Electrochemotherapy Causes Caspase-Independent Necrotic-Like Death in Pancreatic Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Electroporation in the Presence of Bleomycin, Cisplatin and Oxaliplatin Negatively Affects the Ability of Pancreatic Cells to Recover and Proliferate
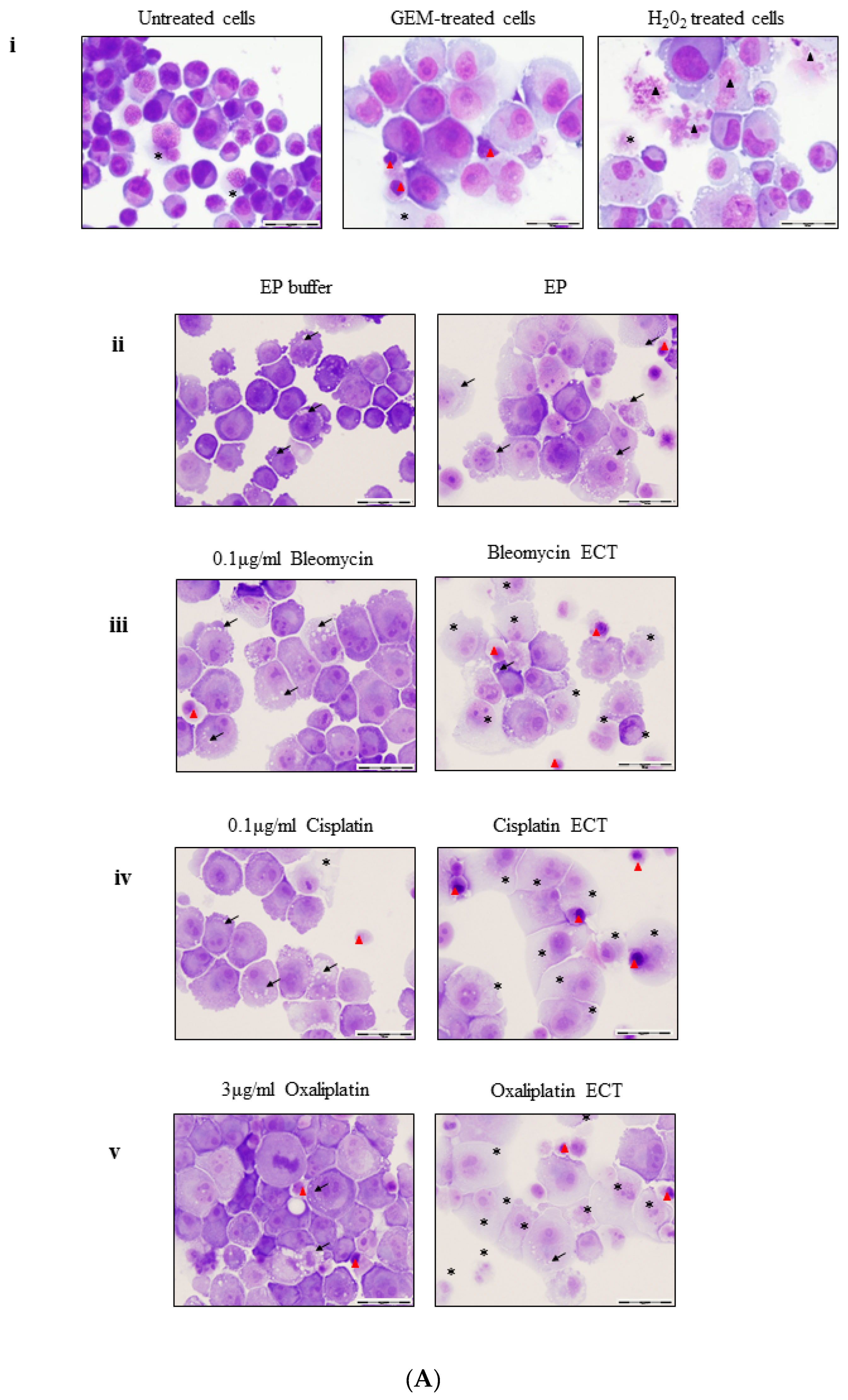
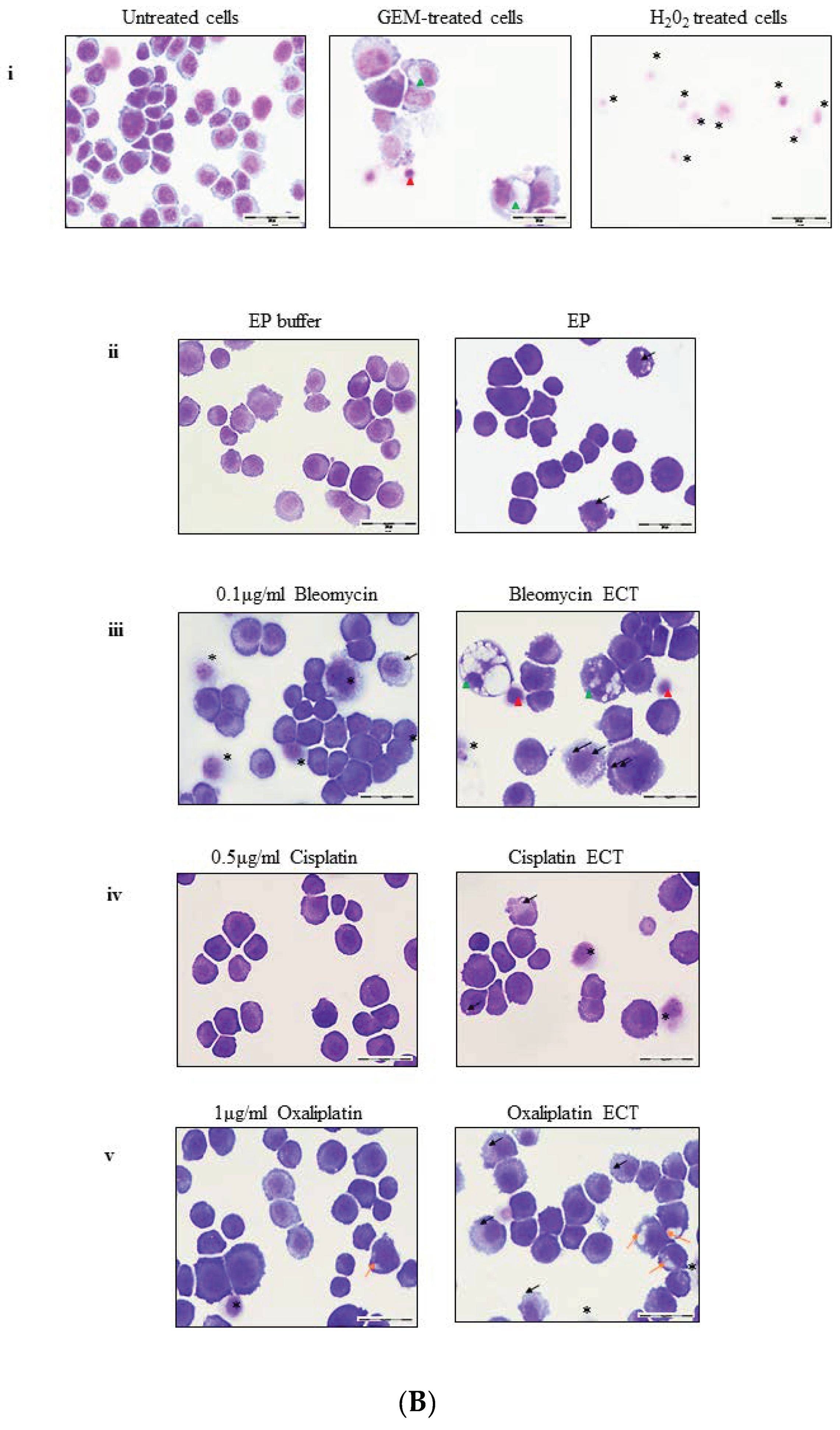
2.2. ECT of Pancreatic Cells Leads to Altered Necrotic-Like Morphology Relative to Drug Alone
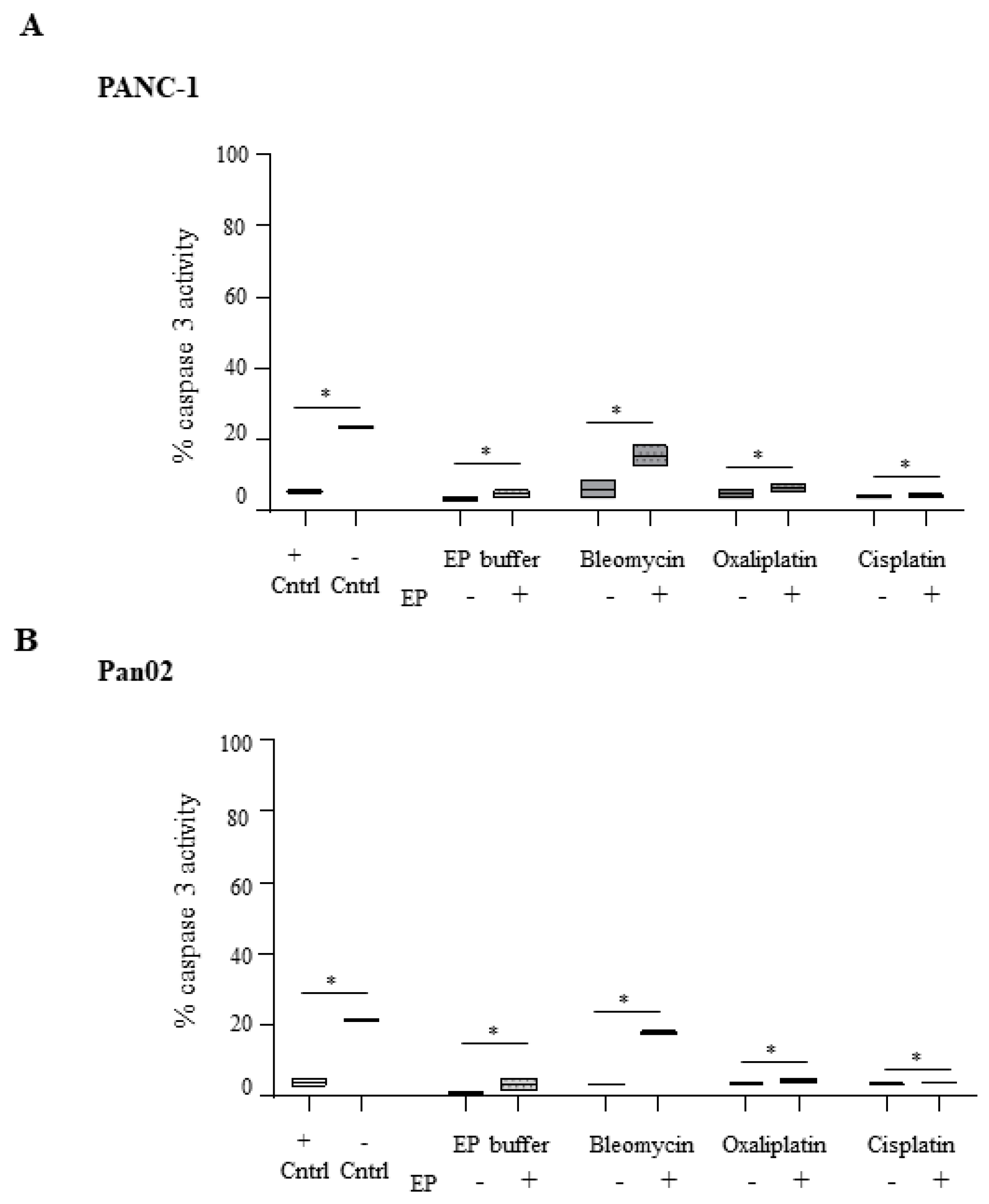
2.3. Electroporation-Delivered Bleomycin But not Cisplatin or Oxaliplatin Leads to Effector Caspase Activation in Pancreatic Cancer Cells
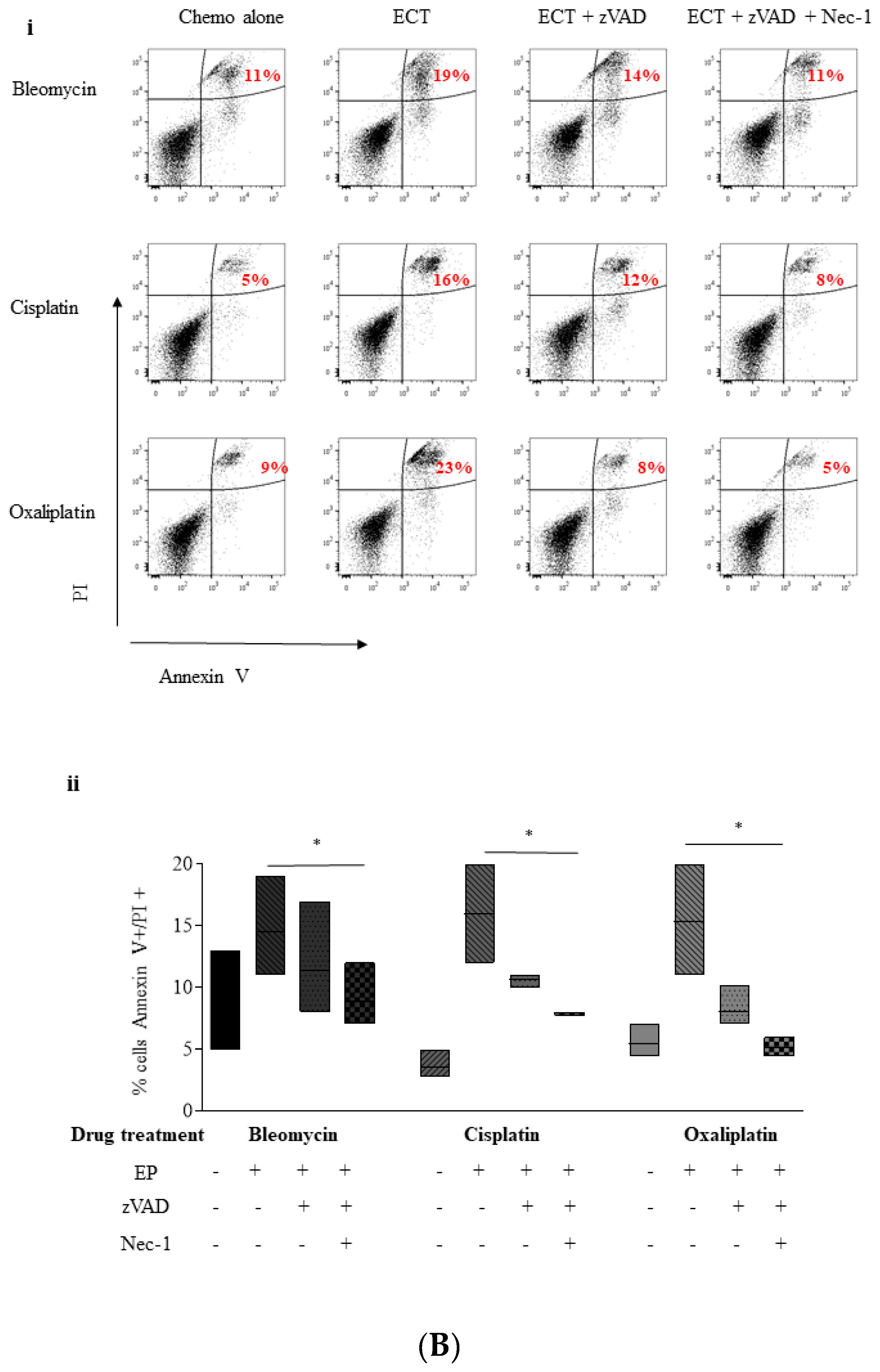
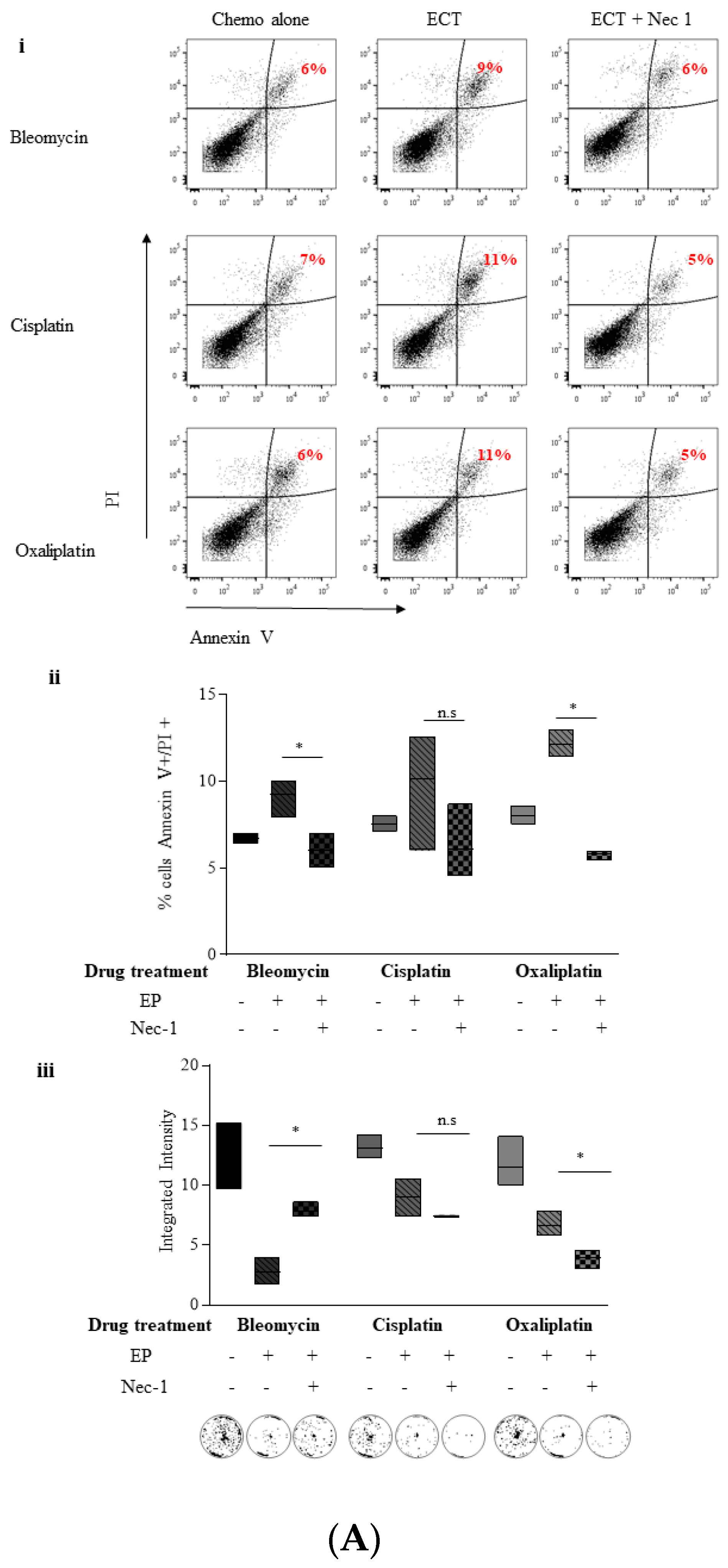
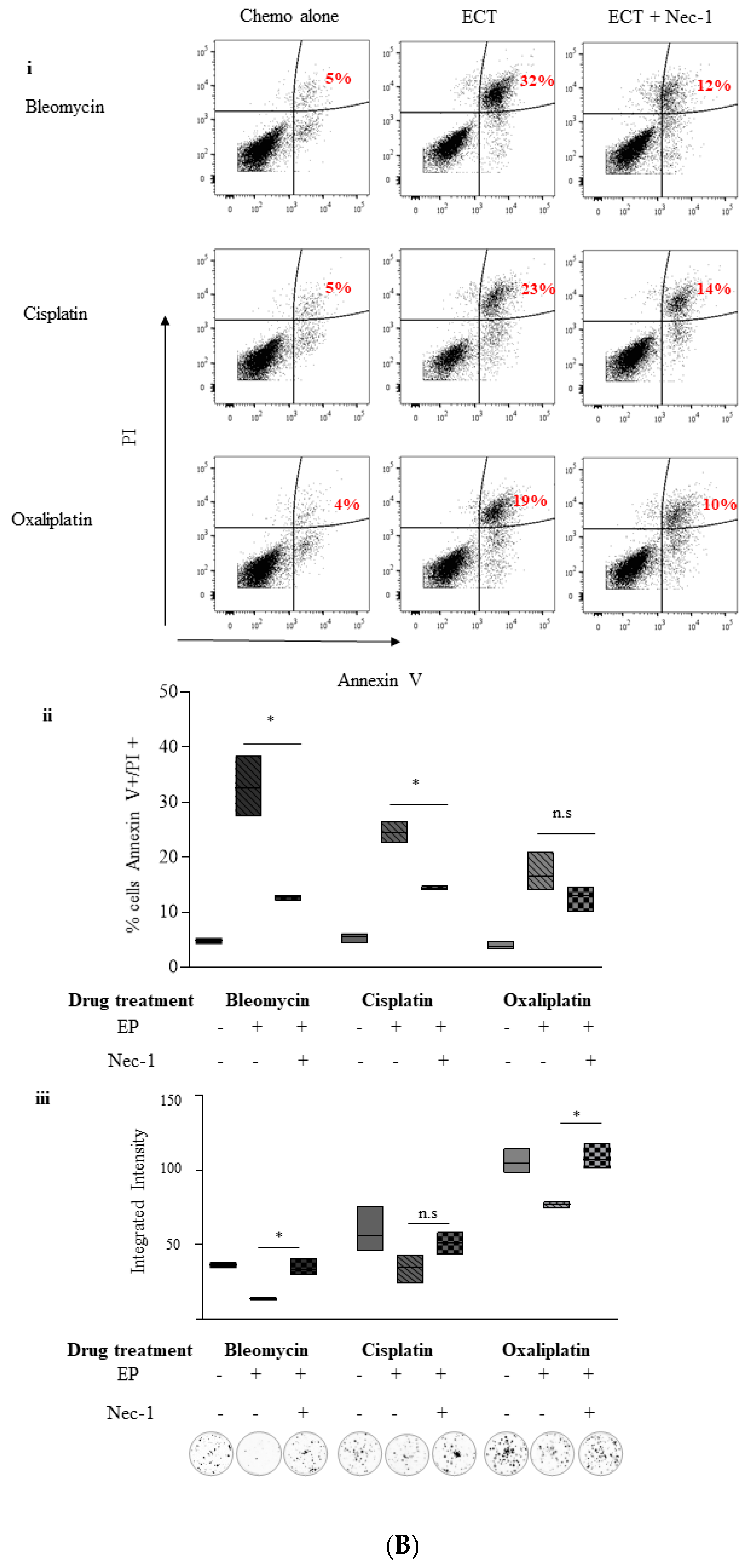
2.4. ECT-Induced Cell Death in Pancreatic Cancer Cells Can Be Rescued with Pre-Incubation with a Pan Caspase Inhibitor and Necrostatin-1
2.5. Inhibition of Necroptosis Combined with ECT Prevents Pancreatic Cell Death and Improves Recovery
2.5.1. Nec-1
2.5.2. Necrosulphonamide (NSA)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. PI Exclusion Assay to Measure Viability
4.3. Electroporation
4.4. Colony Formation Assay
4.5. Evaluation of Caspase-3 Activity
4.6. Evaluation of Morphology
4.7. Analysis of Phosphatidylserine (PS) Exposure Versus Cell Permeability by Flow Cytometry
