Exp. Metastasis 2011, 28, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Vigetti, D.; Viola, M.; Karousou, E.; De Luca, G.; Passi, A. Metabolic control of hyaluronan synthases. Matrix Biol. 2014, 35, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Sironen, R.K.; Tammi, M.; Tammi, R.; Auvinen, P.K.; Anttila, M.; Kosma, V.M. Hyaluronan in human malignancies. Exp. Cell Res. 2011, 317, 383–391. [Google Scholar] [CrossRef]
- Anttila, M.A.; Tammi, R.H.; Tammi, M.I.; Syrjanen, K.J.; Saarikoski, S.V.; Kosma, V.M. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000, 60, 150–155. [Google Scholar]
- Wang, S.J.; Bourguignon, L.Y. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2006, 132, 771–778. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker nanog, STAT-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 2008, 283, 17635–17651. [Google Scholar] [CrossRef] [PubMed]
- Torre, C.; Wang, S.J.; Xia, W.; Bourguignon, L.Y. Reduction of hyaluronan-CD44-mediated growth, migration, and cisplatin resistance in head and neck cancer due to inhibition of rho kinase and pi-3 kinase signaling. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Ween, M.P.; Lokman, N.A.; Tan, I.A.; Pyragius, C.E.; Oehler, M.K. Chemotherapy-induced hyaluronan production: A novel chemoresistance mechanism in ovarian cancer. BMC Cancer 2013, 13, 476. [Google Scholar] [CrossRef] [PubMed]
- Kultti, A.; Pasonen-Seppanen, S.; Jauhiainen, M.; Rilla, K.J.; Karna, R.; Pyoria, E.; Tammi, R.H.; Tammi, M.I. 4-methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 2009, 315, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Krawzak, H.W.; Heistermann, H.P.; Andrejewski, K.; Hohlbach, G. Postprandial bile-duct kinetics under the influence of 4-methylumbelliferone (hymecromone). Int. J. Clin. Pharmacol. Ther. 1995, 33, 569–572. [Google Scholar] [PubMed]
- Piccioni, F.; Malvicini, M.; Garcia, M.G.; Rodriguez, A.; Atorrasagasti, C.; Kippes, N.; Piedra Buena, I.T.; Rizzo, M.M.; Bayo, J.; Aquino, J.; et al. Antitumor effects of hyaluronic acid inhibitor 4-methylumbelliferone in an orthotopic hepatocellular carcinoma model in mice. Glycobiology 2012, 22, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Twarock, S.; Freudenberger, T.; Poscher, E.; Dai, G.; Jannasch, K.; Dullin, C.; Alves, F.; Prenzel, K.; Knoefel, W.T.; Stoecklein, N.H.; et al. Inhibition of oesophageal squamous cell carcinoma progression by in vivo targeting of hyaluronan synthesis. Mol. Cancer 2011, 10, 30. [Google Scholar] [CrossRef]
- Tamura, R.; Yokoyama, Y.; Yoshida, H.; Imaizumi, T.; Mizunuma, H. 4-methylumbelliferone inhibits ovarian cancer growth by suppressing thymidine phosphorylase expression. J. Ovarian Res. 2014, 7, 94. [Google Scholar] [CrossRef]
- Yates, T.J.; Lopez, L.E.; Lokeshwar, S.D.; Ortiz, N.; Kallifatidis, G.; Jordan, A.; Hoye, K.; Altman, N.; Lokeshwar, V.B. Dietary supplement 4-methylumbelliferone: An effective chemopreventive and therapeutic agent for prostate cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Nagy, N.; Kuipers, H.F.; Frymoyer, A.R.; Ishak, H.D.; Bollyky, J.B.; Wight, T.N.; Bollyky, P.L. 4-methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front. Immunol. 2015, 6, 123. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Lokman, N.A.; Sabit, I.; Gunasegaran, K.; Bonner, W.M.; Pyragius, C.E.; Macpherson, A.M.; Oehler, M.K. Novel ex vivo ovarian cancer tissue explant assay for prediction of chemosensitivity and response to novel therapeutics. Cancer Lett. 2018, 421, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lokman, N.A.; Elder, A.S.; Ricciardelli, C.; Oehler, M.K. Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int. J. Mol. Sci. 2012, 13, 9959–9970. [Google Scholar] [CrossRef] [PubMed]
