EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells




Abstract
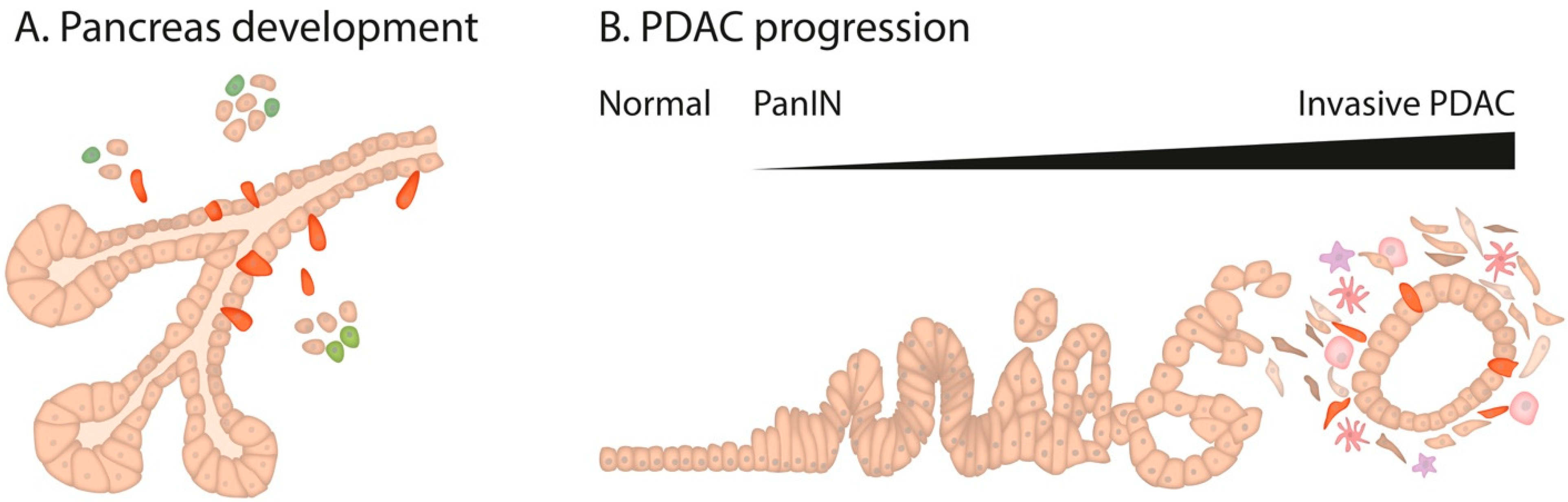
1. Pancreatic Development
2. Pancreatic Cancer
3. (Cancer) Stem Cells
4. EMT in Pancreatic Development and Cancer
5. EMT and CSCs
6. EMT, CTCs and Metastasis
7. EMT and Therapy Resistance
8. Future Directions
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slack, J.M. Developmental biology of the pancreas. Development 1995, 121, 1569–1580. [Google Scholar] [PubMed]
- Pan, F.C.; Brissova, M. Pancreas development in humans. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Kim, S.K.; Melton, D.A. Pancreas development is promoted by cyclopamine, a Hedgehog signaling inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 13036–13041. [Google Scholar] [CrossRef] [PubMed]
- Hebrok, M.; Kim, S.K.; Melton, D.A. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 1998, 12, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Pictet, R.L.; Clark, W.R.; Williams, R.H.; Rutter, W.J. An ultrastructural analysis of the developing embryonic pancreas. Dev. Biol. 1972, 29, 436–467. [Google Scholar] [CrossRef]
- Bürki, K.; Hagenbüchle, O.; Krapp, A.; Knöfler, M.; Ledermann, B.; Berney, C.; Zoerkler, N.; Wellauer, P.K. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998, 12, 3752–3763. [Google Scholar]
- Seymour, P.A.; Freude, K.K.; Tran, M.N.; Mayes, E.E.; Jensen, J.; Kist, R.; Scherer, G.; Sander, M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA 2007, 104, 1865–1870. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Cooper, B.; Gannon, M.; Ray, M.; Macdonald, R.J.; Wright, C.V. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 2002, 32, 128–134. [Google Scholar] [CrossRef]
- Apelqvist, Å.; Li, H.; Sommer, L.; Beatus, P.; Anderson, D.J.; Honjo, T.; De Angelis, M.H.; Lendahl, U.; Edlund, H. Notch signalling controls pancreatic cell differentiation. Nature 1999, 400, 877–881. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Hebrok, M. Control of Cell Identity in Pancreas Development and Regeneration. Gastroenterology 2013, 144, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Rooman, I.; Real, F.X. Pancreatic ductal adenocarcinoma and acinar cells: A matter of differentiation and development? Gut 2012, 61, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Van Der Sijde, F.; Vietsch, E.E.; Mustafa, D.A.M.; Besselink, M.G.; Koerkamp, B.G.; Van Eijck, C.H.J. Circulating Biomarkers for Prediction of Objective Response to Chemotherapy in Pancreatic Cancer Patients. Cancers (Basel) 2019, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Geer, R.J.; Brennan, M.F. Prognostic indicators for survival after resection of pancreatic adenocarcinoma. Am. J. Surg. 1993, 165, 68–73. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA A Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardiere, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef]
- Nowell, P. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Cancer stem cells as ’units of selection’. Evol. Appl. 2013, 6, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin. Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Bruce, W.R.; Van Der Gaag, H. A Quantitative Assay for the Number of Murine Lymphoma Cells capable of Proliferation in vivo. Nature 1963, 199, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M.; Lepelletier, Y.; et al. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genome Res. 2003, 17, 1253–1270. [Google Scholar] [CrossRef]
- Shi, X.; Gipp, J.; Bushman, W. Anchorage-Independent Culture Maintains Prostate Stem Cells. Dev. Biol. 2007, 312, 396–406. [Google Scholar] [CrossRef]
- Suzuki, A.; Oyama, K.; Fukao, K.; Nakauchi, H.; Taniguchi, H. Establishment of Clonal Colony-Forming Assay System for Pancreatic Stem/Progenitor Cells. Cell Transplant. 2002, 11, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Weissman, I.L.; Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L.; Reya, T.; Morrison, S.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Puri, S.; Folias, A.E.; Hebrok, M. Plasticity and dedifferentiation within the pancreas: Development, homeostasis, and disease. Cell Stem Cell 2015, 16, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Valles, A.; Haji, N.; De Gregorio, D.; Matta-Camacho, E.; Eslamizade, M.J.; Popic, J.; Sharma, V.; Cao, R.; Rummel, C.; Tanti, A.; et al. Translational control of depression-like behavior via phosphorylation of eukaryotic translation initiation factor 4E. Nat. Commun. 2018, 9, 2459. [Google Scholar] [CrossRef] [PubMed]
- De Haro-Hernández, R.; Cabrera-Muñoz, L.; Méndez, J.D. Regeneration of β-cells and neogenesis from small ducts or acinar cells promote recovery of endocrine pancreatic function in alloxan-treated rats. Arch. Med. Res. 2004, 35, 114–120. [Google Scholar] [CrossRef]