4.8. Inhibitor Pre-Treatment
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sun, H.; Ma, H.; Hong, G.; Sun, H.; Wang, J. Survival improvement in patients with pancreatic cancer by decade: A period analysis of the SEER database, 1981–2010. Sci. Rep. 2014, 4, 6747. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Gehl, J. Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 2003, 177, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Kristoffersson, S.; Andersson, R.; Bergenfeldt, M. The role of irreversible electroporation (IRE) for locally advanced pancreatic cancer: A systematic review of safety and efficacy. Scand. J. Gastroenterol. 2017, 52, 1165–1171. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Wichtowski, M.; Murawa, D. Electrochemotherapy in the treatment of melanoma. Contemp. Oncol. 2018, 22, 8–13. [Google Scholar] [CrossRef]
- Hamacher, R.; Schmid, R.M.; Saur, D.; Schneider, G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol. Cancer 2008, 7, 64. [Google Scholar] [CrossRef]
- Muerkoster, S.S.; Lust, J.; Arlt, A.; Hasler, R.; Witt, M.; Sebens, T.; Schreiber, S.; Folsch, U.R.; Schafer, H. Acquired chemoresistance in pancreatic carcinoma cells: Induced secretion of IL-1beta and NO lead to inactivation of caspases. Oncogene 2006, 