- Lompardia, S.L.; Papademetrio, D.L.; Mascaro, M.; Alvarez, E.M.; Hajos, S.E. Human leukemic cell lines synthesize hyaluronan to avoid senescence and resist chemotherapy. Glycobiology 2013, 23, 1463–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchakina, O.N.; Ban, H.; Hostetler, B.J.; McKallip, R.J. Inhibition of hyaluronic acid formation sensitizes chronic myelogenous leukemia to treatment with doxorubicin. Glycobiology 2016, 26, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Armstrong, M.A.; Oehler, M.K.; Ricciardelli, C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit. Rev. Oncol. Hematol. 2015, 96, 220–256. [Google Scholar] [CrossRef]
- Cordo Russo, R.I.; Garcia, M.G.; Alaniz, L.; Blanco, G.; Alvarez, E.; Hajos, S.E. Hyaluronan oligosaccharides sensitize lymphoma resistant cell lines to vincristine by modulating p-glycoprotein activity and pi3k/AKT pathway. Int. J. Cancer 2008, 122, 1012–1018. [Google Scholar] [CrossRef]
- Misra, S.; Ghatak, S.; Toole, B.P. Regulation of mdr1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ERBB2. J. Biol. Chem. 2005, 280, 20310–20315. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Xia, W.; Wong, G. Hyaluronan-mediated CD44 interaction with p300 and sirt1 regulates beta-catenin signaling and NFkappaB-specific transcription activity leading to MDR1 and Bcl-Xl gene expression and chemoresistance in breast tumor cells. J. Biol. Chem. 2009, 284, 2657–2671. [Google Scholar] [CrossRef]
- Ohashi, R.; Takahashi, F.; Cui, R.; Yoshioka, M.; Gu, T.; Sasaki, S.; Tominaga, S.; Nishio, K.; Tanabe, K.K.; Takahashi, K. Interaction between CD44 and hyaluronate induces chemoresistance in non-small cell lung cancer cell. Cancer Lett. 2007, 252, 225–234. [Google Scholar] [CrossRef]
- Lokeshwar, V.B.; Lopez, L.E.; Munoz, D.; Chi, A.; Shirodkar, S.P.; Lokeshwar, S.D.; Escudero, D.O.; Dhir, N.; Altman, N. Antitumor activity of hyaluronic acid synthesis inhibitor 4-methylumbelliferone in prostate cancer cells. Cancer Res. 2010, 70, 2613–2623. [Google Scholar] [CrossRef]
- Arai, E.; Nishida, Y.; Wasa, J.; Urakawa, H.; Zhuo, L.; Kimata, K.; Kozawa, E.; Futamura, N.; Ishiguro, N. Inhibition of hyaluronan retention by 4-methylumbelliferone suppresses osteosarcoma cells in vitro and lung metastasis in vivo. Br. J. Cancer 2011, 105, 1839–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakawa, H.; Nishida, Y.; Wasa, J.; Arai, E.; Zhuo, L.; Kimata, K.; Kozawa, E.; Futamura, N.; Ishiguro, N. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int. J. Cancer 2012, 130, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Uchakina, O.N.; Ban, H.; McKallip, R.J. Targeting hyaluronic acid production for the treatment of leukemia: Treatment with 4-methylumbelliferone leads to induction of MAPK-mediated apoptosis in k562 leukemia. Leuk. Res. 2013, 37, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Uchakina, O.; McKallip, R.J. Hyaluronic acid inhibitor 4-methylumbelliferone activates the intrinsic apoptosis pathway in k562 chronic myelogenous leukemia cells. Anticancer Res. 2015, 35, 5231–5240. [Google Scholar] [PubMed]