- Furuyama, K.; Chera, S.; Van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Zulewski, H.; Abraham, E.J.; Gerlach, M.J.; Daniel, P.B.; Moritz, W.; Müller, B.; Vallejo, M.; Thomas, M.K.; Habener, J.F. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001, 50, 521–533. [Google Scholar] [CrossRef]
- Kopp, J.L.; Dubois, C.L.; Schaffer, A.E.; Hao, E.; Shih, H.P.; Seymour, P.A.; Ma, J.; Sander, M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 2011, 138, 653–665. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar] [CrossRef]
- Beer, R.L.; Parsons, M.J.; Rovira, M. Centroacinar cells: At the center of pancreas regeneration. Dev. Biol. 2016, 413, 8–15. [Google Scholar] [CrossRef]
- Hayashi, K.; Takahashi, T.; Kakita, A.; Yamashina, S. Regional differences in the cellular proliferation activity of the regenerating rat pancreas after partial pancreatectomy. Arch. Histol. Cytol. 1999, 62, 337–346. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gasslander, T.; Ihse, I.; Smeds, S. The Importance of the Centroacinar Region in Cerulein-Induced Mouse Pancreatic Growth. Scand. J. Gastroenterol. 1992, 27, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.-M.; Koorstra, J.-B.; RajeshKumar, N.V.; He, X.; et al. Prognostic Significance of Tumorigenic Cells with Mesenchymal Features in Pancreatic Adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Adikrisna, R.; Tanaka, S.; Muramatsu, S.; Aihara, A.; Ban, D.; Ochiai, T.; Irie, T.; Kudo, A.; Nakamura, N.; Yamaoka, S.; et al. Identification of pancreatic cancer stem cells and selective toxicity of chemotherapeutic agents. Gastroenterology 2012, 143, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Lorenzo, I.; Dorado, J.; Lonardo, E.; Alcala, S.; Serrano, A.G.; Clausell-Tormos, J.; Cioffi, M.; Megias, D.; Zagorac, S.; Balic, A.; et al. Intracellular autofluorescence: A biomarker for epithelial cancer stem cells. Nat. Methods 2014, 11, 1161–1169. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Lightfoot, S.A.; Hoskins, A.B.; Lerner, M.; Brackett, D.J.; Postier, R.G.; Ramanujam, R.; Mohammed, A.; Rao, C.V.; et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011, 71, 2328–2338. [Google Scholar] [CrossRef]
- Wang, Z.; Park, H.J.; Carr, J.R.; Chen, Y.-J.; Zheng, Y.; Li, J.; Tyner, A.L.; Costa, R.H.; Bagchi, S.; Raychaudhuri, P. FoxM1 in Tumorigenicity of the Neuroblastoma Cells and Renewal of the Neural Progenitors. Cancer Res. 2011, 71, 4292–4302. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Meier, R.; Furuta, S.; Lenburg, M.E.; Kenny, P.A.; Xu, R.; Bissell, M.J. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J. Clin. Investig. 2012, 122, 3211–3220. [Google Scholar] [CrossRef]
- Lytle, N.K.; Ferguson, L.P.; Rajbhandari, N.; Gilroy, K.; Fox, R.G.; Deshpande, A.; Schürch, C.M.; Hamilton, M.; Robertson, N.; Lin, W.; et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 2019, 177, 572–586. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers 2019, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Suzuki, A.; Kawashimo, K.; Ishikawa, M.; Ohkohchi, N.; Taniguchi, H. Isolation of Mouse Pancreatic Ductal Progenitor Cells Expressing CD133 and c-Met by Flow Cytometric Cell Sorting. Gastroenterology 2007, 132, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Immervoll, H.; Hoem, D.; Steffensen, O.J.; Miletic, H.; Molven, A. Visualization of CD44 and CD133 in normal pancreas and pancreatic ductal adenocarcinomas: Non-overlapping membrane expression in cell populations positive for both markers. J. Histochem. Cytochem. 2011, 59, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Kayali, A.G.; Van Gunst, K.; Campbell, I.L.; Stotland, A.; Kritzik, M.; Liu, G.; Flodström-Tullberg, M.; Zhang, Y.-Q.; Sarvetnick, N. The stromal cell–derived factor-1α/CXCR4 ligand–receptor axis is critical for progenitor survival and migration in the pancreas. J. Cell Biol. 2003, 163, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, V.; Crisa, L.; Beattie, G.; Mally, M.; Lopez, A.; Fannon, A.; Ptasznik, A.; Inverardi, L.; Ricordi, C.; Deerinck, T.; et al. KSA Antigen Ep-CAM Mediates Cell-Cell Adhesion of Pancreatic Epithelial Cells: Morphoregulatory Roles in Pancreatic Islet Development. J. Cell Biol. 1998, 140, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Raanan, C.; Schreiber, L.; Polin, N.; Givol, D. LGR5 and Nanog identify stem cell signature of pancreas beta cells which initiate pancreatic cancer. Biochem. Biophys. Res. Commun. 2013, 433, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Dhayat, S.A.; Traeger, M.M.; Rehkaemper, J.; Stroese, A.J.; Steinestel, K.; Wardelmann, E.; Kabar, I.; Senninger, N. Clinical Impact of Epithelial-to-Mesenchymal Transition Regulating MicroRNAs in Pancreatic Ductal Adenocarcinoma. Cancers 2018, 10, 328. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zöller, M. Exosomes, metastases, and the miracle of cancer stem cell markers. Cancer Metastasis Rev. 2019, 38, 259–295. [Google Scholar] [CrossRef]