25, 3973–3981. [Google Scholar] [CrossRef]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.; Sersa, G.; Garbay, J.R.; Gehl, J.; Collins, C.G.; Snoj, M.; Billard, V.; Geertsen, P.F.; Larkin, J.O.; Miklavcic, D.; et al. Electrochemotherapy—An easy, highly effective and safe treatment of cutaneous and subcutaneous metastases: Results of ESOPE (European Standard Operating Procedures of Electrochemotherapy) study. EJC Suppl. 2006, 4, 3–13. [Google Scholar] [CrossRef]
- Maglietti, F.; Tellado, M.; Olaiz, N.; Michinski, S.; Marshall, G. Combined local and systemic bleomycin administration in electrochemotherapy to reduce the number of treatment sessions. Radiol. Oncol. 2016, 50, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Henry, C.M. Distinguishing between apoptosis, necrosis, necroptosis and other cell death modalities. Methods 2013, 61, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Wu, Y.H.; Levine, Z.A.; Gundersen, M.A.; Vernier, P.T. Water influx and cell swelling after nanosecond electropermeabilization. Biochim. Biophys. Acta 2013, 1828, 1715–1722. [Google Scholar] [CrossRef]
- Harkin, D.G.; Hay, E.D. Effects of electroporation on the tubulin cytoskeleton and directed migration of corneal fibroblasts cultured within collagen matrices. Cell Motil Cytoskeleton 1996, 35, 345–357. [Google Scholar] [CrossRef]