- Nagase, H.; Kudo, D.; Suto, A.; Yoshida, E.; Suto, S.; Negishi, M.; Kakizaki, I.; Hakamada, K. 4-methylumbelliferone suppresses hyaluronan synthesis and tumor progression in SCID mice intra-abdominally inoculated with pancreatic cancer cells. Pancreas 2017, 46, 190. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.; Ota, T.; Zhuo, L.; Urakawa, H.; Kozawa, E.; Hamada, S.; Kimata, K.; Ishiguro, N.; Nishida, Y. Antitumor effects of 4-methylumbelliferone, a hyaluronan synthesis inhibitor, on malignant peripheral nerve sheath tumor. Int. J. Cancer 2017, 140, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.B.; Kohi, S.; Koga, A.; Hirata, K.; Sato, N. Hyaluronan stimulates pancreatic cancer cell motility. Oncotarget 2016, 7, 4829–4840. [Google Scholar]
- Kato, N.; Shibata, K.; Uchigasaki, S.; Kurose, A. Relation between hyaluronan synthesis and cell morphology in ovarian clear cell carcinomas. Pathol. Int. 2016, 66, 218–223. [Google Scholar] [CrossRef]
- Keller, K.E.; Sun, Y.Y.; Vranka, J.A.; Hayashi, L.; Acott, T.S. Inhibition of hyaluronan synthesis reduces versican and fibronectin levels in trabecular meshwork cells. PLoS ONE 2012, 7, e48523. [Google Scholar] [CrossRef]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K.; et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012, 72, 537–547. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Mochizuki, N.; Kongtawelert, P.; Konno, K.; Itano, N. Excessive hyaluronan production promotes acquisition of cancer stem cell signatures through the coordinated regulation of twist and the transforming growth factor beta (TGF-beta)-snail signaling axis. J. Biol. Chem. 2014, 289, 26038–26056. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, H.; Kasai, T.; Khayrani, A.C.; Asakura, M.; Oo, A.K.K.; Du, J.; Vaidyanath, A.; El-Ghlban, S.; Mizutani, A.; Seno, A.; et al. Targeting glioblastoma cells expressing CD44 with liposomes encapsulating doxorubicin and displaying chlorotoxin-igg fc fusion protein. Int. J. Mol. Sci. 2018, 19, 659. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Zhang, X.; Stojkovic, P.; Hyslop, L.; Anyfantis, G.; Herbert, M.; Murdoch, A.P.; Stojkovic, M.; Lako, M. Putative role of hyaluronan and its related genes, HAS2 and RHAMM, in human early preimplantation embryogenesis and embryonic stem cell characterization. Stem Cells 2007, 25, 3045–3057. [Google Scholar] [CrossRef] [PubMed]
- Shiina, M.; Bourguignon, L.Y. Selective activation of cancer stem cells by size-specific hyaluronan in head and neck cancer. Int. J. Cell Biol. 2015, 2015, 989070. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Ito, S.; Nakamura, H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013, 73, 4112–4122. [Google Scholar] [CrossRef] [PubMed]
- He, Q.Z.; Luo, X.Z.; Wang, K.; Zhou, Q.; Ao, H.; Yang, Y.; Li, S.X.; Li, Y.; Zhu, H.T.; Duan, T. Isolation and characterization of cancer stem cells from high-grade serous ovarian carcinomas. Cell. Physiol. Biochem. 2014, 33, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Sukowati, C.H.C.; Anfuso, B.; Fiore, E.; Ie, S.I.; Raseni, A.; Vascotto, F.; Avellini, C.; Mazzolini, G.; Tiribelli, C. Hyaluronic acid inhibition by 4-methylumbelliferone reduces the expression of cancer stem cells markers during hepatocarcinogenesis. Sci. Rep. 2019, 9, 4026. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef]
- Landen, C.N., Jr.; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar] [CrossRef]