- Brandi, J.; Dando, I.; Pozza, E.D.; Biondani, G.; Jenkins, R.; Elliott, V.; Park, K.; Fanelli, G.; Zolla, L.; Costello, E.; et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J. Proteom. 2017, 150, 310–322. [Google Scholar] [CrossRef]
- Brandi, J.; Pozza, E.D.; Dando, I.; Biondani, G.; Robotti, E.; Jenkins, R.; Elliott, V.; Park, K.; Marengo, E.; Costello, E.; et al. Secretome protein signature of human pancreatic cancer stem-like cells. J. Proteom. 2016, 136, 1–12. [Google Scholar] [CrossRef]
- Engle, D.D.; Tiriac, H.; Rivera, K.D.; Pommier, A.; Whalen, S.; Oni, T.E.; Alagesan, B.; Lee, E.J.; Yao, M.A.; Lucito, M.S.; et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019, 364, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Hermann, P.C.; Heeschen, C. Pancreatic cancer stem cells – update and future perspectives. Mol. Oncol. 2010, 4, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; De Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Bez, A.; Corsini, E.; Curti, D.; Biggiogera, M.; Colombo, A.; Nicosia, R.F.; Pagano, S.F.; Parati, E.A. Neurosphere and neurosphere-forming cells: Morphological and ultrastructural characterization. Brain Res. 2003, 993, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Rovira, M.; Scott, S.G.; Liss, A.S.; Jensen, J.; Thayer, S.P.; Leach, S.D. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc. Natl. Acad. Sci. USA 2010, 107, 75–80. [Google Scholar] [CrossRef]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef]
- Vessoni, A.T.; Muotri, A.R.; Okamoto, O.K. Autophagy in Stem Cell Maintenance and Differentiation. Stem Cells Dev. 2012, 21, 513–520. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Ramos, E.B.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Houshmand, G.; Mishra, S.; Fong, G.-H.; Gittes, G.K.; Esni, F. Impaired pancreatic development in Hif2-alpha deficient mice. Biochem. Biophys. Res. Commun. 2010, 399, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2α regulates Oct-4: Effects of hypoxiaon stem cell function, embryonic development, and tumor growth. Genome Res. 2006, 20, 557–570. [Google Scholar]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The Hypoxia-Associated Factor Switches Cells from HIF-1α– to HIF-2α–Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumor heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sainz, B., Jr. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Biol. 2018, 53, 223–231. [Google Scholar] [CrossRef]
- Makohon-Moore, A.P.; Matsukuma, K.; Zhang, M.; Reiter, J.G.; Gerold, J.M.; Jiao, Y.; Sikkema, L.; Attiyeh, M.A.; Yachida, S.; Sandone, C.; et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 2018, 561, 201. [Google Scholar] [CrossRef]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef]
- Ball, C.R.; Oppel, F.; Ehrenberg, K.R.; Dubash, T.D.; Dieter, S.M.; Hoffmann, C.M.; Abel, U.; Herbst, F.; Koch, M.; Werner, J.; et al. Succession of transiently active tumor-initiating cell clones in human pancreatic cancer xenografts. EMBO Mol. Med. 2017, 9, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Li, C.-Y.; Ho, I.-L.; Corti, D.; Loponte, S.; Sapio, L.; Del Poggetto, E.; Yen, E.-Y.; Robinson, F.S.; Peoples, M.; et al. Pre-existing Functional Heterogeneity of Tumorigenic Compartment as the Origin of Chemoresistance in Pancreatic Tumors. Cell Rep. 2019, 26, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Greenburg, G. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nature 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Chiang, M.-K.; A Melton, D. Single-Cell Transcript Analysis of Pancreas Development. Dev. Cell 2003, 4, 383–393. [Google Scholar] [CrossRef]
- Gouzi, M.; Kim, Y.H.; Katsumoto, K.; Johansson, K.; Grapin-Botton, A.; Grapin-Botton, A. Neurogenin3 initiates stepwise delamination of differentiating endocrine cells during pancreas development. Dev. Dyn. 2011, 240, 589–604. [Google Scholar] [CrossRef]
- Reichert, M.; Takano, S.; Von Burstin, J.; Kim, S.-B.; Lee, J.-S.; Ihida-Stansbury, K.; Hahn, C.; Heeg, S.; Schneider, G.; Rhim, A.D.; et al. The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes Dev. 2013, 27, 288–300. [Google Scholar] [CrossRef]
- Scavuzzo, M.A.; Hill, M.C.; Chmielowiec, J.; Yang, D.; Teaw, J.; Sheng, K.; Kong, Y.; Bettini, M.; Zong, C.; Martin, J.F.; et al. Endocrine lineage biases arise in temporally distinct endocrine progenitors during pancreatic morphogenesis. Nat. Commun. 2018, 9, 3356. [Google Scholar] [CrossRef] [PubMed]