- Meulenberg, C.J.W.; Todorovic, V.; Cemazar, M. Differential cellular effects of electroporation and electrochemotherapy in monolayers of human microvascular endothelial cells. PLoS ONE 2012, 7, e52713. [Google Scholar] [CrossRef]
- Fryer, R.A.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar] [PubMed]
- Kim, M.H.; Chung, J.; Chung, S.M.; Kwag, N.H.; Yoo, J.S. Hydrogen peroxide-induced cell death in a human retinal pigment epithelial cell line, ARPE-19. Korean J. Ophthalmol. 2003, 17, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Hollville, E.; Martin, S.J. Measuring apoptosis by microscopy and flow cytometry. Methods 2013, 61, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Juan, J.; Li, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar]
- De Almagro, M.C.; Vucic, D. Necroptosis: Pathway diversity and characteristics. Semin. Cell Dev. Biol. 2015, 39, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Oliver, L.; Meflah, K.; Vallette, F.M. Caspase-3 can be pseudo-activated by a Ca2+-dependent proteolysis at a non-canonical site. FEBS Lett. 2005, 579, 2364–2368. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Ueno, A.; Saga, K.; Fukuzawa, M.; Kaneda, Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014, 74, 1056–1066. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef]
- Sawai, H.; Domae, N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem. Biophys. Res. Commun. 2011, 411, 569–573. [Google Scholar] [CrossRef]
- Lekshmi, A.; Varadarajan, S.N.; Lupitha, S.S.; Indira, D.; Mathew, K.A.; Nair, A.C.; Nair, M.; Prasad, T.; Sekar, H.; Gopalakrishnan, A.K.; et al. A quantitative real-time approach for discriminating apoptosis and necrosis. Cell Death Discov. 2017, 3, 16101. [Google Scholar] [CrossRef] [PubMed]
- Pietkiewicz, S.; Schmidt, J.H.; Lavrik, I.N. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical Annexin V/propidium iodide staining. J. Immunol. Methods 2015, 423, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Zhu, H.; Chow, S.C.; MacFarlane, M.; Nicholson, D.W.; Cohen, G.M. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 1996, 315 Pt 1, 21–24. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Grootjans, S.; Callewaert, N.; Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: Consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013, 20, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Sun, L.; Liu, W.; He, S.; Wang, X.; Lei, X. Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein. MedChemComm 2014, 3, 235–398. [Google Scholar] [CrossRef]