- Wang, Y.C.; Yo, Y.T.; Lee, H.Y.; Liao, Y.P.; Chao, T.K.; Su, P.H.; Lai, H.C. Aldh1-bright epithelial ovarian cancer cells are associated with cd44 expression, drug resistance, and poor clinical outcome. Am. J. Pathol. 2012, 180, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef] [PubMed]
- Ayub, T.H.; Keyver-Paik, M.D.; Debald, M.; Rostamzadeh, B.; Thiesler, T.; Schroder, L.; Barchet, W.; Abramian, A.; Kaiser, C.; Kristiansen, G.; et al. Accumulation of aldh1-positive cells after neoadjuvant chemotherapy predicts treatment resistance and prognosticates poor outcome in ovarian cancer. Oncotarget 2015, 6, 16437–16448. [Google Scholar] [CrossRef] [PubMed]
- Kudo, D.; Kon, A.; Yoshihara, S.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. Effect of a hyaluronan synthase suppressor, 4-methylumbelliferone, on b16f-10 melanoma cell adhesion and locomotion. Biochem. Biophys. Res. Commun. 2004, 321, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Futamura, N.; Urakawa, H.; Arai, E.; Kozawa, E.; Ishiguro, N.; Nishida, Y. Hyaluronan synthesis inhibitor supplements the inhibitory effects of zoledronic acid on bone metastasis of lung cancer. Clin. Exp. Metastasis 2013, 30, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, S.; Kon, A.; Kudo, D.; Nakazawa, H.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. A hyaluronan synthase suppressor, 4-methylumbelliferone, inhibits liver metastasis of melanoma cells. FEBS Lett. 2005, 579, 2722–2726. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, E.; Kudo, D.; Nagase, H.; Shimoda, H.; Suto, S.; Negishi, M.; Kakizaki, I.; Endo, M.; Hakamada, K. Antitumor effects of the hyaluronan inhibitor 4-methylumbelliferone on pancreatic cancer. Oncol. Lett. 2016, 12, 2337–2344. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Lokman, N.A.; Pyragius, C.E.; Ween, M.P.; Macpherson, A.M.; Ruszkiewicz, A.; Hoffmann, P.; Oehler, M.K. Keratin 5 overexpression is associated with serous ovarian cancer recurrence and chemotherapy resistance. Oncotarget 2017, 8, 17819–17832. [Google Scholar] [CrossRef] [Green Version]
- Ricciardelli, C.; Lokman, N.A.; Cheruvu, S.; Tan, I.A.; Ween, M.P.; Pyragius, C.E.; Ruszkiewicz, A.; Hoffmann, P.; Oehler, M.K. Transketolase is upregulated in metastatic peritoneal implants and promotes ovarian cancer cell proliferation. Clin. Exp. Metastasis 2015, 32, 441–455. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Prev. Biomark. 2010, 19, 2611–2622. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lokman, N.A.; Price, Z.K.; Hawkins, E.K.; Macpherson, A.M.; Oehler, M.K.; Ricciardelli, C. 4-Methylumbelliferone Inhibits Cancer Stem Cell Activation and Overcomes Chemoresistance in Ovarian Cancer. Cancers 2019, 11, 1187. https://doi.org/10.3390/cancers11081187
Lokman NA, Price ZK, Hawkins EK, Macpherson AM, Oehler MK, Ricciardelli C. 4-Methylumbelliferone Inhibits Cancer Stem Cell Activation and Overcomes Chemoresistance in Ovarian Cancer. Cancers. 2019; 11(8):1187. https://doi.org/10.3390/cancers11081187
Chicago/Turabian StyleLokman, Noor A., Zoe K. Price, Emily K. Hawkins, Anne M. Macpherson, Martin K. Oehler, and Carmela Ricciardelli. 2019. "4-Methylumbelliferone Inhibits Cancer Stem Cell Activation and Overcomes Chemoresistance in Ovarian Cancer" Cancers 11, no. 8: 1187. https://doi.org/10.3390/cancers11081187
APA StyleLokman, N. A., Price, Z. K., Hawkins, E. K., Macpherson, A. M., Oehler, M. K., & Ricciardelli, C. (2019). 4-Methylumbelliferone Inhibits Cancer Stem Cell Activation and Overcomes Chemoresistance in Ovarian Cancer. Cancers, 11(8), 1187. https://doi.org/10.3390/cancers11081187