- Rukstalis, J.M.; Habener, J.F. Snail2, a mediator of epithelial-mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Expr. Patterns 2007, 7, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Serafimidis, I.; Rodriguez-Aznar, E.; Lesche, M.; Yoshioka, K.; Takuwa, Y.; Dahl, A.; Pan, D.; Gavalas, A. Pancreas lineage allocation and specification are regulated by sphingosine-1-phosphate signalling. PLoS Biol. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Loubat-Casanovas, J.; Pena, R.; Gonzalez, N.; Alba-Castellon, L.; Rosell, S.; Franci, C.; Navarro, P.; Garcia de Herreros, A. Snail1 is required for the maintenance of the pancreatic acinar phenotype. Oncotarget 2016, 7, 4468–4482. [Google Scholar] [CrossRef]
- Gershengorn, M.C.; Hardikar, A.A.; Wei, C.; Geras-Raaka, E.; Marcus-Samuels, B.; Raaka, B.M. Epithelial-to-Mesenchymal Transition Generates Proliferative Human Islet Precursor Cells. Science 2004, 306, 2261–2264. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, K.L.; Eshpeter, A.; Rajotte, R.V.; Korbutt, G.S. Epithelial cells within the human pancreas do not coexpress mesenchymal antigens: Epithelial-mesenchymal transition is an artifact of cell culture. Lab. Invest. 2009, 89, 110–121. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Creighton, C.J. Pan-cancer survey of epithelial-mesenchymal transition markers across the Cancer Genome Atlas. Dev. Dyn. 2018, 247, 555–564. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 Cooperatively Determine the Mammary Stem Cell State. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.-K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional Genomics Reveals a BMP-Driven Mesenchymal-to-Epithelial Transition in the Initiation of Somatic Cell Reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zon, L.I. Resolving the Controversy about N-Cadherin and Hematopoietic Stem Cells. Cell Stem Cell 2010, 6, 199–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, J.; Han, Q.; Peng, T.; Peng, M.; Wei, B.; Li, D.; Wang, X.; Yu, S.; Yang, J.; Cao, S.; et al. The oncogene c-Jun impedes somatic cell reprogramming. Nature 2015, 17, 856–867. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McCallister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nature 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nature 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Ding, Q.; Miyazaki, Y.; Tsukasa, K.; Matsubara, S.; Yoshimitsu, M.; Takao, S. CD133 facilitates epithelial-mesenchymal transition through interaction with the ERK pathway in pancreatic cancer metastasis. Mol. Cancer 2014, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Reichert, M.; Bakir, B.; Das, K.K.; Nishida, T.; Miyazaki, M.; Heeg, S.; Collins, M.A.; Marchand, B.; Hicks, P.D.; et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016, 30, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, O.H.; Córcoles, R.; Fabra, À.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Bajor, D.L.; Norgard, R.J.; Sahmoud, A.; Bhagwat, N.; Pham, M.N.; Cornish, T.C.; Iacobuzio-Donahue, C.A.; Vonderheide, R.H.; Stanger, B.Z. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 2016, 7, 12819. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Alcala, S.; Garcia, E.; Sanchez-Ripoll, Y.; Azevedo, M.M.; Cioffi, M.; Tatari, M.; Miranda-Lorenzo, I.; Hidalgo, M.; López, G.G.; et al. Microenvironmental hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by activating its cancer stem cell compartment. Gut 2015, 64, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Martín, B.; Tatari, M.; Heeschen, C.; Guerra, S.; Moreno, B. ISG15 Is a Critical Microenvironmental Factor for Pancreatic Cancer Stem Cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Carron, E.; Vallespinós, M.; Machado, H.L. Cancer Stem Cells and Macrophages: Implications in Tumor Biology and Therapeutic Strategies. Mediat. Inflamm. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sancho, P.; Cañamero, M.; Martinelli, P.; Madriles, F.; Michl, P.; Gress, T.; De Pascual, R.; Gandia, L.; Guerra, C.; et al. Nicotine Promotes Initiation and Progression of KRAS-Induced Pancreatic Cancer via Gata6-Dependent Dedifferentiation of Acinar Cells in Mice. Gastroenterology 2014, 147, 1119–1133.e4. [Google Scholar] [CrossRef]