- Linecker, M.; Pfammatter, T.; Kambakamba, P.; DeOliveira, M.L. Ablation Strategies for Locally Advanced Pancreatic Cancer. Dig. Surg. 2016, 33, 351–359. [Google Scholar] [CrossRef]
- Probst, U.; Fuhrmann, I.; Beyer, L.; Wiggermann, P. Electrochemotherapy as a New Modality in Interventional Oncology: A Review. Technol. Cancer Res. Treat. 2018, 17, 1533033818785329. [Google Scholar] [CrossRef]
- Gehl, J.; Sersa, G.; Matthiessen, L.W.; Muir, T.; Soden, D.; Occhini, A.; Quaglino, P.; Curatolo, P.; Campana, L.G.; Kunte, C.; et al. Updated standard operating procedures for electrochemotherapy of cutaneous tumours and skin metastases. Acta Oncol. 2018, 57, 874–882. [Google Scholar] [CrossRef]
- Miklavcic, D.; Sersa, G.; Brecelj, E.; Gehl, J.; Soden, D.; Bianchi, G.; Ruggieri, P.; Rossi, C.R.; Campana, L.G.; Jarm, T. Electrochemotherapy: Technological advancements for efficient electroporation-based treatment of internal tumors. Med. Biol. Eng. Comput. 2012, 50, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Girelli, R.; Prejano, S.; Cataldo, I.; Corbo, V.; Martini, L.; Scarpa, A.; Claudio, B. Feasibility and safety of electrochemotherapy (ECT) in the pancreas: A pre-clinical investigation. Radiol. Oncol. 2015, 49, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Bimonte, S.; Leongito, M.; Granata, V.; Barbieri, A.; del Vecchio, V.; Falco, M.; Nasto, A.; Albino, V.; Piccirillo, M.; Palaia, R.; et al. Electrochemotherapy in pancreatic adenocarcinoma treatment: pre-clinical and clinical studies. Radiol Oncol. 2016, 50, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, H.; Shimizu, Y.; Ohsawa, Y.; Kawahara, A.; Uchiyama, Y.; Nagata, S. Necrotic death pathway in Fas receptor signaling. J. Cell Biol. 2000, 151, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Mompean, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, S.; Oppermann, J.; Reichert, S.; Fulda, S.; Rodel, C.; Rodel, F. The SMAC mimetic BV6 sensitizes colorectal cancer cells to ionizing radiation by interfering with DNA repair processes and enhancing apoptosis. Radiat. Oncol. 2015, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; da Silva, R.B.; e Sousa, C.R.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; McQuade, T.; Zhang, H.; Zhang, J.; Chan, F.K.-M. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS ONE 2011, 6, e23209. [Google Scholar] [CrossRef] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Saung, M.T.; Zheng, L. Current Standards of Chemotherapy for Pancreatic Cancer. Clin. Ther. 2017, 39, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (PANC-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Corbett, T.H.; Roberts, B.J.; Leopold, W.R.; Peckham, J.C.; Wilkoff, L.J.; Griswold, D.P., Jr.; Schabel, F.M., Jr. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984, 44, 717–726. [Google Scholar] [PubMed]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; DuHadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012, 3, e437. [Google Scholar] [CrossRef]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, P.; O’Donovan, T.R.; McKenna, S.L.; Forde, P.F. Electrochemotherapy Causes Caspase-Independent Necrotic-Like Death in Pancreatic Cancer Cells. Cancers 2019, 11, 1177. https://doi.org/10.3390/cancers11081177
Fernandes P, O’Donovan TR, McKenna SL, Forde PF. Electrochemotherapy Causes Caspase-Independent Necrotic-Like Death in Pancreatic Cancer Cells. Cancers. 2019; 11(8):1177. https://doi.org/10.3390/cancers11081177
Chicago/Turabian StyleFernandes, Philana, Tracey R. O’Donovan, Sharon L. McKenna, and Patrick F. Forde. 2019. "Electrochemotherapy Causes Caspase-Independent Necrotic-Like Death in Pancreatic Cancer Cells" Cancers 11, no. 8: 1177. https://doi.org/10.3390/cancers11081177
APA StyleFernandes, P., O’Donovan, T. R., McKenna, S. L., & Forde, P. F. (2019). Electrochemotherapy Causes Caspase-Independent Necrotic-Like Death in Pancreatic Cancer Cells. Cancers, 11(8), 1177. https://doi.org/10.3390/cancers11081177