- Poruk, K.E.; Blackford, A.L.; Weiss, M.J.; Cameron, J.L.; He, J.; Goggins, M.; Rasheed, Z.A.; Wolfgang, C.L.; Wood, L.D. Circulating Tumor Cells Expressing Markers of Tumor-Initiating Cells Predict Poor Survival and Cancer Recurrence in Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Rébillard, X.; Riethdorf, S.; Fabbro, M.; Pantel, K.; Alix-Panabières, C.; Pellé, O.; Vendrell, J.-P.; Müller, V. Detection and Characterization of Putative Metastatic Precursor Cells in Cancer Patients. Clin. Chem. 2007, 53, 537–539. [Google Scholar]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ’seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, T. A Case of Cancer in Which Cells Similar to Those in the Tumors Were Seen in the Blood after Death. Aust. Med. J. 1869, 14, 146–149. [Google Scholar]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Janni, W.J.; Rack, B.; Terstappen, L.W.M.M.; Pierga, J.-Y.; Taran, F.-A.; Fehm, T.; Hall, C.; De Groot, M.R.; Bidard, F.-C.; Friedl, T.W.; et al. Pooled Analysis of the Prognostic Relevance of Circulating Tumor Cells in Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 2583–2593. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.J. Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients with Nonmalignant Diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Bidard, F.; Ferrand, F.; Huguet, F.; Hammel, P.; Louvet, C.; Malka, D.; Boige, V.; Ducreux, M.; André, T.; De Gramont, A.; et al. Disseminated and circulating tumor cells in gastrointestinal oncology. Crit. Rev. Oncol. 2012, 82, 103–115. [Google Scholar] [CrossRef]
- Bidard, F.C.; Huguet, F.; Louvet, C.; Mineur, L.; Chibaudel, B.; Artru, P.; Desseigne, F.; Bachet, J.-B.; Pierga, J.Y.; Hammel, P.; et al. Circulating tumor cells in locally advanced pancreatic adenocarcinoma: The ancillary CirCe 07 study to the LAP 07 trial. Ann. Oncol. 2013, 24, 2057–2061. [Google Scholar] [CrossRef]
- Ankeny, J.S.; Court, C.M.; Hou, S.; Li, Q.; Song, M.; Wu, D.; Chen, J.F.; Lee, T.; Lin, M.; Sho, S.; et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br. J. Cancer 2016, 114, 1367–1375. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.; Li, X.-Y.; Lü, N.; An, T.; Liu, Z.-P.; Fu, R.; Lv, W.-C.; Zhang, Y.-W.; Xu, X.-J.; Rowe, R.G.; et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nature 2016, 18, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Lapin, M.; Tjensvoll, K.; Oltedal, S.; Javle, M.; Smaaland, R.; Gilje, B.; Nordgård, O. Single-cell mRNA profiling reveals transcriptional heterogeneity among pancreatic circulating tumour cells. BMC Cancer 2017, 17, 390. [Google Scholar] [CrossRef] [PubMed]
- Stott, S.L.; Hsu, C.-H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nature 2012, 14, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef]
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Jordan, N.V.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-Cell RNA Sequencing Identifies Extracellular Matrix Gene Expression by Pancreatic Circulating Tumor Cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev. Cell 2018, 45, 696–711.e8. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Handler, J.; Cullis, J.; Avanzi, A.; Vucic, E.A.; Bar-Sagi, D. Pre-neoplastic pancreas cells enter a partially mesenchymal state following transient TGF-β exposure. Oncogene 2018, 37, 4334–4342. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; LeBleu, V.S.; Carstens, J.L.; Sugimoto, H.; Zheng, X.; Malasi, S.; Saur, D.; Kalluri, R. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Context-specific roles of EMT programmes in cancer cell dissemination. Nature 2017, 19, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial/mesenchymal plasticity: How have quantitative mathematical models helped improve our understanding? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed]
- A Galván, J.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nature 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.A.; Ebine, K.; Sahai, V.; Kumar, K.; Siddiqui, K.; Hwang, R.F.; Grippo, P.J.; Munshi, H.G. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol. Cancer Res. 2013, 11, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Awaji, M.; Singh, R.K. Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancers (Basel) 2019. [Google Scholar] [CrossRef] [PubMed]
- Messal, H.A.; Alt, S.; Ferreira, R.M.M.; Gribben, C.; Wang, V.M.-Y.; Cotoi, C.G.; Salbreux, G.; Behrens, A. Tissue curvature and apicobasal mechanical tension imbalance instruct cancer morphogenesis. Nature 2019, 566, 126–130. [Google Scholar] [CrossRef]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nature 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018. [Google Scholar] [CrossRef]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016. [Google Scholar] [CrossRef]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.A.; Peeper, D.S. Deregulating EMT and Senescence: Double Impact by a Single Twist. Cancer Cell 2008, 14, 5–7. [Google Scholar] [CrossRef]
- Baygi, M.E.; Soheili, Z.S.; Schmitz, I.; Sameie, S.; Schulz, W.A. Snail regulates cell survival and inhibits cellular senescence in human metastatic prostate cancer cell lines. Cell Biol. Toxicol. 2010, 26, 553–567. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, J.; Feng, H.; Peng, S.; Adams, U.; Bai, Y.; Huang, L.; Li, J.; Huang, J.; Meng, S.; et al. YAP/TEAD-Mediated Transcription Controls Cellular Senescence. Cancer Res. 2013, 73, 3615–3624. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Marchand, B.; Pitarresi, J.R.; Reichert, M.; Suzuki, K.; Laczkó, D.; Rustgi, A.K. PRRX1 isoforms cooperate with FOXM1 to regulate the DNA damage response in pancreatic cancer cells. Oncogene 2019, 38, 4325–4339. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of Epithelial-Mesenchymal Transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of Notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Cao, E.Y.; Kumar, V.; Zhang, X.; Leong, H.S.; Wong, A.M.L.; Ramakrishnan, N.; Hakimullah, M.; Teo, H.M.V.; Chong, F.T.; et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat. Commun. 2018, 9, 4931. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Wojtowicz, K.; Zabel, M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed. Pharmacother. 2013, 67, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Gallmeier, E.; Hermann, P.C.; Mueller, M.T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 2011, 29, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; Mc Dermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.-I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–167. [Google Scholar] [CrossRef]
- D’Errico, G.; Alonso-Nocelo, M.; Vallespinos, M.; Hermann, P.C.; Alcalá, S.; García, C.P.; Martin-Hijano, L.; Valle, S.; Earl, J.; Cassiano, C.; et al. Tumor-associated macrophage-secreted 14-3-3ζ signals via AXL to promote pancreatic cancer chemoresistance. Oncogene 2019, 38, 5469–5485. [Google Scholar] [CrossRef]
- Gittes, G.K. Developmental biology of the pancreas: A comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Belmonte, J.C.I. Stem Cells: A Renaissance in Human Biology Research. Cell 2016, 165, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Hermann, P.C.; Witthauer, J.; Rubio–Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined Targeted Treatment to Eliminate Tumorigenic Cancer Stem Cells in Human Pancreatic Cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Trabulo, S.M.; Sainz, B.; Balić, A.; Garcia, E.; Hahn, S.A.; Vandana, M.; Sahoo, S.K.; Tunici, P.; Bakker, A.; et al. Multimodal Treatment Eliminates Cancer Stem Cells and Leads to Long-Term Survival in Primary Human Pancreatic Cancer Tissue Xenografts. PLoS ONE 2013, 8, e66371. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y.; Yu, Z.; Zyromski, N.; Schipani, E.; Carta, F.; et al. Regulation of HIF1α under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual-Targeting in Patient-Derived 3D Pancreatic Cancer Models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef] [PubMed]
- Firuzi, O.; Che, P.P.; El Hassouni, B.; Buijs, M.; Coppola, S.; Lohr, M.; Funel, N.; Heuchel, R.; Carnevale, I.; Schmidt, T.; et al. Role of c-MET Inhibitors in Overcoming Drug Resistance in Spheroid Models of Primary Human Pancreatic Cancer and Stellate Cells. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdes, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genome Res. 2004, 18, 1131–1143. [Google Scholar] [CrossRef]
- Wu, W.-S.; Heinrichs, S.; Xu, D.; Garrison, S.P.; Zambetti, G.P.; Adams, J.M.; Look, A.T. Slug Antagonizes p53-Mediated Apoptosis of Hematopoietic Progenitors by Repressing puma. Cell 2005, 123, 641–653. [Google Scholar] [CrossRef]
- Inoue, A.; Seidel, M.G.; Wu, W.; Kamizono, S.; Ferrando, A.A.; Bronson, R.T.; Iwasaki, H.; Akashi, K.; Morimoto, A.; Hitzler, J.K.; et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002, 2, 279–288. [Google Scholar] [CrossRef]
- Saxena, M.; A Stephens, M.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Guo, X.; Qian, X.; Wang, H.; Yang, C.; Brinkman, K.L.; Serrano-Gonzalez, M.; Jope, R.S.; Zhou, B.; Engler, D.A.; et al. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J. Mol. Cell Biol. 2012, 4, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nature 2014, 16, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Redon, C.E.; Sethi, T.K.; Burrell, A.S.; Jailwala, P.; Kasoji, M.; Abrams, N.; Merchant, A.; Bonner, W.M. Twist1 and Slug mediate H2AX-regulated epithelial-mesenchymal transition in breast cells. Cell Cycle 2016, 15, 2398–2404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pieraccioli, M.; Nicolai, S.; Antonov, A.; Somers, J.; Malewicz, M.; Melino, G.; Raschella, G. ZNF281 contributes to the DNA damage response by controlling the expression of XRCC2 and XRCC4. Oncogene 2016, 35, 2592–2601. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; A Mathot, R.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Tan, A.C.; Uson, M.; Angenendt, M.; Ma, W.W.; Villaroel, M.C.; Zhao, M.; RajeshKumar, N.V.; Jimeno, A.; Donehower, R.; et al. Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br. J. Cancer 2010, 103, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.-T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Aykut, B.; Teske, C.; Giese, N.A.; Weitz, J.; Welsch, T.; Giese, N.N. Salinomycin inhibits growth of pancreatic cancer and cancer cell migration by disruption of actin stress fiber integrity. Cancer Lett. 2015, 358, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.; Mollenhauer, J.; Brune, K. Selective growth inhibition of ductal pancreatic adenocarcinoma cells by the lysosomotropic agent chloroquine. Cancer Lett. 1989, 44, 61–66. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Balic, A.; Trabulo, S.M.; Sainz, B.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; Heeschen, C.; et al. Chloroquine Targets Pancreatic Cancer Stem Cells via Inhibition of CXCR4 and Hedgehog Signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Daemen, A.; Peterson, D.; Sahu, N.; Mccord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef]
- Su, H.-T.; Weng, C.-C.; Hsiao, P.-J.; Chen, L.-H.; Kuo, T.-L.; Cheng, K.-H. Stem Cell Marker Nestin Is Critical for TGF-β1-Mediated Tumor Progression in Pancreatic Cancer. Mol. Cancer Res. 2013, 11, 768–779. [Google Scholar] [CrossRef]
- Liu, L.; Salnikov, A.V.; Bauer, N.; Aleksandrowicz, E.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Schemmer, P.; Werner, J.; et al. Triptolide reverses hypoxia-induced epithelial-mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. Int. J. Cancer 2014, 134, 2489–2503. [Google Scholar] [CrossRef]
- Yen, W.-C.; Fischer, M.M.; Axelrod, F.; Bond, C.; Cain, J.; Cancilla, B.; Henner, W.R.; Meisner, R.; Sato, A.; Shah, J.; et al. Targeting Notch Signaling with a Notch2/Notch3 Antagonist (Tarextumab) Inhibits Tumor Growth and Decreases Tumor-Initiating Cell Frequency. Clin. Cancer Res. 2015, 21, 2084–2095. [Google Scholar] [CrossRef]
- Verma, R.K.; Yu, W.; Shrivastava, A.; Shankar, S.; Srivastava, R.K. α-Mangostin-encapsulated PLGA nanoparticles inhibit pancreatic carcinogenesis by targeting cancer stem cells in human, and transgenic (KrasG12D, and KrasG12D/tp53R270H) mice. Sci. Rep. 2016, 6, 32743. [Google Scholar] [CrossRef] [PubMed]
- Kirane, A.; Toombs, J.E.; Ostapoff, K.; Carbon, J.G.; Zaknoen, S.; Braunfeld, J.; Schwarz, R.E.; Burrows, F.J.; Brekken, R.A. Apricoxib, a Novel Inhibitor of COX-2, Markedly Improves Standard Therapy Response in Molecularly Defined Models of Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 5031–5042. [Google Scholar] [CrossRef] [PubMed]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol. Cancer 2013, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor β receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A Specific Inhibitor of TGF-β Receptor Kinase, SB-431542, as a Potent Antitumor Agent for Human Cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Al-Ismaeel, Q.; Neal, C.P.; Al-Mahmoodi, H.; Almutairi, Z.; Al-Shamarti, I.; Straatman, K.; Jaunbocus, N.; Irvine, A.; Issa, E.; Moreman, C.; et al. ZEB1 and IL-6/11-STAT3 signalling cooperate to define invasive potential of pancreatic cancer cells via differential regulation of the expression of S100 proteins. Br. J. Cancer 2019, 121, 65–75. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, Y.-Z.; Cheung, M.; Cao, M.; Yu, C.; Chen, L.; Zhan, L.; He, Z.-W.; Sun, C.-Y. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway. Cell Death Dis. 2019, 10, 230. [Google Scholar] [CrossRef]
- Walter, K.; Tiwary, K.; Trajkovic-Arsic, M.; Hidalgo-Sastre, A.; Dierichs, L.; Liffers, S.T.; Gu, J.; Gout, J.; Schulte, L.A.; Münch, J.; et al. MEK Inhibition Targets Cancer Stem Cells and Impedes Migration of Pancreatic Cancer Cells. Stem Cells Int. 2019, 2019, 8475389. [Google Scholar] [CrossRef]
- Dent, P.; Booth, L.; Roberts, J.L.; Liu, J.; Poklepovic, A.; Lalani, A.S.; Tuveson, D.; Martinez, J.; Hancock, J.F. Neratinib inhibits Hippo/YAP signaling, reduces mutant K-RAS expression, and kills pancreatic and blood cancer cells. Oncogene 2019, 38, 5890–5904. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| EMT Regulators in Embryonic Development of the Pancreas | |||
|---|---|---|---|
| Gene | Assay | Description | Ref. |
| Snai1 | expression pattern | in situ hybridization | [99] |
| expression | single cell RNAseq | [101] | |
| Snai2 | expression pattern | in situ hybridization, immunofluorescence | [99,102] |
| expression | single cell RNAseq | [101] | |
| functional analysis | Snail2 electroporation promotes cell delamination | [99] | |
| Prrx1 | expression | microarray | [100] |
| Twist | expression | single cell RNAseq | [101] |
| YAP | expression pattern | Immunofluorescence | [104] |
| functional analysis | Conditional KO shows reduction in endocrine cells | [104] | |
| E-cadherin/Vimentin | expression | Coexpression in Ngn3+ cells shown by single-cell PCR | [98] |
| EMT Regulators in PDAC progression | |||
| Gene | Assay | Description | Ref. |
| Snai1 | Conditional KO in KPC mice | Dispensable for metastasis, promotes chemoresistance | [156] |
| Expression by qPCR, WB | Upregulation of Snai1 in Panc1 spheres enriches for stemness markers | [122] | |
| Snai2 | Cd133 KD in Capan1 M9 cells | Stemness marker CD133 regulates Snail2 expression | [121] |
| Twist | conditional KO in KPC mice | Dispensable for metastasis, promotes chemoresistance | [156] |
| Zeb1 | expression in KPCY mice | Present in CTCs | [118] |
| KD in human cells | Promotes chemoresistance | [181] | |
| Conditional KO in KPC mice | Critical for stemness and metastasis | [120] | |
| Prrx1 | OE and KD | Differential isoform regulation of Sox9-mediated stemness | [100] |
| Inducible OE in KPC mice | Isoform switching regulates EMT states (delamination & metastasis) | [123] | |
| OE in human cells | Limits DNA damage | [182] | |
| Fsp1/aSMA | Lineage tracing in KPC mice | Do not contribute to metastasis | [157] |
| YAP | KD in human cells | YAP1 amplification can promote KRas independent recurrence | [183] |
| EMT signature | Notch KD in human cells | Notch signaling promotes EMT-mediated chemoresistance | [184] |
| Patient CTCs | Single-cell qPCR showed enrichment in mesenchymal markers | [143] | |
| KCYp120ctnwt/+ mice | Mono-allelic p120ctn loss shifts metastatic burden to the lung | [151] | |
| KPCY mice | Tumor cells retain E-cadherin transcripts during EMT | [162] | |
| KPC mouse single-cell RNAseq | Stem cells and mesenchymal signatures show overlap | [60] | |
| Patient-derived cells | EMT signature contributes to metastasis and chemoresistance | [172] | |
| Drugs Used for Targeting EMT in PDAC | ||||
|---|---|---|---|---|
| Type of Drug | Compound | Target | Description | Ref. |
| Epigenetic | Mocetinostat | HDAC | HDAC I inhibitor restores miR-203 expression to downregulate Zeb1 | [212] |
| Antibiotic | Salinomycin | RhoA | Loss of actin stress fibers and reduced metastasis | [213] |
| Anti-malaria | Chloroquine | Autophagy | Blockade of autophagy | [214,215] |
| EMT | CXCR4 and hedgehog signaling inhibition with subsequent EMT inhibition | [216] | ||
| Anti-diabetic | Metformin | OXPHOS | Inhibition of CSCs by Gata6 upregulation and decrease of Snail1 | [130] |
| Metabolism | Glycolytic and Glutaminolytic Inhibitors | EMT signature | Correlation of EMT signature with sensitivity to specific inhibitors | [217] |
| Natural compound | Withaferin-A | Nestin | Suppression of metastasis | [218] |
| Triptolide | NF-kB | Inhibition of hypoxia and Twist2 induced stem-like features | [219] | |
| Monoclonal antibody | Tarextumab | Notch2/3 | Reversed Snail and Twist upregulation mediated by Gemcitabine | [220] |
| Nanoparticles | Mangostin | SHH | Downregulation of Snail, Slug, Zeb1 and N-cadherin | [221] |
| Small molecule inhibitors | Apricoxib | COX2 | Reverses EMT | [222] |
| Erlotinib | EGFR | Suppresses cancer metastasis | [223] | |
| cyclopamine | SHH | Inhibits Snai1 mediated EMT | [224] | |
| LY2109761 | TGFbRI/II | Suppresses cancer metastasis | [225] | |
| SB-431542 | TRKI | Attenuates TGF-beta-induced EMT | [226] | |
| VS-4718 | FAK | Reduction of ALDH and CD44 together with metastasis | [227] | |
| Stattic | STAT3 | Reduced migration and invasion | [228] | |
| LY294002 | PI3K/Akt | Decreased expression of vimentin, Snail1 and Snail2 | [229] | |
| PD0325901 | MEK | Attenuates TGF-beta-induced EMT | [230] | |
| Neratinib | ERBB1/2/4 | Translocation of YAP to the cytosol | [231] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez-Aznar, E.; Wiesmüller, L.; Sainz, B., Jr.; Hermann, P.C. EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. https://doi.org/10.3390/cancers11081136
Rodriguez-Aznar E, Wiesmüller L, Sainz B Jr., Hermann PC. EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells. Cancers. 2019; 11(8):1136. https://doi.org/10.3390/cancers11081136
Chicago/Turabian StyleRodriguez-Aznar, Eva, Lisa Wiesmüller, Bruno Sainz, Jr., and Patrick C. Hermann. 2019. "EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells" Cancers 11, no. 8: 1136. https://doi.org/10.3390/cancers11081136
APA StyleRodriguez-Aznar, E., Wiesmüller, L., Sainz, B., Jr., & Hermann, P. C. (2019). EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells. Cancers, 11(8), 1136. https://doi.org/10.3390/cancers11081136